
Complex Roles of NEIL1 and OGG1: Insights Gained from Murine Knockouts and Human Polymorphic Variants

Oregon Institute of Occupational Health Sciences, Department of Molecular and Medical Genetics, Oregon Health & Science University, Portland, OR 97239, USA
DNA 2022, 2(4), 279-301; https://doi.org/10.3390/dna2040020
Submission received: 2 October 2022
/
Revised: 17 November 2022
/
Accepted: 17 November 2022
/
Published: 1 December 2022
(This article belongs to the Special Issue From Mutation and Repair to Therapeutics)
Abstract
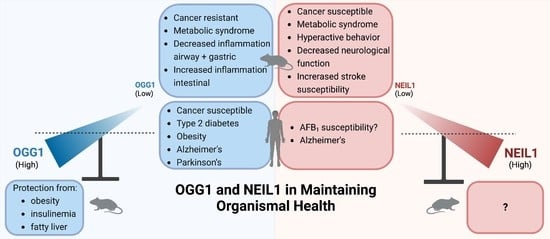
:DNA glycosylases promote genomic stability by initiating base excision repair (BER) in both the nuclear and mitochondrial genomes. Several of these enzymes have overlapping substrate recognition, through which a degree of redundancy in lesion recognition is achieved. For example, OGG1 and NEIL1 both recognize and release the imidazole-ring-fragmented guanine, FapyGua as part of a common overall pathway to cleanse the genome of damaged bases. However, these glycosylases have many differences, including their differential breadth of substrate specificity, the contrasting chemistries through which base release occurs, the subsequent steps required to complete the BER pathway, and the identity of specific protein-binding partners. Beyond these differences, the complexities and differences of their in vivo biological roles have been primarily elucidated in studies of murine models harboring a knockout of Neil1 or Ogg1, with the diversity of phenotypic manifestations exceeding what might have been anticipated for a DNA glycosylase deficiency. Pathologies associated with deficiencies in nuclear DNA repair include differential cancer susceptibilities, where Ogg1-deficient mice are generally refractory to carcinogenesis, while deficiencies in Neil1-deficient mice confer cancer susceptibility. In contrast to NEIL1, OGG1 functions as a key transcription factor in regulating inflammation and other complex gene cascades. With regard to phenotypes attributed to mitochondrial repair, knockout of either of these genes results in age- and diet-induced metabolic syndrome. The adverse health consequences associated with metabolic syndrome can be largely overcome by expression of a mitochondrial-targeted human OGG1 in both wild-type and Ogg1-deficient mice. The goal of this review is to compare the roles that NEIL1 and OGG1 play in maintaining genomic integrity, with emphasis on insights gained from not only the diverse phenotypes that are manifested in knockout and transgenic mice, but also human disease susceptibility associated with polymorphic variants.
{kind=link}
{kind=link}
{kind=link}
1. Introduction
The identification of enzymes associated with the initiation of BER of oxidatively induced DNA damage was first established in Escherichia coli, comprising formamidopyrimidine DNA glycosylase (Fpg), endonuclease III (Nth1), endonuclease VIII (Nei), and adenine:guanine DNA glycosylase (MutY) (reviewed in [1,2]). In humans, three genes have been described for encoding NEI-like glycosylases (NEIL) (reviewed in [3]), and their initial characterizations were concurrently described in multiple laboratories [4,5,6,7]. The first eukaryotic functional equivalent of E. coli Fpg was discovered by functional complementation of a strain deficient in both Fpg and MutY. This strategy identified the Saccharomyces cerevisiae OGG1 gene (yOGG1), which facilitated the initial biochemical characterization of this enzyme [8]. Building on this discovery and characterization, the human BER enzyme that initiates repair of 8-oxoG (hOGG1) was concurrently reported in multiple laboratories [9,10,11,12,13,14,15]. This review focuses on in vitro investigations of human NEIL1 and OGG1, as well as in vivo phenotypic consequences following knockout of murine Neil1 and Ogg1, including organelle-specific complementation. When possible, extrapolations are made toward disease implications associated with deficiencies in human OGG1 and NEIL1.
2. Target Site Location and Substrate Recognition
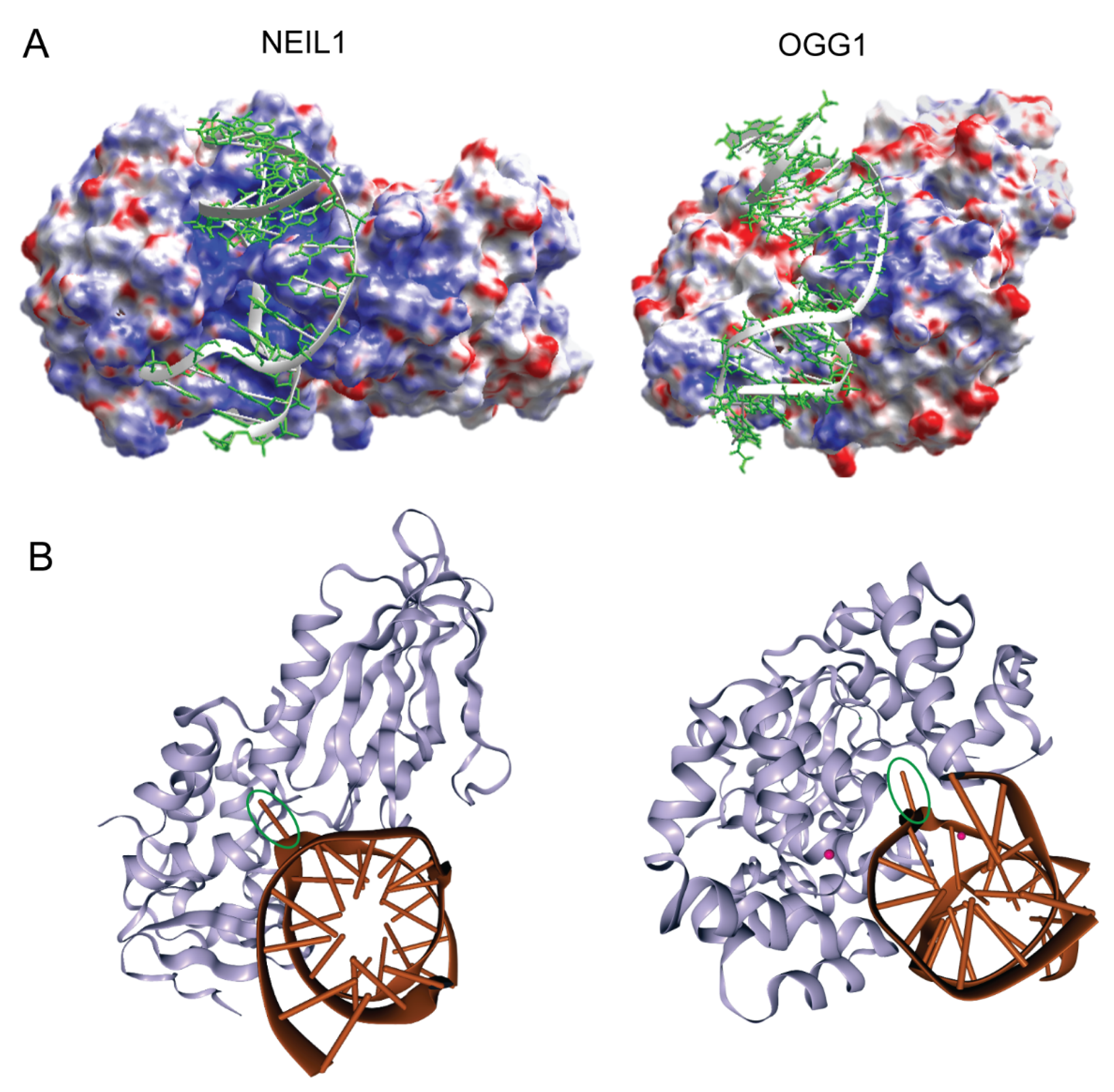
As is common among DNA glycosylases, X-ray-derived crystal structures of both the apoenzymes and the DNA co-crystal complexes of NEIL1 and OGG1 reveal that these relatively low-molecular-weight proteins share an asymmetric surface charge distribution through which nonspecific DNA binding and processivity are achieved (Figure 1A) [16,17,18,19,20,21,22,23]. This differential surface charge distribution is believed to guide short-range enzyme–DNA orientation geometries, leading to productive—albeit transient—nonspecific DNA binding. Once properly oriented on DNA, non-directional sliding along DNA provides the context through which alterations in base structure and helical stability can be detected through partial nucleotide flipping. This mechanism of nonspecific DNA sliding and the biological importance of this nonspecific binding was first described for the DNA glycosylase known as T4 pyrimidine dimer glycosylase (T4-pdg) [24,25,26,27], and it has subsequently been described for many DNA glycosylases (reviewed in [28,29,30,31,32]). Moreover, as revealed by various biophysical techniques and structure–function analyses, these enzymes create a flexible active site into which damaged nucleotides can be flipped and accommodated between globular and active site-forming domains (Figure 1B) [17,19,20,22,23]. Specific binding is achieved through this complex set of molecular motions, bringing active site residues in close proximity to the damage-containing nucleotides to initiate the chemistry of glycosyl bond scission. The specific chemical mechanisms for base release generally dictate whether a glycosylase will only catalyze base release, leaving an abasic (AP) site with the phosphodiester backbone intact, or whether both base release and phosphodiester incision are mechanistically coupled. These are discussed in the following section, in which the chemical mechanisms of OGG1 and NEIL1 are contrasted.
Investigations reporting the initial cloning, expression, and characterization of NEIL1, NEIL2, and NEIL3 generally utilize synthetic oligodeoxynucleotides containing site-specific base damage—including dihydrouracil, thymine glycol, and other saturated pyrimidines—to identify substrate specificities of NEIL1 [4,5,6,7]. Based on data demonstrating that NEIL1 expression was correlated well with the S-phase of the cell cycle [5,35,36], it was hypothesized that it may possess activity at sites of stalled replication, as modelled through DNA bubble structures. In agreement with this hypothesis, in vitro biochemical studies demonstrated that NEIL1 did not require fully double-stranded DNAs as substrates, but that it can catalyze incision on substrates located within designed bubble or forked structures as well as single-stranded DNA [37,38,39,40,41,42,43]. In some cases, rates of incision were accelerated using these substrate designs. NEIL1’s association with proteins associated with replication complexes is described below in Section 4. Although these investigations using synthetic oligodeoxynucleotides provide a framework from which to evaluate the base specificity of NEIL1, investigations by Dr. Miral Dizdaroglu and collaborators have generated the most comprehensive insights into the specificity of NEIL1 by using DNA damaged with γ-rays followed by GC/MS analyses [5,44,45,46]. This experimental design measures base release following incubation of purified NEIL1 with irradiated, high-molecular-weight genomic DNA. These data reveal that, in addition to various saturated pyrimidines—including thymine glycol, 5-OH-cytosine, 5,6-dihydrouracil, and 5-OH uracil—the most preferred substrates for NEIL1 are the two ring-fragmented purines 2-6-diamino-4-hydroxy-5-formamidopyrimidine (FapyG) and 4,6-diamino-5-formamidopyrimidine (FapyAde). In subsequent years, NEIL1 has been shown to recognize and incise DNA at all tested alkyl-modified Fapy-dG substrates, including methyl Fapy-dG, nitrogen mustard Fapy-dG, and aflatoxin B1 (AFB1)-Fapy-dG [43,46,47,48,49]. In addition to ring-fragmented and alkyl-FapyGua purines, a number of studies have demonstrated that the DNA substrates with the highest catalytic efficiencies for NEIL1 are the secondary oxidation products of 8-oxodG—spiroiminohydantoin (Sp) and 5-guanidinohydantoin (Gh)—as well as a variety of Sp-amine adducts, including Sp-lysine, Sp-glucosamine (GlcN), and tetra- and pentapeptides [40,41,47,50,51,52,53]. NEIL1 also catalyzes the incision of DNA containing 5-carboxylcytosine as part of a substrate redundancy for thymine DNA glycosylase (TDG), in a repair process associated with active DNA demethylation [54]. Furthermore, NEIL1 has also been demonstrated to initiate repair of a variety of psoralen-induced interstrand DNA crosslinks [55,56,57].
In contrast to the extremely broad substrate range of NEIL1, to date, the only ionizing-radiation-induced base damages recognized by OGG1 are 7,8-dihydro-8-oxo-2′-guanine (8-oxoGua) and FapyGua [5,58,59]. The initial characterizations of human OGG1 demonstrated that there was a clear preference for the nucleotide opposite 8-oxo-dG, with near-exclusive incision when a dC was opposite the site of the lesion [9,10,11,12,13,14,15]. Given this dominant base-pair recognition preference, it is anticipated that OGG1 will not incise damage within single-stranded DNAs. Furthermore, rates of incision of synthetic oligodeoxynucleotides vary greatly depending on sequence context, methylation status, and the presence of mismatched nucleotides [60]. The substrate specificity of OGG1 can be modulated by carrying out reactions in the presence of the AP endonuclease APEX1, where OGG1 can release 8-oxoAde, 8-oxoIno, and 6-methoxy-8-oxoGua [61]. Since NEIL1 does not catalyze the release of 8-oxoGua, these enzymes only share a single substrate in common: FapyGua. Measurements of the catalytic efficiencies of the removal of FapyGua from duplex DNA are comparable: 9.3 ± 0.6 × 10−5 and 9.0 ± 0.2 × 10−5 min−1 mmol−1 L for NEIL1 and OGG1, respectively [45,62,63].
Overall, although NEIL1 and OGG1 only share a single common substrate, their combined spectrum of recognition includes the most abundant lesions and covers a major portion of the total oxidatively induced base damage, as can be inferred from the data using ionizing radiation as a source of oxidation. However, even though this review focuses on these two glycosylases, human cells have multiple DNA glycosylases that provide built-in redundancy in the recognition and repair of DNA base damage, including NEIL2, NEIL3, and NTH1. A rigorous analysis of the overlapping substrate specificities of these glycosylases, as well as several non-human glycosylases, has previously been reviewed [45].
3. Mechanisms of Chemical Catalysis with Implications for the Completion of the BER Pathway
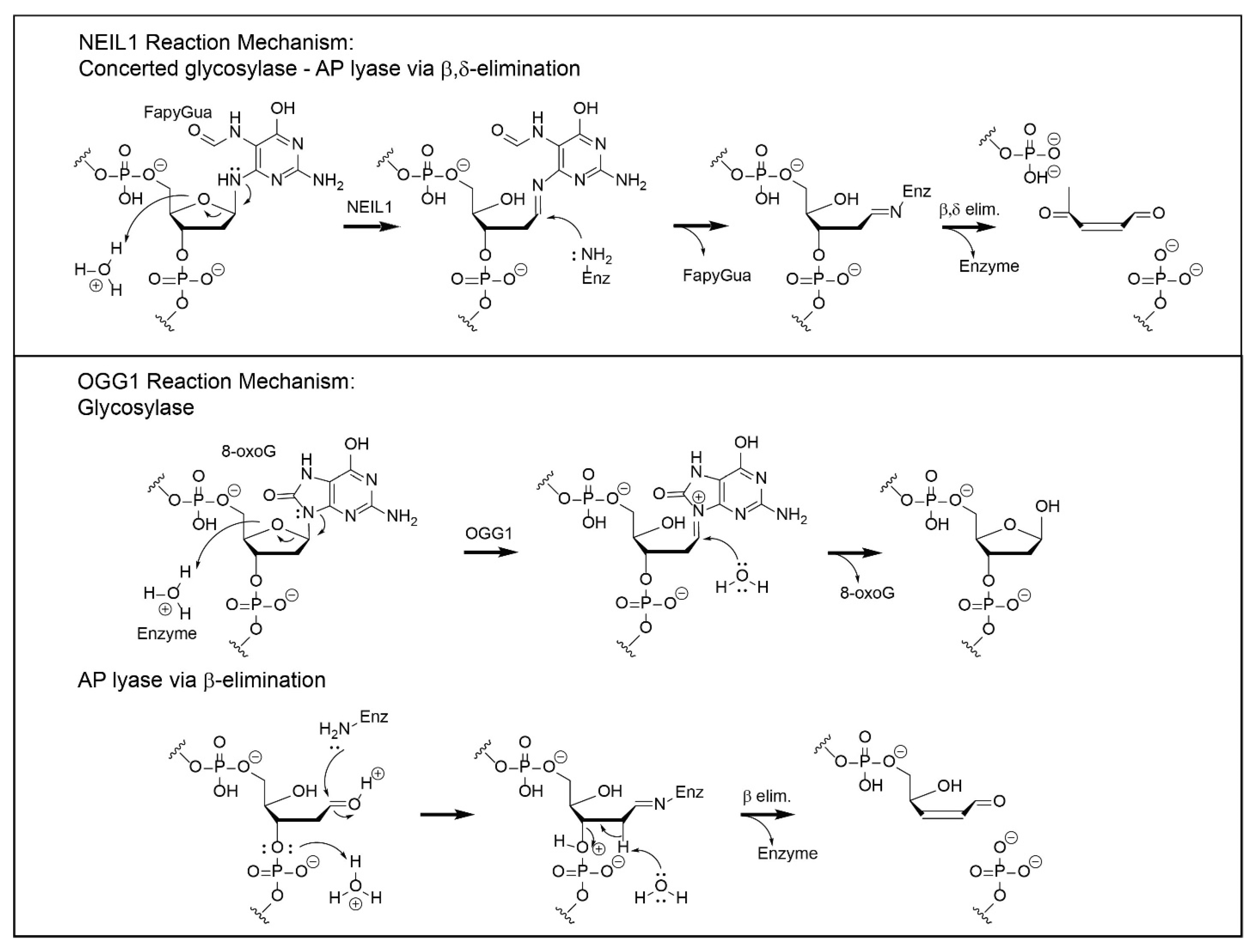
Although both NEIL1 and OGG1 are DNA glycosylases, the chemical mechanisms by which they catalyze base release are distinct (Figure 2). DNA glycosylases that also have an associated AP site lyase activity utilize either a primary or secondary amine to form a covalent imine intermediate with the C1′ of the deoxyribose that is associated with the damaged base. This mechanism was first established for T4-pdg, in which the enzyme–DNA intermediate could be trapped by a concomitant reaction with sodium borohydride [64,65,66,67]. Such covalent intermediates are stable under further biophysical analyses, facilitating the mapping of active site nucleophiles for all glycosylases with AP lyase activities [68,69]. Enzymes that catalyze a β-elimination reaction all use a primary amine (e.g., a lysine or an N-terminal α-amino group), while enzymes that catalyze β,δ-elimination reactions—such as NEIL1 and NEIL2—use the secondary amine of an N-terminal proline (P2). For NEIL1, the adjacent glutamic acid (E3) is essential for this reaction as a proton donor [4,23]. Site-directed mutagenesis studies of NEIL1 have also implicated Lys54 and Arg339 in the β- and δ-elimination reactions [37,70]. Additional insights into critical portions of the enzyme for catalysis have been generated by site-directed mutagenesis and in vitro characterization. Mutagenesis at R277 to R277A resulted in decreased glycosylase activity, while full lyase function was retained [16]. Characterization of two of the human polymorphic variants of NEIL1—Gly83Asp and Cys136Arg—showed no glycosylase activity [44,71]. The loss of function of Gly83Asp was attributed to the introduction of an aspartic acid residue at the entrance of the nucleotide-flipping pocket. The Cys136Arg variant is located in the flexible hinge region of NEIL1, where changes can affect the dynamic opening and closing of the substrate-recognition pocket.
OGG1 was originally characterized as a bifunctional glycosylase-AP lyase, based on sodium borohydride trapping of an enzyme–DNA intermediate. However, structure–function analyses have demonstrated that the glycosylase reaction is catalyzed by an activated water molecule, coordinated by Asp268 [72]. Thus, the glycosylase reaction is mechanistically independent of the β-elimination reaction, which occurs via the subsequent reaction of an opportunistically positioned lysine residue (K249) in close proximity to the AP site. This separation of function was confirmed by the creation of the site-directed mutants K249C/C253K, which retain full glycosylase activity while exhibiting no lyase function, and C253W, which is devoid of glycosylase activity but retains full AP lyase function. Thus, to be consistent with its actual catalytic mechanism, OGG1 should be referred to as a monofunctional glycosylase, with the capacity to also catalyze nicking at AP sites. Intracellularly, data have been reported that support the conclusion that OGG1 functions as a glycosylase without concomitant lyase activity. This was explicitly tested by engineering shuttle vectors containing site-specific 8-oxo-dG lesions that were flanked by either 5′ or 3′ nuclease-resistant phosphorothioate linkages [73]. This study revealed that, in cells, OGG1 did not catalyze strand incision, and that cleavage of the phosphodiester backbone 5′ to the lesion was carried out by APE1. This is consistent with prior in vitro biochemical studies that demonstrated that physiologically relevant concentrations of magnesium preferentially abrogated the AP lyase activity of OGG1 [74]. Additional support for OGG1 functioning as a monofunctional glycosylase is based on analyses of data from the infection of OGG1-proficient gastric epithelial cells with Helicobacter pylori, in which there were increased levels of AP sites, while suppression of OGG1 in the same experimental design resulted in decreased levels of AP sites [75].
Additional structure–function investigations have been reported concerning the potential roles of all cysteine residues in OGG1 in the context of its varied functions within cells [76]. These studies revealed that the majority of mutants affected either the glycosylase activity (C146 and C255), the lyase activity (C140, C163, C241, and C253), or the structural stability (C253), with mutagenesis at only two cysteine residues resulting in no discernable loss of function (C28 and C75). These data suggest that differential site-specific oxidation of cysteine residues in OGG1 could play a major regulatory role in switching between repair and transcriptional regulation.
Although cancer and cancer-associated variants are discussed below, a recent report by Popov et al. [77] utilized molecular dynamics analyses of 20 clinically observed variants of OGG1, combined with successful expression and structure–function analyses of 13 of these variants. Two of the variants—I145M and G202C—were severely catalytically compromised by >10-fold, while an additional three—R161W, V267M, and S292N—were reduced by 1.5–2-fold. It is also of interest that two of the variants exceeded WT rates, with H111R and R206C showing a 2.0–2.5-fold increase. Overall, their experimental data were in good agreement with the molecular dynamics simulations, emphasizing the utility of this strategy in large structure–function analyses and the potential identification of pathogenic variants of DNA glycosylases.
The individual downstream steps in BER are significantly different for repair initiated by NEIL1 and OGG1. As elaborated below, these differences are dictated by the nature of the chemistry through which base release and phosphodiester bond scission occur. Downstream repair of NEIL1-initiated repair builds on the single-nucleotide gapped DNA, with both 5′ and 3′ phosphates. Unlike monofunctional glycosylases, or glycosylase-AP lyases, there is no requirement for the activity of an AP endonuclease (APE1); rather, the 3′-phosphate removal is catalyzed by the activity of DNA kinase/phosphatase (PNKP), creating a 3′-OH [78]. In fact, APE1 is unable to remove a 3′phosphate. Thus, PNKP activity generates the ideal substrate for the filling of single-nucleotide gaps by DNA polymerase β and subsequent ligation by DNA ligase 3 [79].
In contrast, OGG1-initiated repair will result in either an AP site within fully duplex DNA or base loss and β-elimination generating a 3′-dRp. Both of these intermediates require processing by APE1 to create the 3′-OH and subsequent reaction(s) by DNA polymerase β to remove the 5′ sugar moiety via its 8 kDa AP lyase domain if OGG1 functions as a glycosylase alone. The resulting gap is filled and sealed as described above.
4. Cell-Cycle Specificity and Protein-Binding Partners
Early characterization of NEIL1-mediated repair revealed a predominant S-phase cell-cycle specificity, suggesting an involvement and association with genomic replication [5,35,36]. This hypothesis was supported by biochemical studies that showed the ability of NEIL1 to excise base damage from bubble and forked structures [37,38,39,40,41,42,43]. A large number of proteins have been identified as associated with NEIL1, including replication- and repair-associated proteins, e.g., PCNA, RPA, RFC, heterogeneous nuclear ribonucleoprotein U, Werner syndrome protein, PARP1, FEN1, DNA ligase 3, polymerase δ, polymerase β, NEIL2, TDG, HDAC1, and the Rad9–Rad1–Hus1 complex [38,39,42,54,78,80,81,82,83].
Unlike NEIL1, on which there is an abundance of experimental evidence supporting cell-cycle regulation, the literature concerning cell-cycle regulation of OGG1 is conflicting. As part of the original cloning and characterization of the essential promoter of OGG1, it was shown that there were no expression differences across any phases of the cell cycle and that there were equivalent levels of 8-oxodG nicking activity across all phases [84]. Additionally, Western blot analyses revealed no changes in OGG1 levels throughout the cell cycle [85]. However, these conclusions are at odds with another study showing a cell-cycle dependence of expression of the two major isoforms of OGG1 and a dynamic re-localization to the nucleolus during the S-phase [86]. It is not clear what accounts for this underlying discrepancy.
As was the case for substrate specificities, the protein-binding partners of OGG1 share almost no overlap with the NEIL1-associated proteins but include the repair-associated proteins PARP1, XRCC1, NEIL1, APE1, RECQL4, UV-DDB, CUX1, CUX2, SATB1, and the Rad9–Rad1–Hus1 complex [61,87,88,89,90,91,92,93,94,95,96,97,98,99]. The protein-binding partners of OGG1 and the significance of these interactions have been comprehensively reviewed [100]. Recently, a role for OGG1 has been discovered in the regulation of Myc-controlled genes, in which direct binding of Myc to OGG1 resulted in an inhibition of OGG1 incision activity and recruitment of Myc to its promoters [101]. These data are consistent with the hypothesis that catalytically inactive forms of OGG1 bind to 8-oxo-dG sites in promoter regions, thereby influencing the activation of inducible transcriptional networks. This is discussed in more detail in Section 5 below.
In addition to their nuclear-localized binding partners, both NEIL1 and OGG1 interact with mitochondrial-imported proteins that are associated with repair and replication. mtNEIL1 binds to the tetrameric form of mitochondrial single-stranded binding protein (mtSSB) in the presence of duplex DNA, but in the absence of DNA the complex is reduced to a NEIL1–mtSSB complex [102]. Furthermore, mtNEIL1 also interacts with mitochondrial transcription factor A (TFAM) [103]. A proposed biological role for this interaction is that in the absence of NEIL1, TFAM-transcribed mitochondrial genes are significantly downregulated.
ERCC6 (CSA) and ERCC8 (CSB) also bind mitochondrial OGG1 [104]. The interactions of mtOGG1 with CSA and CSB protect against age- and stress-induced mtDNA mutations, as well as apoptosis-mediated loss of subcutaneous fat.
5. Phenotypes in Knockout Mice
Given the central roles that NEIL1 and OGG1 play in both nuclear and mitochondrial genome maintenance, it was anticipated that the loss of these critical functions would result in an increased frequency of cellular transformation and carcinogenesis. Although not originally anticipated, changes and/or reductions in mitochondrial genome integrity resulted in metabolic disorders and disruptions in inflammatory responses in oxidatively stressed organs such as the lungs, brain, liver, stomach, and colon. Several decades of investigations have laid the basic foundation for developing a clearer understanding of the adverse consequences that result from the loss of these enzymes.
5.1. Cancers
Among the most perplexing observations using murine knockout models of NEIL1 and OGG1 deficiencies are the differences in cancer susceptibilities. Ogg1−/− mice are refractory to spontaneous carcinogenesis. These data are extraordinary in that the loss of BER-initiated repair of 8-oxo-dG lesions and subsequent replication bypass of residual 8-oxo-dG lead to high-frequency misincorporation of dA opposite the oxidized G. However, if the misincorporated dA is not removed by the adenine DNA glycosylase MutY-homologue (MUTYH), the subsequent round of replication generates a stable, inheritable G-to-T transversion. Thus, it was not surprising that knockout of both Ogg1−/− and Myh−/− in murine models led to increased lung and small intestinal cancers [105,106]. In addition to the lack of spontaneous carcinogenesis, even under the most extreme oxidizing conditions associated with potassium bromate ingestion through supplementation of drinking water and 8-oxo-dG levels increasing >1000-fold over baseline, no carcinogenesis was observed in Ogg1−/− mice [107]. Furthermore, in our own laboratory’s experience with two independently derived Ogg1−/− mouse strains, no tumors were ever noted, whether in mice raised under conditions that induce many other physiological alterations—such as a high-fat diet—or in mice that achieved ages > 20 months. This absence of a carcinogenic phenotype with concomitant mutagenesis has not been adequately rationalized but may be related to the absence of an induction of NF-κB-associated inflammation.
In contrast to the results described above, and given the substrate specificity of OGG1, it is of interest that Ogg1−/− mice showed enhanced multiorgan carcinogenesis when challenged with combinations of a variety of DNA-alkylating agents, including nitrosamines and nitrosourea [108]. Although it was speculated that these alkylating agents enhanced oxidative stress, thereby accounting for increased carcinogenesis in multiple sites, this was not explicitly tested. Furthermore, Ogg1−/− mice have also been shown to have increased susceptibility to phenobarbital- and UVB-induced carcinogenesis [109,110].
In contrast to the lack of spontaneous carcinogenesis observed in Ogg−/− mice, Neil1−/− mice show age- and gender-dependent spontaneous carcinogenesis—primarily in the liver and lungs [111,112]. In mice aged over a year, spontaneous formation of lung adenomas and adenocarcinomas was observed in 12% of the males and 0% of females, while hepatocellular carcinomas were found in 16% of males and 11% of females [112]. Creation of a double knockout of the Neil1 and Nth1 genes resulted in significant increases in the spontaneous frequency and distribution of tumors in both male and female mice relative to either individual knockout [112]. In males, lung adenomas and adenocarcinomas were observed in 74% and 41% of males and females, respectively, while hepatocellular carcinomas increased to 47% and 17% in males and females, respectively. These studies suggest an underlying genomic instability in Neil1−/− mice that can result in carcinogenic endpoints. However, the studies described above have not been adequately reconciled with independently constructed knockouts of Neil1, double knockouts of Neil1 and Neil2, or the triple knockout of all of the murine Neil genes, in which all three sets of knockouts showed no increases in cancers, mutations, or oxidatively induced base damage [113]. Although there are no studies resolving these differences in spontaneous carcinogenesis, it is possible that additional factors—such as differences in the intestinal microbiome—could influence these rates.
As previously described, the substrate specificity of NEIL1 encompasses a variety of alkylated FapyGua adducts, including AFB1-FapyGua [43]. To test the in vivo significance of Neil1-initiated BER of aflatoxin-induced DNA damage, Neil1−/− mice and control C57Bl6 mice were challenged with two concentrations of AFB1, and their adduct accumulation was measured at 6 and 48 h post-exposure, while induction of hepatocellular carcinogenesis was measured at 15 months [43]. These analyses revealed a threefold increase in the accumulation of AFB1-FapyGua adducts in Neil1−/− mice versus control C57Bl6 mice at 48 h post-injection of aflatoxin. This differential accumulation was attributed to the loss of BER-initiated repair, even though NER repair was active. As hypothesized for the carcinogenesis portion of this study, and in agreement with the elevated levels of AFB1-FapyGua accumulation in Neil1−/− mice, the frequency of hepatocellular carcinoma (HCC) was greatly increased in these mice relative to their wild-type counterparts. Using tumor burden as a relative risk assessment, Neil1−/− mice displayed a 3.4-fold increase in HCC formation relative to control mice receiving comparable doses of aflatoxin. Previously, in a separate investigation, the cancer risk associated with the loss of nucleotide excision repair following aflatoxin exposure was measured in XPA−/− mice [114]. Comparable analyses of HCC formation in aflatoxin-treated XPA−/− versus control wild-type mice revealed a modest 1.2-fold increase in liver carcinogenesis. Thus, at least in murine models, BER-initiated repair appears to be significantly more important in suppressing aflatoxin-induced HCCs than NER.
Extending studies on cancer susceptibility in Neil1−/− mice, and given NEIL1’s role in the recognition and repair of oxidatively induced base damage, it was hypothesized that deficiencies in Neil1 could confer increased sensitivity to UVB-induced carcinogenesis via increased levels of reactive oxygen species [115]. In response to long-term but low-level UVB exposure, Neil1−/− mice showed much greater levels of cutaneous tissue destruction and enhanced chronic inflammatory responses as measured by cytokine message levels, along with higher levels of neutrophil infiltration, relative to age-matched controls. However, in this study, differential carcinogenesis assessments could not be performed, due to the severity of tissue loss in the Neil1−/− mice.
5.2. Metabolic Syndrome
The term metabolic syndrome, which is also called insulin resistance syndrome, refers to a group of conditions that, on aggregate, raise an individual’s risk of coronary heart disease, type 2 diabetes, stroke, and other diseases. It is characterized by abdominal obesity, high blood pressure, high blood sugars and triglycerides, and low HLD cholesterol.
Although our laboratory’s original justification for creating a whole-body knockout of Neil1 was in anticipation of elevated mutagenesis and carcinogenesis due to decreased repair of oxidatively induced DNA damage, the most dominant phenotype in the Neil1−/− mice was the development of midlife metabolic syndrome [111]. Even on a standard low-fat chow diet, male mice older than 9 months of age were preferentially susceptible to the accumulation of massive amounts of fat within the subcutis, retroperitoneum, mediastinum, and abdomen. Livers were enlarged, granulated, and pale yellow, with >50% of the cellular contents being a mixture of micro- and macrovesicular fat globules. The most prominent areas in the liver for fat accumulation were the centrilobular regions. Furthermore, livers showed no overt indication of significant inflammation or fibrosis. Although the increase in weight gain was readily discernable beyond 9 months, analyses of circulating leptin levels were clearly evident at 3 months of age, proving to be a strong predictor of future weight gains and fatty liver disease. Since Neil1 is differentially spliced for both nuclear and mitochondrial localization, we hypothesized that a potential driver of metabolic syndrome in these mice could be the accumulation of mitochondrial DNA (mtDNA) damage and deletions. Steady-state levels of mtDNA damage were assessed by long-range PCR, in which PCR-blocking lesions were calculated by reductions in full-length (16 kb) mtDNA, and deletions (less than full-length mtDNAs) were visualized by reducing the time of the PCR extension reactions to selectively amplify these molecules. For both criteria, Neil1−/− liver samples showed consistent evidence of increased damage and progressive loss of mtDNA integrity [111]. Such a loss in mitochondrial genomic integrity would be expected to compromise oxidative phosphorylation efficiencies which, in turn, would further amplify the production of reactive oxygen species and increase the DNA damage burden. Although not explicitly tested in these studies, these results suggest that correction of the mitochondrial repair deficiency by way of transgenic expression of a mitochondrial-targeted NEIL1 could prevent metabolic syndrome in Neil1−/− mice.
The previous investigation exclusively focused on phenotypes in aged Neil1−/− mice that consumed a low-fat diet. In a follow-up study, young Neil1−/− and wild-type mice were challenged with a high-fat diet, and weight gain and other readouts of metabolic syndrome were analyzed [116]. Consistent with the conclusions from the original report, Neil1−/− mice gained significantly more weight and had more severe hepatic steatosis relative to control mice and showed decreased mtDNA and protein content. The high-fat-diet-fed Neil1−/− mice also showed elevated inflammatory gene expression profiles relative to their wild-type counterparts, which may also be an indicator of this genotype having a lowered threshold for tolerance of cellular oxidative stress.
Since Neil1−/− mice showed hallmarks of susceptibility to the development of metabolic syndrome, we also investigated whether this phenotype was restricted to Neil1−/− mice or whether Ogg1−/− mice would also manifest a similar phenotype. Consistent with the phenotypes observed in Neil1−/− mice, on a low-fat diet, aged Ogg1−/− mice were significantly fatter and heavier than their control counterparts [117]. Also consistent with prior studies, a 10-week high-fat diet challenge of young, age-matched mice revealed that the Ogg1−/− mice also experienced greater weight gain, increased adiposity, and fatty livers, even though their food intake and physical activity were not significantly different. This weight gain was accompanied by concomitant hyperinsulinemia and impaired glucose tolerance, suggesting the development of insulin resistance and, potentially, a prediabetic condition. Indirect calorimetric measurements revealed that the Ogg1−/− mice showed characteristic changes from fat oxidation towards carbohydrate utilization—a result that was consistent with their obese phenotype. These data were supported by gene expression profiles indicating decreased hepatic fat oxidation. While these studies focused on hepatic alterations associated with metabolic syndrome, follow-up investigations examined whether skeletal muscle was comparably affected [118]. The Ogg1−/− mice showed significant increases in the accumulation of triacylglycerol, cholesterol esters, and diacylglycerols in the skeletal muscle. Unlike the analyses of the liver, the skeletal muscle did not show decreased levels of mtDNA or increased 8-oxo-dG. This investigation also revealed that Ogg1−/− mice showed a large decrease in grip strength and reduced treadmill endurance.
In addition to tissue-specific control and manifestations of fat accumulation, there is an abundance of literature that associates changes in the composition of the intestinal microbiome with the development of obesity phenotypes [119]. In contrast to wild-type mice, Ogg1−/− mice fed either a normal chow or a high-fat diet showed a significantly greater microbial diversity on either diet [120]. These analyses revealed that Ogg1−/− mice retained a microbiota that was consistent with increased energy harvest—a result that is highly consistent with the previously described obesity phenotype. In addition, the microbiome of the Ogg1−/− mice also showed significant increases in pro-inflammatory microbes, even though the airway pathogenesis indicated a severe reduction in the inflammatory gene cascades. To test this further, Ogg1−/− and control mice were subjected to an acute intestinal inflammatory challenge using dextran-sulfate-sodium-supplemented drinking water. In contrast to the lung pathology, the Ogg1−/− mice showed a much more severe response than the wild-type controls, with greater weight loss, lethargy, diarrhea, and histology, consistent with severe ulcerative colitis. Consistent with the loss of functional Ogg1, GCMS analyses revealed significantly elevated levels of FapyGua, 8-oxoGua, and FapyAde [120]. Given the tissue-specific differences in inflammatory responses between the lungs and colon in Ogg1−/− mice, additional investigations are warranted to harmonize these findings.
Since data from both the Ogg1−/− and Neil1−/− mouse models indicated that BER-mediated repair in mitochondria likely plays an integral role in regulating energy balance, we hypothesized that in contrast to decreased repair levels, increased expression of a mitochondrial-targeted human OGG1 might render mice more resistant to obesity and metabolic syndrome [121,122,123]. These studies utilized a transgenic OGG1 model (OGG1Tg) developed in the lab of Dr. Lars Eide, in which OGG1Tg mice constitutively expressed the human OGG1a gene coupled with the mitochondrial targeting sequence of the manganese superoxide dismutase gene. These mice display modest, 2–3-fold increases in mitochondrial OGG1 activity above that observed in wild-type mice. This level of increase in the expression of the mitochondrial-targeted OGG1 is likely significant, because greatly elevated expression of OGG1 (or NTH1) increases cytotoxicity in lymphoblastoid cells [124,125]. These mice were challenged with the previously described high-fat diet, but extended for a total of 12 weeks. The OGG1Tg mice were highly refractory to the development of obesity, insulin resistance, fatty liver, and adipose tissue inflammation, even though there was a significant increase in food intake in the OGG1Tg mice. This reduction in body weight relative to wild-type mice was well correlated with increased energy expenditure in the OGG1Tg mice. Additionally, examination of the histology of adipocytes in epididymal fat deposits in the OGG1Tg mice versus controls revealed significant reductions in size, with concomitant large increases in the expression of genes regulating fatty acid oxidation and lipolytic processes. These changes in gene expression were restricted to white adipose tissue and strongly suggest that this organ is primarily responsible for the phenotypic changes associated with the OGG1Tg mice. Thus, it is likely that the beneficial effects were achieved via a modest increase in the expression of mitochondrial-targeted OGG1, while a global large overexpression is detrimental [124,125].
While all of the above studies utilized a high-fat diet to accelerate the manifestations of metabolic syndrome, yellow Agouti mice (Ay/α) represent a genetic model of obesity driven by hyperphagia. To test whether global expression of the human OGG1 transgene could also confer resistance to weight gain in these mice, the Ay/α were crossed with OGG1Tg mice and selected Ay/αTg [122]. Even though there were trends for increased food intake in the Ay/αTg versus the Ay/α, both weight and fat mass were significantly decreased in Ay/αTg males and females. Consistent with the previous findings in the C57Bl6 background, the Ay/αTg mice exhibited increased energy expenditure and mitochondrial content, without increased voluntary physical activity. Interestingly, the resistance to obesity and the increase in mitochondrial content in white adipose tissue were shown to be dependent on maternal transmission of the transgene, such that paternal transmission conferred no improvement in these parameters. These findings clearly suggest a major role for mitochondrial quality in determining resistance to obesity in offspring.
5.3. Inflammation: The Role of OGG1 as a Regulator of Transcriptional Responses to Oxidatively Induced Base Damage
In addition to its integral functions in the initiation of BER, an unanticipated role for OGG1 has emerged from in-depth analyses of phenotypes in the Ogg1−/− mice relative to responses to inflammation-driven oxidative stress. A series of investigations using Ogg1−/− mice revealed greatly reduced, selective airway gene expression levels in response to allergens [126,127] and resistance to lipopolysaccharide challenge, as well as resistance to tumor necrosis factor alpha (TNF-α)-induced inflammation [128,129]. Analyzed collectively, these data suggest that in the absence of Ogg1, this deficiency confers resistance to normal pro-inflammatory gene activation cascades. Key studies designed to understand the molecular basis of these observations have shown that under conditions of oxidative stress, 8-oxoG lesions preferentially form in promoter and gene-regulatory regions. Comparison of the sites of OGG1 and NF-κB binding following TNF-α mediated challenge revealed a major overlap [130]. This relationship is supported by data demonstrating that knockdown of OGG1 significantly downregulates NF-κB-dependent gene expression profiles [127,131], suggesting that binding of OGG1 to sites of 8-oxoG does not initiate the chemistry of base release. This finding is consistent with data demonstrating that increased levels of 8-oxoG in GC-rich Sp1 DNA-binding sites are well correlated with the degree of gene expression enhancement in adipose tissue [132]. The mechanisms through which OGG1 locates and binds to damaged sites within genomic DNA have been extensively reviewed [31]. Furthermore, in the overall control of estrogen-responsive genes, chromatin binding and activation of the LSD1 demethylase produced localized oxidatively induced DNA damage which, in turn, recruited OGG1 and topoisomerase IIβ to these sites, thereby regulating estrogen-induced transcription [133].
The majority of these data are highly consistent with a model in which a catalytically incompetent form of OGG1 binds to an 8-oxoG site, and the lack of incision is achieved by either the modulation of the local DNA structure, rapid redox-driven post-translational modification, or association with other proteins. As discussed above, the potential roles of cysteine residues in OGG1’s function relative to its redox status have been recently addressed [76]. This non-catalytically active structure induces a sharp bend in DNA, facilitating the recruitment of NF-κB and its associated transcription initiation complex. Similar conclusions are likely germane for Sp1-regulated binding sites. Such non-catalytically productive binding of OGG1 to promoter and regulatory regions serves as the initiating step in activation by these master regulators. The re-establishment of pre-induction gene regulation in these regions could be achieved through a return to redox homeostasis and activation of OGG1-initiated BER.
In addition to the aforementioned examples of regulation in gene expression, several investigations have shown that the formation and processing of 8-oxoG lesions are key components in regions of the genome containing potential G-quadruplex-forming, Z-DNA-forming, and i-motif-forming sequences [53,134,135,136,137]. Interestingly, control of the expression of many DNA-repair genes is regulated in a strand-specific manner through structural alterations of potential G-quadruplex-DNA-forming sequences. Transitions in gene expression reflect transitions between the native, undamaged DNA and those containing 8-oxoG lesions that are processed by OGG1 and, subsequently, APE1.
Over the past 7 years, there have been significant advances in the development of small-molecule inhibitors of OGG1, and their use has substantiated conclusions drawn from genetic knockout models and potentially identified novel strategies for targeting OGG1 in disease prevention and treatment [138,139,140,141]. The lead compound developed within the Helleday laboratory—TH5487—which prevents binding of OGG1 to sites of base damage, has been successfully used to efficiently suppress allergen-induced inflammation and pro-inflammatory gene expression [140], attenuate pulmonary fibrosis [142], suppress polycystic ovarian syndrome by inhibiting NF-κB signaling [143], potentiate the anticancer efficacy of methotrexate [144], trigger synthetic lethality of olaparib in BRCA1 deficiencies [145], and limit inflammatory diseases [146]. Thus, it is evident that there are broad applications of OGG1 inhibitors in disease treatment, and more in-depth analyses are warranted to understand which function of OGG1 is responsible for these encouraging biological findings.
5.4. Roles of OGG1 and NEIL1 in Huntington’s Disease
Huntington’s disease is an autosomal-dominant, late-onset neurological disorder that is caused by expansion of CAG trinucleotide repeats in exon 1 of the huntingtin gene, resulting in increases in the length of a polyglutamine track from ~35 repeats in the wild type to >42 in fully penetrant disease (reviewed in [147,148]. Although oxidatively induced damage has been associated with various neurological diseases, as well as the aging process, work from Cynthia McMurray’s group reported the initial correlation between active repair of 8-oxo-dG lesions by OGG1 and an age-dependent expansion of CAG repeats—a process that they described as a “toxic oxidation” model [149]. In the development of this model, it was first established that there was an age-dependent, selective accumulation of oxidatively induced DNA damage in the brain and liver, even though repair levels did not appreciably decrease. Using hydrogen peroxide as an oxidative-damage-inducing treatment, it was demonstrated that there was a dose-dependent increase in single-stranded breaks, resulting in concomitant and selective expansion of the CAG repeats, while no expansion was detected in the TTC/GAA repeats. The essential role of OGG1 in initiating this process was established by demonstrating that CAG expansion was greatly reduced or delayed in most Ogg1−/− mice relative to wild-type controls or Nth1−/− or Agg−/− mice. The key role of OGG1 in catalyzing the initiation of the trinucleotide repeat was confirmed using in vitro BER reconstitution assays, in which expansion occurred via repair-initiated strand displacement and slippage. Building on these investigations, Goula et al. [150] mechanistically expanded the role of BER in promoting triplet expansion by showing that both flap endonuclease 1 (FEN1) and a cofactor (HMGB1) were reduced in the striatum—a disease-susceptible brain tissue—while they were not reduced in disease-spared cerebellar regions. These data further established a mechanistic link between OGG1-initiated repair and CAG expansion in affected brain tissues.
As mentioned above, since triplet expansion was not completely abolished in Ogg1−/− mice relative to wild-type controls, additional backup pathways have been examined for promoting the repair of oxidatively induced base damage. Specifically, Kovtun et al. [151] examined a potential role for the Cockayne syndrome B protein in transcription-coupled repair of such damage, along with knockouts of the mismatch repair proteins Msh2 and Msh3. Inclusion of analyses of Msh2 and Msh3 was well justified based on the role of mismatch repair also being well correlated with triplet expansion, with deficiencies in Msh2 and Msh3 leading to decreases in triplet repeats (reviewed in [152]). Csb−/− mice showed a large reduction in deletions at CAG repeat tracts, while manifesting a fivefold increase in expansions. Msh2−/− mice showed the mirror image of the Csb−/− mice. Evidence was presented that CSB protects against CAG expansion by antagonizing the activity of Ogg1 and tract-length reduction during parent–child transmission.
At present, there are no therapeutically responsive treatments for Huntington’s patients, and there are ongoing questions as to whether inheritance of prior triplet expansions or somatic expansion is the primary driver of disease manifestation. To address this, murine models were created in which the CAG repeat in the murine huntingtin locus was expanded to contain a 150-repeat tract in the backgrounds of Ogg1 proficiency and deficiency [153]. These findings demonstrated that while the size of the inherited alleles did not influence somatic expansion, the loss of Ogg1 suppressed somatic expansion and the age of disease onset. Moreover, pharmacological intervention via the addition of a mitochondrial-targeted free-radical-scavenging agent suppressed somatic triplet expansion, with delays in the onset and progression of disease. These data provide the foundation from which novel therapeutic strategies can be developed.
Germane to Neil1’s role in maintaining genomic stability, expansion of trinucleotide repeats that are associated with the development of Huntington’s disease was significantly reduced in Neil1−/− mice relative to controls [154]. Furthermore, this study demonstrated that Neil1 loss reduced germline expansion in males, concluding that Neil1 plays a role in germline and Huntington’s disease trinucleotide repeat expansion.
5.5. Neurodegeneration in Neil1 Deficiency
Due to the terminal replication status of neuronal cells, the potential adverse impacts of DNA damage are significant, with repair being the only viable option. In the aging brain, increased levels of oxidative stress and decreased repair can lead to cognitive declines and the manifestation of diseases such as Alzheimer’s disease (reviewed in [155]). Thus, it was anticipated that deficiencies in the repair of oxidatively induced damage in murine brains could be associated with various manifestations of brain dysfunction. For example, the Neil1−/− mice showed impaired spatial memory retention and both increased brain damage and poorer outcomes in a focal ischemia/reperfusion stroke model [156]. Neil1−/− mice also have overall deficits in olfactory function, as measured by decreased performances in multiple behavioral tests [157]. Further manifestations of neuronal deficits in these mice include the presentation of hyperactive, anxiety-mediated behaviors in open-field tests and deficiencies in cognitive memory as measured by novel object recognition [158,159]. To extend the scope of effects arising from Neil1 deficiency, sublethal ionizing radiation was administered to Neil1−/− and control mice and tissues collected at different times [158]. Although both sets of mice lost weight post-radiation relative to non-irradiated controls, the decreases in the Neil1−/− mice were more severe. Analyses of gene expression profiles in the brain cortex revealed that neuronal functional pathways were downregulated in radiation-treated versus sham-treated Neil1−/− mice, while the opposite trend was evident in the wild-type mice. These irradiated Neil1−/− mice showed decreased neurogenesis as measured by bromodeoxyuridine labeling. Non-irradiated Neil1−/− mice displayed increased densities of microglia and astrocytes, suggestive of chronic neuroinflammation, as well as abnormal neuroinflammatory responses post-irradiation. Collectively, these data reveal an important role of Neil1 in brain development as well as the responsiveness of the brain to oxidatively induced stress, as evidenced by decreased neurogenesis, recovery from neuroinflammation, and behavioral deficits.
6. Extrapolation to Human Investigations
6.1. OGG1-Associated Cancer Variants
In contrast to the lack of carcinogenesis in murine models, epidemiological studies in human cancers show a positive correlation with the dominant human polymorphic variant of OGG1: Ser326Cys. In Asian and Caucasian populations, this variant is widely distributed, being present at 40–60% and 25–40%, respectively [160]. Biochemical characterization of the Ser326Cys OGG1 variant displays reduced catalytic efficiency, which may be partially attributable to its ability to form dimeric Cys-Cys disulfide bonds [161,162,163]. A significant number of adverse health effects have been reported in individuals carrying monoallelic Ser/Cys and biallelic Cys/Cys variants. These analyses have shown positive correlations with a variety of tumors, including colorectal cancer [164], breast cancer [165], oropharyngeal squamous-cell carcinoma [166], acute myeloid leukemia [167], endometrial cancer [168], prostate cancer [169,170], and lung cancer [171].
In addition to the S326C variant, R131Q—a catalytically inactive variant—has been associated with small-cell lung carcinoma [172]. Furthermore, R229Q has been reported to be associated with leukemia, with this variant demonstrating thermal lability and rendering cells sensitive to radiation [173,174].
6.2. Low-Level Expression of OGG1 in Therapeutically Responsive Subtypes of Human Acute Myeloid Leukemia (AML)
Within the diverse subtypes of human AML, there are two subtypes—RUNX1-RUNX1T1 fusion and CBFB-MYH11—that are significantly more therapeutically responsive than the remaining subtypes [175]. This increased efficacy is manifested in significantly longer disease-free survival. Analyses of the gene expression profiles of 404 genes (318 DNA-damage-response genes and another 87 additional genes related to DNA replication, the cell cycle, apoptosis, and Ara-C import and metabolism) revealed a unique gene (OGG1) that was downregulated in the cytarabine-responsive AML, but normal or elevated in the poor-response subtypes [176]. This investigation demonstrated that OGG1-deficient AML cell lines showed enhanced sensitivity to cytarabine treatment, with increased strand breaks occurring primarily at common fragile sites, along with enhanced oxidatively induced DNA damage. Insights into the mechanism of this enhanced lethality were guided by results that demonstrated that this preferential lethality could not be conferred by other replication-stress-inducing agents. However, increased cell killing was well correlated with a mechanism in which DNA polymerase δ inserted cytarabine opposite 8-oxo-dG, resulting in termination of DNA synthesis. The overall conclusion of this investigation suggested that the selective toxicity and therapeutic efficacy of cytarabine treatment in OGG1-deficient AML occurred via its incorporation opposite unrepaired 8-oxo-dG. These studies also suggest that selective small-molecule inhibitors of OGG1 could be used in combination chemotherapies for AML patients who are poorly responsive to standard-of-care chemotherapy.
6.3. NEIL1-Associated Cancers
It is currently unknown whether specific single-nucleotide polymorphic variants of NEIL1 are well correlated with increased carcinogenesis. Extrapolation of our investigations with Neil1−/− mice, which displayed increased susceptibility to aflatoxin-induced carcinogenesis and increased DNA adduct levels, suggests that deficiencies in human NEIL1 might render individuals more susceptible to aflatoxin-induced HCCs. In this regard, a significant number of single-nucleotide variants of NEIL1 have been characterized for alterations in biochemical activities [44,47,71,177,178]. Directly relevant to HCC formation in portions of the world where aflatoxin exposure is very high, the three most common NEIL1 variants in East Asia—A51V, P68H, and G245R—have been biochemically characterized [178]. Although the catalytic efficiency of G245R was comparable to that of the wild-type enzyme, the activity of P68H was compromised—especially on the AFB1-FapyGua adduct. The A51V variant showed the ability to excise DNA adducts, but it was inactivated at 37 °C, with a half-life of ~5 min. Thus, A51V is predicted to be catalytically dead in a cellular context. The potential impact of these NEIL1 variants on susceptibility to early-onset HCC formation was addressed by genome sequencing of 49 aflatoxin-associated HCC patients in whom each of the three NEIL1 variant alleles (i.e., A51V, P68H, and G245R) were identified, even though they were only present at 0.17%, 1.4%, and 0.65%, respectively, in this geographical region. To test the significance of these observations, expanded population-based studies are needed to ascertain whether there is an association between rare genetic variations in the NEIL1 gene and susceptibility to HCC. Comparable analyses also need to be performed in other regions of the world that have endemic aflatoxin exposure and variants of NEIL1 that compromise catalytic efficiency and stability. Such investigations are warranted in sub-Saharan Africa, Turkey, and Central America (including Mexico).
6.4. Potential Applications of Agonists of OGG1 in Disease Prevention
In addition to the numerous correlations between the Ser326Cys OGG1 variant and cancer susceptibility, there are epidemiological studies implicating decreased OGG1 function in obesity and type 2 diabetes [179,180], Parkinson’s and Alzheimer’s diseases [181,182], and cardiovascular disease [183]. Collectively, these data suggest that there is therapeutic potential for the development of agonists of OGG1 in the treatment of human diseases. This suggestion is reinforced by the studies described above, demonstrating the health benefits in murine models with enhanced mtOGG1 expression. In this regard, the initial report of an agonist of OGG1 identified 8-bromoGua, which showed significant stimulation of the OGG1 lyase activity [74,184]. Additional studies have reported the identification of novel OGG1 agonists [163], with recent follow-up studies determining the underlying mechanisms of the AP lyase catalytic function and the benefits of enhanced mitochondrial repair [185]. Recently, another potential activator of OGG1 has been described in which the AP lyase activity of OGG1 was stimulated via small-molecule screening [186]. Overall, the development of agonists for OGG1 (and potentially other DNA glycosylases) is in very early development, with the identification of potential applications of these agonists still in an exploratory phase. In the coming years, the potential druggability of OGG1 for enhanced catalytic efficiency will likely be developed, and with those applications there should be a greater appreciation of the potential benefits of enhanced BER of oxidatively induced DNA damage.
7. Perspectives
Having the privilege of working in the field of BER—with particular emphasis on the DNA glycosylases—for over 40 years, it has been very gratifying to witness the progressive understanding of the function and diverse biological roles that these enzymes play in organismal health. This evolving perspective includes the target search mechanisms and chemistry through which base removal occurs, the ability to correlate catalytic efficiencies with glycosylase structure–function studies, the discovery of unanticipated phenotypes in murine knockout and transgenic models, and the potential applications of linking structure–function studies of single-nucleotide polymorphic variants with human disease. Despite all of this progress, there are still major challenges and unanswered questions within these areas of investigation. One such issue that deserves ongoing investigation concerns inter-laboratory discrepancies in murine phenotypes, including differences in spontaneous carcinogenesis in Neil1-deficient mice. A potential clue that may help resolve this challenge is the status and influence of the intestinal microbiome on organismal health, and how this differs between laboratories. Another area that deserves further investigation is organ-specific disease manifestations with respect to inflammation. Specifically, in the Ogg1−/− model, the initiation of inflammatory cascades in these mice in response to oxidative stressors is significantly suppressed in the airway epithelial tissues and stomach, but enhanced in intestinal tissues. Developing a deeper understanding underlying these observations should result in a greater appreciation for the role(s) that OGG1 plays in the regulation of gene expression. In addition to the challenges, there are multiple areas representing opportunities in this field. This is especially germane with regard to clinical applications for personalized medicine related to disease susceptibility and the ability to create pharmacologically active small-molecule inhibitors and agonists of DNA glycosylases. Analyses of large-scale population- and disease-based sequencing, combined with systematic characterization of single-nucleotide variants, provide the opportunity to propose personalized medicine strategies to delay or prevent disease. This could entail dietary and lifestyle modifications, as well as vaccination when applicable. Finally, over the past 7 years, multiple laboratories and pharmaceutical companies have initiated screenings of different-sized libraries for inhibitors and, more recently, agonists of DNA glycosylases. As described herein, inhibitors of OGG1 may have broad applications in the treatment of human diseases, including asthma and acute myeloid leukemia. The potential health benefits associated with OGG1 agonists include suppression of weight gain and associated pathologies, as well as neurological diseases such as Parkinson’s and Alzheimer’s diseases. Overall, the robust history of past discoveries emanating from glycosylase-centric investigations bodes well for both fundamental basic scientific discoveries and potential applications with profound therapeutic benefits.
Funding
This work was supported by the following current NIH grants: National Institutes of Health (NIH) grants R01 ES-031086, R01 CA-55678, R01 ES-029357, and P01 CA-160032. Work in our laboratories was also supported by the Oregon Institute of Occupational Health Sciences at Oregon Health & Science University via funds from the Division of Consumer and Business Services of the State of Oregon (ORS 656.630).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Acknowledgments
I wish to express gratitude to Amanda McCullough and Irina Minko for their valuable contributions and recommendations concerning this review. I thank Michael Luzadder for input and construction of the graphical abstract created in BioRender.com (last accessed date: 9th November 2022). For the portions of the work represented in this review that emanated from our (Lloyd and McCullough) laboratories, we wish to thank Irina Minko, Vladimir Vartanian, Harini Sampath, ML Dodson, Nichole Owen, Nate Donley, Marcus Calkins, Elliott Gruskin, Robert Schrock, Diane Dowd, and Bin Sun for their ingenuity and dedication. We also thank Miral Dizdaroglu and his group for our long-term collaborations on the substrate specificities of NEIL1, its variants, and other projects.
Conflicts of Interest
R.S.L. is a founding member of Luciole Pharmaceuticals, Inc., which is developing agonists for OGG1.
References
- Kow, Y.W. Base excision repair in E. coli—An overview. Ann. N. Y. Acad. Sci. 1994, 726, 178–180. [Google Scholar] [CrossRef] [PubMed]
- Laval, J. Role of DNA repair enzymes in the cellular resistance to oxidative stress. Pathol. Biol. 1996, 44, 14–24. [Google Scholar] [PubMed]
- Beard, W.A.; Horton, J.K.; Prasad, R.; Wilson, S.H. Eukaryotic base excision repair: New approaches shine light on mechanism. Annu. Rev. Biochem. 2019, 88, 137–162. [Google Scholar] [CrossRef] [PubMed]
- Bandaru, V.; Sunkara, S.; Wallace, S.S.; Bond, J.P. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair 2002, 1, 517–529. [Google Scholar] [CrossRef]
- Hazra, T.K.; Izumi, T.; Boldogh, I.; Imhoff, B.; Kow, Y.W.; Jaruga, P.; Dizdaroglu, M.; Mitra, S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 3523–3528. [Google Scholar] [CrossRef] [Green Version]
- Morland, I.; Rolseth, V.; Luna, L.; Rognes, T.; Bjoras, M.; Seeberg, E. Human DNA glycosylases of the bacterial Fpg/MutM superfamily: An alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res. 2002, 30, 4926–4936. [Google Scholar] [CrossRef]
- Rosenquist, T.A.; Zaika, E.; Fernandes, A.S.; Zharkov, D.O.; Miller, H.; Grollman, A.P. The novel DNA glycosylase, NEIL1, protects mammalian cells from radiation-mediated cell death. DNA Repair 2003, 2, 581–591. [Google Scholar] [CrossRef]
- van der Kemp, P.A.; Thomas, D.; Barbey, R.; de Oliveira, R.; Boiteux, S. Cloning and expression in Escherichia coli of the OGG1 gene of Saccharomyces cerevisiae, which codes for a DNA glycosylase that excises 7,8-dihydro-8-oxoguanine and 2,6-diamino-4-hydroxy-5-N-methylformamidopyrimidine. Proc. Natl. Acad. Sci. USA 1996, 93, 5197–5202. [Google Scholar] [CrossRef] [Green Version]
- Aburatani, H.; Hippo, Y.; Ishida, T.; Takashima, R.; Matsuba, C.; Kodama, T.; Takao, M.; Yasui, A.; Yamamoto, K.; Asano, M. Cloning and characterization of mammalian 8-hydroxyguanine-specific DNA glycosylase/apurinic, apyrimidinic lyase, a functional mutM homologue. Cancer Res. 1997, 57, 2151–2156. [Google Scholar]
- Arai, K.; Morishita, K.; Shinmura, K.; Kohno, T.; Kim, S.R.; Nohmi, T.; Taniwaki, M.; Ohwada, S.; Yokota, J. Cloning of a human homolog of the yeast OGG1 gene that is involved in the repair of oxidative DNA damage. Oncogene 1997, 14, 2857–2861. [Google Scholar] [CrossRef] [Green Version]
- Bjoras, M.; Luna, L.; Johnsen, B.; Hoff, E.; Haug, T.; Rognes, T.; Seeberg, E. Opposite base-dependent reactions of a human base excision repair enzyme on DNA containing 7,8-dihydro-8-oxoguanine and abasic sites. EMBO J. 1997, 16, 6314–6322. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Nash, H.M.; Verdine, G.L. A mammalian DNA repair enzyme that excises oxidatively damaged guanines maps to a locus frequently lost in lung cancer. Curr. Biol. 1997, 7, 397–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radicella, J.P.; Dherin, C.; Desmaze, C.; Fox, M.S.; Boiteux, S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1997, 94, 8010–8015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roldan-Arjona, T.; Wei, Y.F.; Carter, K.C.; Klungland, A.; Anselmino, C.; Wang, R.P.; Augustus, M.; Lindahl, T. Molecular cloning and functional expression of a human cDNA encoding the antimutator enzyme 8-hydroxyguanine-DNA glycosylase. Proc. Natl. Acad. Sci. USA 1997, 94, 8016–8020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenquist, T.A.; Zharkov, D.O.; Grollman, A.P. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc. Natl. Acad. Sci. USA 1997, 94, 7429–7434. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doublie, S.; Bandaru, V.; Bond, J.P.; Wallace, S.S. The crystal structure of human endonuclease VIII-like 1 (NEIL1) reveals a zincless finger motif required for glycosylase activity. Proc. Natl. Acad. Sci. USA 2004, 101, 10284–10289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, A.; Yang, W.; Karplus, M.; Verdine, G.L. Structure of a repair enzyme interrogating undamaged DNA elucidates recognition of damaged DNA. Nature 2005, 434, 612–618. [Google Scholar] [CrossRef]
- Imamura, K.; Wallace, S.S.; Doublie, S. Structural characterization of a viral NEIL1 ortholog unliganded and bound to abasic site-containing DNA. J. Biol. Chem. 2009, 284, 26174–26183. [Google Scholar] [CrossRef] [Green Version]
- Imamura, K.; Averill, A.; Wallace, S.S.; Doublie, S. Structural characterization of viral ortholog of human DNA glycosylase NEIL1 bound to thymine glycol or 5-hydroxyuracil-containing DNA. J. Biol. Chem. 2012, 287, 4288–4298. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.; Lu, L.; Zhang, J.; Yue, Z.; Song, J.; Zong, S.; Liu, M.; Stovicek, O.; Gao, Y.Q.; Yi, C. Tautomerization-dependent recognition and excision of oxidation damage in base-excision DNA repair. Proc. Natl. Acad. Sci. USA 2016, 113, 7792–7797. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Endutkin, A.V.; Bergonzo, C.; Fu, L.; Grollman, A.; Zharkov, D.O.; Simmerling, C. DNA deformation-coupled recognition of 8-oxoguanine: Conformational kinetic gating in human DNA glycosylase. J. Am. Chem. Soc. 2017, 139, 2682–2692. [Google Scholar] [CrossRef]
- Shigdel, U.K.; Ovchinnikov, V.; Lee, S.J.; Shih, J.A.; Karplus, M.; Nam, K.; Verdine, G.L. The trajectory of intrahelical lesion recognition and extrusion by the human 8-oxoguanine DNA glycosylase. Nat. Commun. 2020, 11, 4437. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, J.; Zhu, C.; Zhang, X.; Xiao, W.; Yan, Y.; Liu, L.; Zeng, H.; Gao, Y.Q.; Yi, C. DNA repair glycosylase hNEIL1 triages damaged bases via competing interaction modes. Nat. Commun. 2021, 12, 4108. [Google Scholar] [CrossRef]
- Lloyd, R.S.; Hanawalt, P.C.; Dodson, M.L. Processive action of T4 endonuclease V on ultraviolet-irradiated DNA. Nucleic Acids Res. 1980, 8, 5113–5127. [Google Scholar] [CrossRef] [Green Version]
- Gruskin, E.A.; Lloyd, R.S. The DNA scanning mechanism of T4 endonuclease V. Effect of NaCl concentration on processive nicking activity. J. Biol. Chem. 1986, 261, 9607–9613. [Google Scholar] [CrossRef]
- Dowd, D.R.; Lloyd, R.S. Site-directed mutagenesis of the T4 endonuclease V gene: The role of arginine-3 in the target search. Biochemistry 1989, 28, 8699–8705. [Google Scholar] [CrossRef]
- Dowd, D.R.; Lloyd, R.S. Biological significance of facilitated diffusion in protein-DNA interactions. Applications to T4 endonuclease V-initiated DNA repair. J. Biol. Chem. 1990, 265, 3424–3431. [Google Scholar] [CrossRef]
- Zharkov, D.O.; Grollman, A.P. The DNA trackwalkers: Principles of lesion search and recognition by DNA glycosylases. Mutat. Res. 2005, 577, 24–54. [Google Scholar] [CrossRef]
- Prakash, A.; Doublie, S.; Wallace, S.S. The Fpg/Nei family of DNA glycosylases: Substrates, structures, and search for damage. Prog. Mol. Biol. Transl. Sci. 2012, 110, 71–91. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.J.; Wallace, S.S. Hide and seek: How do DNA glycosylases locate oxidatively damaged DNA bases amidst a sea of undamaged bases? Free Radic. Biol. Med. 2017, 107, 170–178. [Google Scholar] [CrossRef]
- D’Augustin, O.; Huet, S.; Campalans, A.; Radicella, J.P. Lost in the crowd: How does human 8-oxoguanine DNA glycosylase 1 (OGG1) find 8-oxoguanine in the genome? Int. J. Mol. Sci. 2020, 21, 8360. [Google Scholar] [CrossRef]
- Kong, M.; Beckwitt, E.C.; Van Houten, B. Dynamic action of DNA repair proteins as revealed by single molecule techniques: Seeing is believing. DNA Repair 2020, 93, 102909. [Google Scholar] [CrossRef]
- Rose, A.S.; Bradley, A.R.; Valasatava, Y.; Duarte, J.M.; Prlic, A.; Rose, P.W. NGL viewer: Web-based molecular graphics for large complexes. Bioinformatics 2018, 34, 3755–3758. [Google Scholar] [CrossRef] [Green Version]
- McCullough, A.K.; Minko, I.G.; Nilsen, A.; Nagarajan, S.; Lloyd, R.S. Modulation of DNA glycosylase activities via small molecules. In DNA Damage, DNA Repair and Disease; Dizdaroglu, M., Lloyd, R.S., Eds.; The Royal Society of Chemistry: Cambridge, UK, 2021; Volume 1, pp. 323–347. [Google Scholar]
- Hegde, M.L.; Hegde, P.M.; Bellot, L.J.; Mandal, S.M.; Hazra, T.K.; Li, G.M.; Boldogh, I.; Tomkinson, A.E.; Mitra, S. Prereplicative repair of oxidized bases in the human genome is mediated by NEIL1 DNA glycosylase together with replication proteins. Proc. Natl. Acad. Sci. USA 2013, 110, E3090–E3099. [Google Scholar] [CrossRef] [Green Version]
- Hegde, P.M.; Dutta, A.; Sengupta, S.; Mitra, J.; Adhikari, S.; Tomkinson, A.E.; Li, G.M.; Boldogh, I.; Hazra, T.K.; Mitra, S.; et al. The C-terminal domain (CTD) of human DNA glycosylase NEIL1 is required for forming BERosome repair complex with DNA replication proteins at the replicating genome: Dominant negative function of the CTD. J. Biol. Chem. 2015, 290, 20919–20933. [Google Scholar] [CrossRef] [Green Version]
- Dou, H.; Mitra, S.; Hazra, T.K. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J. Biol. Chem. 2003, 278, 49679–49684. [Google Scholar] [CrossRef] [Green Version]
- Das, A.; Boldogh, I.; Lee, J.W.; Harrigan, J.A.; Hegde, M.L.; Piotrowski, J.; de Souza Pinto, N.; Ramos, W.; Greenberg, M.M.; Hazra, T.K.; et al. The human Werner syndrome protein stimulates repair of oxidative DNA base damage by the DNA glycosylase NEIL1. J. Biol. Chem. 2007, 282, 26591–26602. [Google Scholar] [CrossRef] [Green Version]
- Hegde, M.L.; Theriot, C.A.; Das, A.; Hegde, P.M.; Guo, Z.; Gary, R.K.; Hazra, T.K.; Shen, B.; Mitra, S. Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1. J. Biol. Chem. 2008, 283, 27028–27037. [Google Scholar] [CrossRef] [Green Version]
- Yeo, J.; Goodman, R.A.; Schirle, N.T.; David, S.S.; Beal, P.A. RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc. Natl. Acad. Sci. USA 2010, 107, 20715–20719. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Krishnamurthy, N.; Burrows, C.J.; David, S.S. Mutation versus repair: NEIL1 removal of hydantoin lesions in single-stranded, bulge, bubble, and duplex DNA contexts. Biochemistry 2010, 49, 1658–1666. [Google Scholar] [CrossRef] [Green Version]
- Hegde, M.L.; Banerjee, S.; Hegde, P.M.; Bellot, L.J.; Hazra, T.K.; Boldogh, I.; Mitra, S. Enhancement of NEIL1 protein-initiated oxidized DNA base excision repair by heterogeneous nuclear ribonucleoprotein U (hnRNP-U) via direct interaction. J. Biol. Chem. 2012, 287, 34202–34211. [Google Scholar] [CrossRef]
- Vartanian, V.; Minko, I.G.; Chawanthayatham, S.; Egner, P.A.; Lin, Y.C.; Earley, L.F.; Makar, R.; Eng, J.R.; Camp, M.T.; Li, L.; et al. NEIL1 protects against aflatoxin-induced hepatocellular carcinoma in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 4207–4212. [Google Scholar] [CrossRef] [Green Version]
- Roy, L.M.; Jaruga, P.; Wood, T.G.; McCullough, A.K.; Dizdaroglu, M.; Lloyd, R.S. Human polymorphic variants of the NEIL1 DNA glycosylase. J. Biol. Chem. 2007, 282, 15790–15798. [Google Scholar] [CrossRef] [Green Version]
- Dizdaroglu, M.; Coskun, E.; Jaruga, P. Repair of oxidatively induced DNA damage by DNA glycosylases: Mechanisms of action, substrate specificities and excision kinetics. Mutat. Res. Rev. Mutat. Res. 2017, 771, 99–127. [Google Scholar] [CrossRef]
- Minko, I.G.; Vartanian, V.L.; Tozaki, N.N.; Coskun, E.; Coskun, S.H.; Jaruga, P.; Yeo, J.; David, S.S.; Stone, M.P.; Egli, M.; et al. Recognition of DNA adducts by edited and unedited forms of DNA glycosylase NEIL1. DNA Repair 2020, 85, 102741. [Google Scholar] [CrossRef]
- Prakash, A.; Carroll, B.L.; Sweasy, J.B.; Wallace, S.S.; Doublie, S. Genome and cancer single nucleotide polymorphisms of the human NEIL1 DNA glycosylase: Activity, structure, and the effect of editing. DNA Repair 2014, 14, 17–26. [Google Scholar] [CrossRef] [Green Version]
- Minko, I.G.; Christov, P.P.; Li, L.; Stone, M.P.; McCullough, A.K.; Lloyd, R.S. Processing of N5-substituted formamidopyrimidine DNA adducts by DNA glycosylases NEIL1 and NEIL3. DNA Repair 2019, 73, 49–54. [Google Scholar] [CrossRef]
- Tomar, R.; Minko, I.G.; Kellum, A.H., Jr.; Voehler, M.W.; Stone, M.P.; McCullough, A.K.; Lloyd, R.S. DNA sequence modulates the efficiency of NEIL1-catalyzed excision of the aflatoxin B1-induced formamidopyrimidine guanine adduct. Chem. Res. Toxicol. 2021, 34, 901–911. [Google Scholar] [CrossRef]
- Hailer, M.K.; Slade, P.G.; Martin, B.D.; Rosenquist, T.A.; Sugden, K.D. Recognition of the oxidized lesions spiroiminodihydantoin and guanidinohydantoin in DNA by the mammalian base excision repair glycosylases NEIL1 and NEIL2. DNA Repair 2005, 4, 41–50. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, N.; Zhao, X.; Burrows, C.J.; David, S.S. Superior removal of hydantoin lesions relative to other oxidized bases by the human DNA glycosylase hNEIL1. Biochemistry 2008, 47, 7137–7146. [Google Scholar] [CrossRef] [Green Version]
- McKibbin, P.L.; Fleming, A.M.; Towheed, M.A.; Van Houten, B.; Burrows, C.J.; David, S.S. Repair of hydantoin lesions and their amine adducts in DNA by base and nucleotide excision repair. J. Am. Chem. Soc. 2013, 135, 13851–13861. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Liu, M.; Fleming, A.M.; Burrows, C.J.; Wallace, S.S. Neil3 and NEIL1 DNA glycosylases remove oxidative damages from quadruplex DNA and exhibit preferences for lesions in the telomeric sequence context. J. Biol. Chem. 2013, 288, 27263–27272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slyvka, A.; Mierzejewska, K.; Bochtler, M. Nei-like 1 (NEIL1) excises 5-carboxylcytosine directly and stimulates TDG-mediated 5-formyl and 5-carboxylcytosine excision. Sci. Rep. 2017, 7, 9001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couve-Privat, S.; Mace, G.; Rosselli, F.; Saparbaev, M.K. Psoralen-induced DNA adducts are substrates for the base excision repair pathway in human cells. Nucleic Acids Res. 2007, 35, 5672–5682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Couve, S.; Mace-Aime, G.; Rosselli, F.; Saparbaev, M.K. The human oxidative DNA glycosylase NEIL1 excises psoralen-induced interstrand DNA cross-links in a three-stranded DNA structure. J. Biol. Chem. 2009, 284, 11963–11970. [Google Scholar] [CrossRef] [Green Version]
- Martin, P.R.; Couve, S.; Zutterling, C.; Albelazi, M.S.; Groisman, R.; Matkarimov, B.T.; Parsons, J.L.; Elder, R.H.; Saparbaev, M.K. The human DNA glycosylases NEIL1 and NEIL3 excise psoralen-induced DNA-DNA cross-links in a four-stranded DNA structure. Sci. Rep. 2017, 7, 17438. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; de Souza-Pinto, N.C.; Haraguchi, K.; Hogue, B.A.; Jaruga, P.; Greenberg, M.M.; Dizdaroglu, M.; Bohr, V.A. Repair of formamidopyrimidines in DNA involves different glycosylases: Role of the OGG1, NTH1, and NEIL1 enzymes. J. Biol. Chem. 2005, 280, 40544–40551. [Google Scholar] [CrossRef] [Green Version]
- Sidorenko, V.S.; Grollman, A.P.; Jaruga, P.; Dizdaroglu, M.; Zharkov, D.O. Substrate specificity and excision kinetics of natural polymorphic variants and phosphomimetic mutants of human 8-oxoguanine-DNA glycosylase. FEBS J. 2009, 276, 5149–5162. [Google Scholar] [CrossRef] [Green Version]
- Sassa, A.; Beard, W.A.; Prasad, R.; Wilson, S.H. DNA sequence context effects on the glycosylase activity of human 8-oxoguanine DNA glycosylase. J. Biol. Chem. 2012, 287, 36702–36710. [Google Scholar] [CrossRef] [Green Version]
- Sidorenko, V.S.; Nevinsky, G.A.; Zharkov, D.O. Specificity of stimulation of human 8-oxoguanine-DNA glycosylase by AP endonuclease. Biochem. Biophys. Res. Commun. 2008, 368, 175–179. [Google Scholar] [CrossRef]
- Dherin, C.; Radicella, J.P.; Dizdaroglu, M.; Boiteux, S. Excision of oxidatively damaged DNA bases by the human a-hOgg1 protein and the polymorphic a-hOgg1(Ser326Cys) protein which is frequently found in human populations. Nucleic Acids Res. 1999, 27, 4001–4007. [Google Scholar] [CrossRef] [PubMed]
- Dizdaroglu, M. Base-excision repair of oxidative DNA damage by DNA glycosylases. Mutat. Res. 2005, 591, 45–59. [Google Scholar] [CrossRef] [PubMed]
- Schrock, R.D., 3rd; Lloyd, R.S. Reductive methylation of the amino terminus of endonuclease V eradicates catalytic activities. Evidence for an essential role of the amino terminus in the chemical mechanisms of catalysis. J. Biol. Chem. 1991, 266, 17631–17639. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.L.; Schrock, R.D., 3rd; Lloyd, R.S. Evidence for an imino intermediate in the T4 endonuclease V reaction. Biochemistry 1993, 32, 8284–8290. [Google Scholar] [CrossRef]
- Schrock, R.D., 3rd; Lloyd, R.S. Site-directed mutagenesis of the NH2 terminus of T4 endonuclease V. The position of the aNH2 moiety affects catalytic activity. J. Biol. Chem. 1993, 268, 880–886. [Google Scholar] [CrossRef]
- Golan, G.; Zharkov, D.O.; Grollman, A.P.; Dodson, M.L.; McCullough, A.K.; Lloyd, R.S.; Shoham, G. Structure of T4 pyrimidine dimer glycosylase in a reduced imine covalent complex with abasic site-containing DNA. J. Mol. Biol. 2006, 362, 241–258. [Google Scholar] [CrossRef]
- Dodson, M.L.; Michaels, M.L.; Lloyd, R.S. Unified catalytic mechanism for DNA glycosylases. J. Biol. Chem. 1994, 269, 32709–32712. [Google Scholar] [CrossRef]
- Sun, B.; Latham, K.A.; Dodson, M.L.; Lloyd, R.S. Studies on the catalytic mechanism of five DNA glycosylases. Probing for enzyme-DNA imino intermediates. J. Biol. Chem. 1995, 270, 19501–19508. [Google Scholar] [CrossRef] [Green Version]
- Vik, E.S.; Alseth, I.; Forsbring, M.; Helle, I.H.; Morland, I.; Luna, L.; Bjoras, M.; Dalhus, B. Biochemical mapping of human NEIL1 DNA glycosylase and AP lyase activities. DNA Repair 2012, 11, 766–773. [Google Scholar] [CrossRef]
- Forsbring, M.; Vik, E.S.; Dalhus, B.; Karlsen, T.H.; Bergquist, A.; Schrumpf, E.; Bjoras, M.; Boberg, K.M.; Alseth, I. Catalytically impaired hMYH and NEIL1 mutant proteins identified in patients with primary sclerosing cholangitis and cholangiocarcinoma. Carcinogenesis 2009, 30, 1147–1154. [Google Scholar] [CrossRef] [Green Version]
- Dalhus, B.; Forsbring, M.; Helle, I.H.; Vik, E.S.; Forstrom, R.J.; Backe, P.H.; Alseth, I.; Bjoras, M. Separation-of-function mutants unravel the dual-reaction mode of human 8-oxoguanine DNA glycosylase. Structure 2011, 19, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Allgayer, J.; Kitsera, N.; Bartelt, S.; Epe, B.; Khobta, A. Widespread transcriptional gene inactivation initiated by a repair intermediate of 8-oxoguanine. Nucleic Acids Res. 2016, 44, 7267–7280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morland, I.; Luna, L.; Gustad, E.; Seeberg, E.; Bjoras, M. Product inhibition and magnesium modulate the dual reaction mode of hOgg1. DNA Repair 2005, 4, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Kidane, D.; Murphy, D.L.; Sweasy, J.B. Accumulation of abasic sites induces genomic instability in normal human gastric epithelial cells during Helicobacter pylori infection. Oncogenesis 2014, 3, e128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Maayah, M.; Sweasy, J.B.; Alnajjar, K.S. The role of cysteines in the structure and function of OGG1. J. Biol. Chem. 2021, 296, 100093. [Google Scholar] [CrossRef]
- Popov, A.V.; Endutkin, A.V.; Yatsenko, D.D.; Yudkina, A.V.; Barmatov, A.E.; Makasheva, K.A.; Raspopova, D.Y.; Diatlova, E.A.; Zharkov, D.O. Molecular dynamics approach to identification of new OGG1 cancer-associated somatic variants with impaired activity. J. Biol. Chem. 2021, 296, 100229. [Google Scholar] [CrossRef]
- Wiederhold, L.; Leppard, J.B.; Kedar, P.; Karimi-Busheri, F.; Rasouli-Nia, A.; Weinfeld, M.; Tomkinson, A.E.; Izumi, T.; Prasad, R.; Wilson, S.H.; et al. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 2004, 15, 209–220. [Google Scholar] [CrossRef]
- Whitehouse, C.J.; Taylor, R.M.; Thistlethwaite, A.; Zhang, H.; Karimi-Busheri, F.; Lasko, D.D.; Weinfeld, M.; Caldecott, K.W. XRCC1 stimulates human polynucleotide kinase activity at damaged DNA termini and accelerates DNA single-strand break repair. Cell 2001, 104, 107–117. [Google Scholar] [CrossRef] [Green Version]
- Guan, X.; Bai, H.; Shi, G.; Theriot, C.A.; Hazra, T.K.; Mitra, S.; Lu, A.L. The human checkpoint sensor Rad9-Rad1-Hus1 interacts with and stimulates NEIL1 glycosylase. Nucleic Acids Res. 2007, 35, 2463–2472. [Google Scholar] [CrossRef]
- Dou, H.; Theriot, C.A.; Das, A.; Hegde, M.L.; Matsumoto, Y.; Boldogh, I.; Hazra, T.K.; Bhakat, K.K.; Mitra, S. Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes. J. Biol. Chem. 2008, 283, 3130–3140. [Google Scholar] [CrossRef] [Green Version]
- Theriot, C.A.; Hegde, M.L.; Hazra, T.K.; Mitra, S. RPA physically interacts with the human DNA glycosylase NEIL1 to regulate excision of oxidative DNA base damage in primer-template structures. DNA Repair 2010, 9, 643–652. [Google Scholar] [CrossRef]
- Noren Hooten, N.; Fitzpatrick, M.; Kompaniez, K.; Jacob, K.D.; Moore, B.R.; Nagle, J.; Barnes, J.; Lohani, A.; Evans, M.K. Coordination of DNA repair by NEIL1 and PARP-1: A possible link to aging. Aging 2012, 4, 674–685. [Google Scholar] [CrossRef] [PubMed]
- Dhenaut, A.; Boiteux, S.; Radicella, J.P. Characterization of the hOGG1 promoter and its expression during the cell cycle. Mutat. Res. 2000, 461, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Boldogh, I.; Milligan, D.; Lee, M.S.; Bassett, H.; Lloyd, R.S.; McCullough, A.K. hMYH cell cycle-dependent expression, subcellular localization and association with replication foci: Evidence suggesting replication-coupled repair of adenine:8-oxoguanine mispairs. Nucleic Acids Res. 2001, 29, 2802–2809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luna, L.; Rolseth, V.; Hildrestrand, G.A.; Otterlei, M.; Dantzer, F.; Bjoras, M.; Seeberg, E. Dynamic relocalization of hOGG1 during the cell cycle is disrupted in cells harbouring the hOGG1-Cys326 polymorphic variant. Nucleic Acids Res. 2005, 33, 1813–1824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, J.W.; Hazra, T.K.; Izumi, T.; Mitra, S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: Potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001, 29, 430–438. [Google Scholar] [CrossRef] [PubMed]
- Vidal, A.E.; Hickson, I.D.; Boiteux, S.; Radicella, J.P. Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: Bypass of the AP lyase activity step. Nucleic Acids Res. 2001, 29, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Marsin, S.; Vidal, A.E.; Sossou, M.; Menissier-de Murcia, J.; Le Page, F.; Boiteux, S.; de Murcia, G.; Radicella, J.P. Role of XRCC1 in the coordination and stimulation of oxidative DNA damage repair initiated by the DNA glycosylase hOGG1. J. Biol. Chem. 2003, 278, 44068–44074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokkapati, S.K.; Wiederhold, L.; Hazra, T.K.; Mitra, S. Stimulation of DNA glycosylase activity of OGG1 by NEIL1: Functional collaboration between two human DNA glycosylases. Biochemistry 2004, 43, 11596–11604. [Google Scholar] [CrossRef] [PubMed]
- Park, M.J.; Park, J.H.; Hahm, S.H.; Ko, S.I.; Lee, Y.R.; Chung, J.H.; Sohn, S.Y.; Cho, Y.; Kang, L.W.; Han, Y.S. Repair activities of human 8-oxoguanine DNA glycosylase are stimulated by the interaction with human checkpoint sensor Rad9-Rad1-Hus1 complex. DNA Repair 2009, 8, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Noren Hooten, N.; Kompaniez, K.; Barnes, J.; Lohani, A.; Evans, M.K. Poly(ADP-ribose) polymerase 1 (PARP-1) binds to 8-oxoguanine-DNA glycosylase (OGG1). J. Biol. Chem. 2011, 286, 44679–44690. [Google Scholar] [CrossRef] [PubMed]
- Ramdzan, Z.M.; Vadnais, C.; Pal, R.; Vandal, G.; Cadieux, C.; Leduy, L.; Davoudi, S.; Hulea, L.; Yao, L.; Karnezis, A.N.; et al. RAS transformation requires CUX1-dependent repair of oxidative DNA damage. PLoS Biol. 2014, 12, e1001807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pal, R.; Ramdzan, Z.M.; Kaur, S.; Duquette, P.M.; Marcotte, R.; Leduy, L.; Davoudi, S.; Lamarche-Vane, N.; Iulianella, A.; Nepveu, A. CUX2 protein functions as an accessory factor in the repair of oxidative DNA damage. J. Biol. Chem. 2015, 290, 22520–22531. [Google Scholar] [CrossRef] [Green Version]
- Ramdzan, Z.M.; Pal, R.; Kaur, S.; Leduy, L.; Berube, G.; Davoudi, S.; Vadnais, C.; Nepveu, A. The function of CUX1 in oxidative DNA damage repair is needed to prevent premature senescence of mouse embryo fibroblasts. Oncotarget 2015, 6, 3613–3626. [Google Scholar] [CrossRef] [Green Version]
- Kaur, S.; Coulombe, Y.; Ramdzan, Z.M.; Leduy, L.; Masson, J.Y.; Nepveu, A. Special AT-rich sequence-binding protein 1 (SATB1) functions as an accessory factor in base excision repair. J. Biol. Chem. 2016, 291, 22769–22780. [Google Scholar] [CrossRef] [Green Version]
- Jang, S.; Kumar, N.; Beckwitt, E.C.; Kong, M.; Fouquerel, E.; Rapic-Otrin, V.; Prasad, R.; Watkins, S.C.; Khuu, C.; Majumdar, C.; et al. Damage sensor role of UV-DDB during base excision repair. Nat. Struct. Mol. Biol. 2019, 26, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Han, X.; Akbari, M.; Croteau, D.L.; Rasmussen, L.J.; Bohr, V.A. Interaction between RECQL4 and OGG1 promotes repair of oxidative base lesion 8-oxoG and is regulated by SIRT1 deacetylase. Nucleic Acids Res. 2020, 48, 6530–6546. [Google Scholar] [CrossRef]
- Kumar, N.; Theil, A.F.; Roginskaya, V.; Ali, Y.; Calderon, M.; Watkins, S.C.; Barnes, R.P.; Opresko, P.L.; Pines, A.; Lans, H.; et al. Global and transcription-coupled repair of 8-oxoG is initiated by nucleotide excision repair proteins. Nat. Commun. 2022, 13, 974. [Google Scholar] [CrossRef]
- Ba, X.; Boldogh, I. 8-Oxoguanine DNA glycosylase 1: Beyond repair of the oxidatively modified base lesions. Redox Biol. 2018, 14, 669–678. [Google Scholar] [CrossRef]
- Bangalore, D.M.; Tessmer, I. Direct hOGG1-Myc interactions inhibit hOGG1 catalytic activity and recruit Myc to its promoters under oxidative stress. Nucleic Acids Res. 2022, 50, 10385–10398. [Google Scholar] [CrossRef]
- Sharma, N.; Chakravarthy, S.; Longley, M.J.; Copeland, W.C.; Prakash, A. The C-terminal tail of the NEIL1 DNA glycosylase interacts with the human mitochondrial single-stranded DNA binding protein. DNA Repair 2018, 65, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Thompson, M.K.; Arrington, J.F.; Terry, D.M.; Chakravarthy, S.; Prevelige, P.E.; Prakash, A. Novel interaction interfaces mediate the interaction between the NEIL1 DNA glycosylase and mitochondrial transcription factor A. Front. Cell Dev. Biol. 2022, 10, 893806. [Google Scholar] [CrossRef] [PubMed]
- Kamenisch, Y.; Fousteri, M.; Knoch, J.; von Thaler, A.K.; Fehrenbacher, B.; Kato, H.; Becker, T.; Dolle, M.E.; Kuiper, R.; Majora, M.; et al. Proteins of nucleotide and base excision repair pathways interact in mitochondria to protect from loss of subcutaneous fat, a hallmark of aging. J. Exp. Med. 2010, 207, 379–390. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.T.; De Luca, G.; Degan, P.; Parlanti, E.; Dogliotti, E.; Barnes, D.E.; Lindahl, T.; Yang, H.; Miller, J.H.; Bignami, M. Accumulation of the oxidative base lesion 8-hydroxyguanine in DNA of tumor-prone mice defective in both the Myh and Ogg1 DNA glycosylases. Cancer Res. 2004, 64, 4411–4414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, Y.; Yang, H.; Cunanan, C.; Okamoto, K.; Shibata, D.; Pan, J.; Barnes, D.E.; Lindahl, T.; McIlhatton, M.; Fishel, R.; et al. Deficiencies in mouse Myh and Ogg1 result in tumor predisposition and G to T mutations in codon 12 of the K-ras oncogene in lung tumors. Cancer Res. 2004, 64, 3096–3102. [Google Scholar] [CrossRef] [Green Version]
- Arai, T.; Kelly, V.P.; Minowa, O.; Noda, T.; Nishimura, S. The study using wild-type and Ogg1 knockout mice exposed to potassium bromate shows no tumor induction despite an extensive accumulation of 8-hydroxyguanine in kidney DNA. Toxicology 2006, 221, 179–186. [Google Scholar] [CrossRef]
- Kakehashi, A.; Ishii, N.; Okuno, T.; Fujioka, M.; Gi, M.; Wanibuchi, H. Enhanced susceptibility of Ogg1 mutant mice to multiorgan carcinogenesis. Int. J. Mol. Sci. 2017, 18, 1801. [Google Scholar] [CrossRef] [Green Version]
- Kunisada, M.; Sakumi, K.; Tominaga, Y.; Budiyanto, A.; Ueda, M.; Ichihashi, M.; Nakabeppu, Y.; Nishigori, C. 8-Oxoguanine formation induced by chronic UVB exposure makes Ogg1 knockout mice susceptible to skin carcinogenesis. Cancer Res. 2005, 65, 6006–6010. [Google Scholar] [CrossRef] [Green Version]
- Kakehashi, A.; Ishii, N.; Okuno, T.; Fujioka, M.; Gi, M.; Fukushima, S.; Wanibuchi, H. Progression of hepatic adenoma to carcinoma in Ogg1 mutant mice induced by phenobarbital. Oxid. Med. Cell. Longev. 2017, 2017, 8541064. [Google Scholar] [CrossRef] [Green Version]
- Vartanian, V.; Lowell, B.; Minko, I.G.; Wood, T.G.; Ceci, J.D.; George, S.; Ballinger, S.W.; Corless, C.L.; McCullough, A.K.; Lloyd, R.S. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc. Natl. Acad. Sci. USA 2006, 103, 1864–1869. [Google Scholar] [CrossRef] [Green Version]
- Chan, M.K.; Ocampo-Hafalla, M.T.; Vartanian, V.; Jaruga, P.; Kirkali, G.; Koenig, K.L.; Brown, S.; Lloyd, R.S.; Dizdaroglu, M.; Teebor, G.W. Targeted deletion of the genes encoding NTH1 and NEIL1 DNA N-glycosylases reveals the existence of novel carcinogenic oxidative damage to DNA. DNA Repair 2009, 8, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Rolseth, V.; Luna, L.; Olsen, A.K.; Suganthan, R.; Scheffler, K.; Neurauter, C.G.; Esbensen, Y.; Kusnierczyk, A.; Hildrestrand, G.A.; Graupner, A.; et al. No cancer predisposition or increased spontaneous mutation frequencies in NEIL DNA glycosylases-deficient mice. Sci. Rep. 2017, 7, 4384. [Google Scholar] [CrossRef]
- Takahashi, Y.; Nakatsuru, Y.; Zhang, S.; Shimizu, Y.; Kume, H.; Tanaka, K.; Ide, F.; Ishikawa, T. Enhanced spontaneous and aflatoxin-induced liver tumorigenesis in xeroderma pigmentosum group A gene-deficient mice. Carcinogenesis 2002, 23, 627–633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calkins, M.J.; Vartanian, V.; Owen, N.; Kirkali, G.; Jaruga, P.; Dizdaroglu, M.; McCullough, A.K.; Lloyd, R.S. Enhanced sensitivity of Neil1−/− mice to chronic UVB exposure. DNA Repair 2016, 48, 43–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampath, H.; Batra, A.K.; Vartanian, V.; Carmical, J.R.; Prusak, D.; King, I.B.; Lowell, B.; Earley, L.F.; Wood, T.G.; Marks, D.L.; et al. Variable penetrance of metabolic phenotypes and development of high-fat diet-induced adiposity in NEIL1-deficient mice. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E724–E734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampath, H.; Vartanian, V.; Rollins, M.R.; Sakumi, K.; Nakabeppu, Y.; Lloyd, R.S. 8-Oxoguanine DNA glycosylase (OGG1) deficiency increases susceptibility to obesity and metabolic dysfunction. PLoS ONE 2012, 7, e51697. [Google Scholar] [CrossRef]
- Vartanian, V.; Tumova, J.; Dobrzyn, P.; Dobrzyn, A.; Nakabeppu, Y.; Lloyd, R.S.; Sampath, H. 8-oxoguanine DNA glycosylase (OGG1) deficiency elicits coordinated changes in lipid and mitochondrial metabolism in muscle. PLoS ONE 2017, 12, e0181687. [Google Scholar] [CrossRef] [Green Version]
- Aron-Wisnewsky, J.; Warmbrunn, M.V.; Nieuwdorp, M.; Clement, K. Metabolism and metabolic disorders and the microbiome: The intestinal microbiota associated with obesity, lipid metabolism, and metabolic health-pathophysiology and therapeutic strategies. Gastroenterology 2021, 160, 573–599. [Google Scholar] [CrossRef]
- Simon, H.; Vartanian, V.; Wong, M.H.; Nakabeppu, Y.; Sharma, P.; Lloyd, R.S.; Sampath, H. OGG1 deficiency alters the intestinal microbiome and increases intestinal inflammation in a mouse model. PLoS ONE 2020, 15, e0227501. [Google Scholar] [CrossRef] [Green Version]
- Komakula, S.S.B.; Tumova, J.; Kumaraswamy, D.; Burchat, N.; Vartanian, V.; Ye, H.; Dobrzyn, A.; Lloyd, R.S.; Sampath, H. The DNA repair protein OGG1 protects against obesity by altering mitochondrial energetics in white adipose tissue. Sci. Rep. 2018, 8, 14886. [Google Scholar] [CrossRef] [Green Version]
- Burchat, N.; Sharma, P.; Ye, H.; Komakula, S.S.B.; Dobrzyn, A.; Vartanian, V.; Lloyd, R.S.; Sampath, H. Maternal transmission of human OGG1 protects mice against genetically- and diet-Induced obesity through increased tissue mitochondrial content. Front. Cell Dev. Biol. 2021, 9, 718962. [Google Scholar] [CrossRef] [PubMed]
- Komakula, S.S.B.; Blaze, B.; Ye, H.; Dobrzyn, A.; Sampath, H. A Novel Role for the DNA Repair Enzyme 8-Oxoguanine DNA Glycosylase in Adipogenesis. Int. J. Mol. Sci. 2021, 22, 1152. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Galick, H.; Wallace, S.S. Attempted base excision repair of ionizing radiation damage in human lymphoblastoid cells produces lethal and mutagenic double strand breaks. DNA Repair 2004, 3, 1323–1334. [Google Scholar] [CrossRef] [PubMed]
- Yang, N.; Chaudhry, M.A.; Wallace, S.S. Base excision repair by hNTH1 and hOGG1: A two edged sword in the processing of DNA damage in g-irradiated human cells. DNA Repair 2006, 5, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Yuan, K.; Yan, C.; Fox, J., 3rd; Gaid, M.; Breitwieser, W.; Bansal, A.K.; Zeng, H.; Gao, H.; Wu, M. 8-Oxoguanine-DNA glycosylase 1 deficiency modifies allergic airway inflammation by regulating STAT6 and IL-4 in cells and in mice. Free Radic. Biol. Med. 2012, 52, 392–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ba, X.; Aguilera-Aguirre, L.; Rashid, Q.T.; Bacsi, A.; Radak, Z.; Sur, S.; Hosoki, K.; Hegde, M.L.; Boldogh, I. The role of 8-oxoguanine DNA glycosylase-1 in inflammation. Int. J. Mol. Sci. 2014, 15, 16975–16997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabley, J.G.; Pacher, P.; Deb, A.; Wallace, R.; Elder, R.H.; Szabo, C. Potential role for 8-oxoguanine DNA glycosylase in regulating inflammation. FASEB J. 2005, 19, 290–292. [Google Scholar] [CrossRef]
- Touati, E.; Michel, V.; Thiberge, J.M.; Ave, P.; Huerre, M.; Bourgade, F.; Klungland, A.; Labigne, A. Deficiency in OGG1 protects against inflammation and mutagenic effects associated with H. pylori infection in mouse. Helicobacter 2006, 11, 494–505. [Google Scholar] [CrossRef]
- Yang, J.; Mitra, A.; Dojer, N.; Fu, S.; Rowicka, M.; Brasier, A.R. A probabilistic approach to learn chromatin architecture and accurate inference of the NF-kB/RelA regulatory network using ChIP-Seq. Nucleic Acids Res. 2013, 41, 7240–7259. [Google Scholar] [CrossRef] [Green Version]
- Pan, L.; Zhu, B.; Hao, W.; Zeng, X.; Vlahopoulos, S.A.; Hazra, T.K.; Hegde, M.L.; Radak, Z.; Bacsi, A.; Brasier, A.R.; et al. Oxidized guanine base lesions function in 8-oxoguanine DNA glycosylase-1-mediated epigenetic regulation of nuclear factor kB-driven gene expression. J. Biol. Chem. 2016, 291, 25553–25566. [Google Scholar] [CrossRef] [Green Version]
- Park, J.W.; Han, Y.I.; Kim, S.W.; Kim, T.M.; Yeom, S.C.; Kang, J.; Park, J. 8-OxoG in GC-rich Sp1 binding sites enhances gene transcription in adipose tissue of juvenile mice. Sci. Rep. 2019, 9, 15618. [Google Scholar] [CrossRef]
- Perillo, B.; Ombra, M.N.; Bertoni, A.; Cuozzo, C.; Sacchetti, S.; Sasso, A.; Chiariotti, L.; Malorni, A.; Abbondanza, C.; Avvedimento, E.V. DNA oxidation as triggered by H3K9me2 demethylation drives estrogen-induced gene expression. Science 2008, 319, 202–206. [Google Scholar] [CrossRef]
- Fleming, A.M.; Burrows, C.J. G-quadruplex folds of the human telomere sequence alter the site reactivity and reaction pathway of guanine oxidation compared to duplex DNA. Chem. Res. Toxicol. 2013, 26, 593–607. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A.M.; Zhu, J.; Ding, Y.; Visser, J.A.; Zhu, J.; Burrows, C.J. Human DNA repair genes possess potential G-quadruplex sequences in their promoters and 5′-untranslated regions. Biochemistry 2018, 57, 991–1002. [Google Scholar] [CrossRef]
- Rogers, R.A.; Fleming, A.M.; Burrows, C.J. Rapid screen of potential i-motif forming sequences in DNA repair gene promoters. ACS Omega 2018, 3, 9630–9635. [Google Scholar] [CrossRef] [Green Version]
- Fleming, A.M.; Zhu, J.; Ding, Y.; Esders, S.; Burrows, C.J. Oxidative modification of guanine in a potential Z-DNA-forming sequence of a gene promoter impacts gene expression. Chem. Res. Toxicol. 2019, 32, 899–909. [Google Scholar] [CrossRef]
- Donley, N.; Jaruga, P.; Coskun, E.; Dizdaroglu, M.; McCullough, A.K.; Lloyd, R.S. Small molecule inhibitors of 8-oxoguanine DNA glycosylase-1 (OGG1). ACS Chem. Biol. 2015, 10, 2334–2343. [Google Scholar] [CrossRef] [Green Version]
- Tahara, Y.K.; Auld, D.; Ji, D.; Beharry, A.A.; Kietrys, A.M.; Wilson, D.L.; Jimenez, M.; King, D.; Nguyen, Z.; Kool, E.T. Potent and selective inhibitors of 8-oxoguanine DNA glycosylase. J. Am. Chem. Soc. 2018, 140, 2105–2114. [Google Scholar] [CrossRef] [Green Version]
- Visnes, T.; Cazares-Korner, A.; Hao, W.; Wallner, O.; Masuyer, G.; Loseva, O.; Mortusewicz, O.; Wiita, E.; Sarno, A.; Manoilov, A.; et al. Small-molecule inhibitor of OGG1 suppresses proinflammatory gene expression and inflammation. Science 2018, 362, 834–839. [Google Scholar] [CrossRef] [Green Version]
- Tahara, Y.K.; Kietrys, A.M.; Hebenbrock, M.; Lee, Y.; Wilson, D.L.; Kool, E.T. Dual inhibitors of 8-oxoguanine surveillance by OGG1 and NUDT1. ACS Chem. Biol. 2019, 14, 2606–2615. [Google Scholar] [CrossRef]
- Ling, H.; Song, C.; Fang, Y.; Yin, Y.; Wu, Z.; Wang, Y.; Xu, Z.; Gao, S.; Li, A.; Liu, G. TH5487, a small molecule inhibitor of OGG1, attenuates pulmonary fibrosis by NEDD4L-mediated OGG1 degradation. Chem. Biol. Interact. 2022, 362, 109999. [Google Scholar] [CrossRef] [PubMed]
- Xia, J.; Wu, S.; Wu, G.; Yang, J. Inhibition of 8-oxoguanine DNA glycosylase (OGG1) expression suppresses polycystic ovarian syndrome via the NF-kB signaling pathway. Reprod. Biol. 2022, 22, 100679. [Google Scholar] [CrossRef] [PubMed]
- Baquero, J.M.; Benitez-Buelga, C.; Rajagopal, V.; Zhenjun, Z.; Torres-Ruiz, R.; Muller, S.; Hanna, B.M.F.; Loseva, O.; Wallner, O.; Michel, M.; et al. Small molecule inhibitor of OGG1 blocks oxidative DNA damage repair at telomeres and potentiates methotrexate anticancer effects. Sci. Rep. 2021, 11, 3490. [Google Scholar] [CrossRef] [PubMed]
- Baquero, J.M.; Marchena-Perea, E.; Mirabet, R.; Torres-Ruiz, R.; Blanco-Aparicio, C.; Rodriguez-Perales, S.; Helleday, T.; Benitez-Buelga, C.; Benitez, J.; Osorio, A. OGG1 inhibition triggers synthetic lethality and enhances the effect of PARP inhibitor olaparib in BRCA1-deficient TNBC cells. Front. Oncol. 2022, 12, 888810. [Google Scholar] [CrossRef]
- Karsten, S. Targeting the DNA repair enzymes MTH1 and OGG1 as a novel approach to treat inflammatory diseases. Basic Clin. Pharmacol. Toxicol. 2022, 131, 95–103. [Google Scholar] [CrossRef]
- Gatto, E.M.; Rojas, N.G.; Persi, G.; Etcheverry, J.L.; Cesarini, M.E.; Perandones, C. Huntington disease: Advances in the understanding of its mechanisms. Clin. Park. Relat. Disord. 2020, 3, 100056. [Google Scholar] [CrossRef]
- Monckton, D.G. The contribution of somatic expansion of the CAG repeat to symptomatic development in Huntington’s disease: A historical perspective. J. Huntingtons. Dis. 2021, 10, 7–33. [Google Scholar] [CrossRef]
- Kovtun, I.V.; Liu, Y.; Bjoras, M.; Klungland, A.; Wilson, S.H.; McMurray, C.T. OGG1 initiates age-dependent CAG trinucleotide expansion in somatic cells. Nature 2007, 447, 447–452. [Google Scholar] [CrossRef] [Green Version]
- Goula, A.V.; Berquist, B.R.; Wilson, D.M., 3rd; Wheeler, V.C.; Trottier, Y.; Merienne, K. Stoichiometry of base excision repair proteins correlates with increased somatic CAG instability in striatum over cerebellum in Huntington’s disease transgenic mice. PLoS Genet. 2009, 5, e1000749. [Google Scholar] [CrossRef]
- Kovtun, I.V.; Johnson, K.O.; McMurray, C.T. Cockayne syndrome B protein antagonizes OGG1 in modulating CAG repeat length in vivo. Aging 2011, 3, 509–514. [Google Scholar] [CrossRef] [Green Version]
- Iyer, R.R.; Pluciennik, A. DNA mismatch repair and its role in Huntington’s disease. J. Huntingtons. Dis. 2021, 10, 75–94. [Google Scholar] [CrossRef] [PubMed]
- Budworth, H.; Harris, F.R.; Williams, P.; Lee, D.Y.; Holt, A.; Pahnke, J.; Szczesny, B.; Acevedo-Torres, K.; Ayala-Pena, S.; McMurray, C.T. Suppression of somatic expansion delays the onset of pathophysiology in a mouse model of Huntington’s disease. PLoS Genet. 2015, 11, e1005267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollersen, L.; Rowe, A.D.; Illuzzi, J.L.; Hildrestrand, G.A.; Gerhold, K.J.; Tveteras, L.; Bjolgerud, A.; Wilson, D.M., 3rd; Bjoras, M.; Klungland, A. Neil1 is a genetic modifier of somatic and germline CAG trinucleotide repeat instability in R6/1 mice. Hum. Mol. Genet. 2012, 21, 4939–4947. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Canugovi, C.; Yoon, J.S.; Feldman, N.H.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. Endonuclease VIII-like 1 (NEIL1) promotes short-term spatial memory retention and protects from ischemic stroke-induced brain dysfunction and death in mice. Proc. Natl. Acad. Sci. USA 2012, 109, 14948–14953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canugovi, C.; Misiak, M.; Scheibye-Knudsen, M.; Croteau, D.L.; Mattson, M.P.; Bohr, V.A. Loss of NEIL1 causes defects in olfactory function in mice. Neurobiol. Aging 2015, 36, 1007–1012. [Google Scholar] [CrossRef] [Green Version]
- Yang, B.; Figueroa, D.M.; Hou, Y.; Babbar, M.; Baringer, S.L.; Croteau, D.L.; Bohr, V.A. NEIL1 stimulates neurogenesis and suppresses neuroinflammation after stress. Free Radic. Biol. Med. 2019, 141, 47–58. [Google Scholar] [CrossRef]
- Hildrestrand, G.A.; Rolseth, V.; Kunath, N.; Suganthan, R.; Jensen, V.; Bugaj, A.M.; Fernandez-Berrocal, M.S.; Sikko, S.B.; Vetlesen, S.; Kusnierczyk, A.; et al. NEIL1 and NEIL2 DNA glycosylases modulate anxiety and learning in a cooperative manner in mice. Commun. Biol. 2021, 4, 1354. [Google Scholar] [CrossRef]
- Hung, R.J.; Hall, J.; Brennan, P.; Boffetta, P. Genetic polymorphisms in the base excision repair pathway and cancer risk: A HuGE review. Am. J. Epidemiol. 2005, 162, 925–942. [Google Scholar] [CrossRef] [Green Version]
- Hill, J.W.; Evans, M.K. Dimerization and opposite base-dependent catalytic impairment of polymorphic S326C OGG1 glycosylase. Nucleic Acids Res. 2006, 34, 1620–1632. [Google Scholar] [CrossRef] [Green Version]
- Bravard, A.; Vacher, M.; Moritz, E.; Vaslin, L.; Hall, J.; Epe, B.; Radicella, J.P. Oxidation status of human OGG1-S326C polymorphic variant determines cellular DNA repair capacity. Cancer Res. 2009, 69, 3642–3649. [Google Scholar] [CrossRef]
- Baptiste, B.A.; Katchur, S.R.; Fivenson, E.M.; Croteau, D.L.; Rumsey, W.L.; Bohr, V.A. Enhanced mitochondrial DNA repair of the common disease-associated variant, Ser326Cys, of hOGG1 through small molecule intervention. Free Radic. Biol. Med. 2018, 124, 149–162. [Google Scholar] [CrossRef]
- Kabzinski, J.; Walczak, A.; Dziki, A.; Mik, M.; Majsterek, I. Impact of the Ser326Cys polymorphism of the OGG1 gene on the level of oxidative DNA damage in patients with colorectal cancer. Pol. Przegl. Chir. 2018, 90, 13–15. [Google Scholar] [CrossRef] [Green Version]
- Alanazi, M.; Pathan, A.A.K.; Shaik, J.P.; Alhadheq, A.; Khan, Z.; Khan, W.; Al Naeem, A.; Parine, N.R. The hOGG1 Ser326Cys gene polymorphism and breast cancer risk in Saudi population. Pathol. Oncol. Res. 2017, 23, 525–535. [Google Scholar] [CrossRef]
- Costa, E.F.; Santos, E.S.; Liutti, V.T.; Leal, F.; Santos, V.C.; Rinck-Junior, J.A.; Mariano, F.V.; Coutinho-Camillo, C.M.; Altemani, A.; Lima, C.S.; et al. Association between polymorphisms in genes related to DNA base-excision repair with risk and prognosis of oropharyngeal squamous cell carcinoma. J. Cancer Res. Clin. Oncol. 2016, 142, 1917–1926. [Google Scholar] [CrossRef]
- Gotoh, N.; Saitoh, T.; Takahashi, N.; Kasamatsu, T.; Minato, Y.; Lobna, A.; Oda, T.; Hoshino, T.; Sakura, T.; Shimizu, H.; et al. Association between OGG1 S326C CC genotype and elevated relapse risk in acute myeloid leukemia. Int. J. Hematol. 2018, 108, 246–253. [Google Scholar] [CrossRef]
- Smolarz, B.; Michalska, M.M.; Samulak, D.; Wojcik, L.; Romanowicz, H. Studies of correlations between single nucleotide polymorphisms of DNA repair genes and endometrial cancer in Polish women. Anticancer Res. 2018, 38, 5223–5229. [Google Scholar] [CrossRef]
- Xu, J.; Zheng, S.L.; Turner, A.; Isaacs, S.D.; Wiley, K.E.; Hawkins, G.A.; Chang, B.L.; Bleecker, E.R.; Walsh, P.C.; Meyers, D.A.; et al. Associations between hOGG1 sequence variants and prostate cancer susceptibility. Cancer Res. 2002, 62, 2253–2257. [Google Scholar]
- Chen, L.; Elahi, A.; Pow-Sang, J.; Lazarus, P.; Park, J. Association between polymorphism of human oxoguanine glycosylase 1 and risk of prostate cancer. J. Urol. 2003, 170, 2471–2474. [Google Scholar] [CrossRef]
- Le Marchand, L.; Donlon, T.; Lum-Jones, A.; Seifried, A.; Wilkens, L.R. Association of the hOGG1 Ser326Cys polymorphism with lung cancer risk. Cancer Epidemiol. Biomark. Prev. 2002, 11, 409–412. [Google Scholar]
- Geer, L.Y.; Marchler-Bauer, A.; Geer, R.C.; Han, L.; He, J.; He, S.; Liu, C.; Shi, W.; Bryant, S.H. The NCBI BioSystems database. Nucleic Acids Res. 2010, 38, D492–D496. [Google Scholar] [CrossRef] [Green Version]
- Hyun, J.W.; Choi, J.Y.; Zeng, H.H.; Lee, Y.S.; Kim, H.S.; Yoon, S.H.; Chung, M.H. Leukemic cell line, KG-1 has a functional loss of hOGG1 enzyme due to a point mutation and 8-hydroxydeoxyguanosine can kill KG-1. Oncogene 2000, 19, 4476–4479. [Google Scholar] [CrossRef] [Green Version]
- Hyun, J.W.; Cheon, G.J.; Kim, H.S.; Lee, Y.S.; Choi, E.Y.; Yoon, B.H.; Kim, J.S.; Chung, M.H. Radiation sensitivity depends on OGG1 activity status in human leukemia cell lines. Free Radic. Biol. Med. 2002, 32, 212–220. [Google Scholar] [CrossRef]
- Dohner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Buchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A.; et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017, 129, 424–447. [Google Scholar] [CrossRef] [Green Version]
- Owen, N.; Minko, I.G.; Moellmer, S.A.; Cammann, S.K.; Lloyd, R.S.; McCullough, A.K. Enhanced cytarabine-induced killing in OGG1-deficient acute myeloid leukemia cells. Proc. Natl. Acad. Sci. USA 2021, 118, e2016833118. [Google Scholar] [CrossRef]
- Galick, H.A.; Marsden, C.G.; Kathe, S.; Dragon, J.A.; Volk, L.; Nemec, A.A.; Wallace, S.S.; Prakash, A.; Doublie, S.; Sweasy, J.B. The NEIL1 G83D germline DNA glycosylase variant induces genomic instability and cellular transformation. Oncotarget 2017, 8, 85883–85895. [Google Scholar] [CrossRef] [Green Version]
- Minko, I.G.; Vartanian, V.L.; Tozaki, N.N.; Linde, O.K.; Jaruga, P.; Coskun, S.H.; Coskun, E.; Qu, C.; He, H.; Xu, C.; et al. Characterization of rare NEIL1 variants found in East Asian populations. DNA Repair 2019, 79, 32–39. [Google Scholar] [CrossRef]
- Thameem, F.; Puppala, S.; Lehman, D.M.; Stern, M.P.; Blangero, J.; Abboud, H.E.; Duggirala, R.; Habib, S.L. The Ser(326)Cys polymorphism of 8-oxoguanine glycosylase 1 (OGG1) is associated with type 2 diabetes in Mexican Americans. Hum. Hered. 2010, 70, 97–101. [Google Scholar] [CrossRef] [Green Version]
- Anderson, S.M.; Naidoo, R.N.; Ramkaran, P.; Asharam, K.; Muttoo, S.; Chuturgoon, A.A. OGG1 Ser326Cys polymorphism, HIV, obesity and air pollution exposure influences adverse birth outcome susceptibility, within South African Women. Reprod. Toxicol. 2018, 79, 8–15. [Google Scholar] [CrossRef]
- Sliwinska, A.; Kwiatkowski, D.; Czarny, P.; Toma, M.; Wigner, P.; Drzewoski, J.; Fabianowska-Majewska, K.; Szemraj, J.; Maes, M.; Galecki, P.; et al. The levels of 7,8-dihydrodeoxyguanosine (8-oxoG) and 8-oxoguanine DNA glycosylase 1 (OGG1)—A potential diagnostic biomarkers of Alzheimer’s disease. J. Neurol. Sci. 2016, 368, 155–159. [Google Scholar] [CrossRef]
- Sanders, L.H.; Paul, K.C.; Howlett, E.H.; Lawal, H.; Boppana, S.; Bronstein, J.M.; Ritz, B.; Greenamyre, J.T. Editor’s highlight: Base excision repair variants and pesticide exposure increase Parkinson’s disease risk. Toxicol. Sci. 2017, 158, 188–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corella, D.; Ramirez-Sabio, J.B.; Coltell, O.; Ortega-Azorin, C.; Estruch, R.; Martinez-Gonzalez, M.A.; Salas-Salvado, J.; Sorli, J.V.; Castaner, O.; Aros, F.; et al. Effects of the Ser326Cys polymorphism in the DNA repair OGG1 gene on cancer, cardiovascular, and all-cause mortality in the PREDIMED study: Modulation by diet. J. Acad. Nutr. Diet. 2018, 118, 589–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fromme, J.C.; Bruner, S.D.; Yang, W.; Karplus, M.; Verdine, G.L. Product-assisted catalysis in base-excision DNA repair. Nat. Struct. Biol. 2003, 10, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Katchur, S.R.; Jiang, Y.; Briand, J.; Schaber, M.; Kreatsoulas, C.; Schwartz, B.; Thrall, S.; Davis, A.M.; Duvall, S.; et al. Small molecule-mediated allosteric activation of the base excision repair enzyme 8-oxoguanine DNA glycosylase and its impact on mitochondrial function. Sci. Rep. 2022, 12, 14685. [Google Scholar] [CrossRef]
- Michel, M.; Benitez-Buelga, C.; Calvo, P.A.; Hanna, B.M.F.; Mortusewicz, O.; Masuyer, G.; Davies, J.; Wallner, O.; Sanjiv, K.; Albers, J.J.; et al. Small-molecule activation of OGG1 increases oxidative DNA damage repair by gaining a new function. Science 2022, 376, 1471–1476. [Google Scholar] [CrossRef]
Figure 1.
Charge distribution on the surface (A) and nucleotide flipping (B) in co-crystal structures of DNA glycosylases: The crystallographic coordinates of NEIL1 and OGG1 bound to lesion-containing DNA were retrieved from the Protein Data Bank (rcsb.org, last accessed date: 27th February 2020; PDB IDs 5ITX [20] and 1YQR [17], respectively). The images of Poisson–Boltzmann electrostatic potential surface maps were generated using the Schrödinger package (schrodinger.com). The surface is colored according to electrostatic potential (positive: blue; negative: red). The images of glycosylase–DNA complexes were created with NGL Viewer [33] at the RCSB PDB site (rcsb.org). The damaged nucleotides (encircled) are at extrahelical positions. This figure was adapted from [34].
Figure 1.
Charge distribution on the surface (A) and nucleotide flipping (B) in co-crystal structures of DNA glycosylases: The crystallographic coordinates of NEIL1 and OGG1 bound to lesion-containing DNA were retrieved from the Protein Data Bank (rcsb.org, last accessed date: 27th February 2020; PDB IDs 5ITX [20] and 1YQR [17], respectively). The images of Poisson–Boltzmann electrostatic potential surface maps were generated using the Schrödinger package (schrodinger.com). The surface is colored according to electrostatic potential (positive: blue; negative: red). The images of glycosylase–DNA complexes were created with NGL Viewer [33] at the RCSB PDB site (rcsb.org). The damaged nucleotides (encircled) are at extrahelical positions. This figure was adapted from [34].

Figure 2.
Chemistry of the mechanisms of NEIL1 and OGG1.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Lloyd, R.S. Complex Roles of NEIL1 and OGG1: Insights Gained from Murine Knockouts and Human Polymorphic Variants. DNA 2022, 2, 279-301. https://doi.org/10.3390/dna2040020
AMA Style
Lloyd RS. Complex Roles of NEIL1 and OGG1: Insights Gained from Murine Knockouts and Human Polymorphic Variants. DNA. 2022; 2(4):279-301. https://doi.org/10.3390/dna2040020
Chicago/Turabian StyleLloyd, R. Stephen. 2022. "Complex Roles of NEIL1 and OGG1: Insights Gained from Murine Knockouts and Human Polymorphic Variants" DNA 2, no. 4: 279-301. https://doi.org/10.3390/dna2040020