
Contributing Factors for Mutagenic DNA Lesion Bypass by DNA Polymerase Eta (polη)

Department of Pharmaceutical Sciences, School of Pharmacy, University of Connecticut, Storrs, CT 06269, USA
DNA 2022, 2(4), 205-220; https://doi.org/10.3390/dna2040015
Submission received: 19 August 2022
/
Revised: 16 September 2022
/
Accepted: 20 September 2022
/
Published: 28 September 2022
(This article belongs to the Special Issue From Mutation and Repair to Therapeutics)
Abstract
:The integrity of DNA replication is under constant threat from various exogenous and endogenous factors along with some epigenetic factors. When there is damage to the genome, cells respond to the damage in two major ways, DNA damage repair and DNA damage tolerance. One of the major mechanisms for DNA damage tolerance is DNA lesion bypass, which is performed by specific DNA polymerases called Y-family DNA polymerases including DNA polymerase eta (polη). Ever since the discovery of polη’s unique role in bypassing cyclobutane pyrimidine dimer (CPD), a wide range of DNA lesions have been experimentally shown to be bypassed by polη. The structural study of polη was greatly boosted by the first elucidation of the N-terminal catalytic domain of polη by X-ray crystallography in 2010. Ever since, a lot of polη catalytic domain crystal structures have been published, which were complexed with an incoming nucleotide and a lesion containing DNA including pyrimidine dimers, cisplatin GpG adduct, 8-oxoguanine (oxoG), 8-oxoadenine (oxoA), N7-methylguanine (N7mG), O6-methylguanine (O6mG), hypoxanthine (HX), and many others. Though polη’s active site is known to be rigid with few conformational changes, there are several contributing factors that could facilitate the lesion bypass such as catalytic metals, syn–anti conformational equilibrium, tautomerization, and specific residues of polη. Each of these components are discussed in detail in this review.
1. Introduction
Our cells are under threat from constant challenges from both endogenous and exogenous DNA damaging agents. Among them are DNA alkylating agents that cause the formation of various alkylated lesions such as O6-, N3-, and N7-alkylated guanine or adenine [1,2,3,4], reactive oxygen species (ROS) that can generate 8-oxoguanine, 8-oxoadenine, or inter/intrastrand crosslinks [5,6,7,8], spontaneous/induced deamination that forms uracil, hypoxanthine, or xanthine [9,10,11], and many others. These lesions, unless repaired or bypassed properly, can cause the halting of DNA replication that can lead to cell death [12,13]. There are a wide range of DNA lesions in terms of bulkiness ranging from small lesions that do not add any extra functional group, such as hypoxanthine from adenine or uracil from cytosine, to bulky lesions including nitrogen half-mustard guanine (NHMG) [14] or phenanthriplatin, (cisplatin analog) adduct [15]. Different lesions can be repaired by various DNA repair mechanisms such as base excision repair (BER) [16], nucleotide excision repair (NER) [17], mismatch repair [18], homologous recombination (HR) [19], and non-homologous DNA end joining [20]. When a timely repair of a DNA lesion is not feasible, DNA damage tolerance mechanisms kick in to resolve the replication stall. HR is known to be involved in the DNA damage tolerance [21] as well. Another DNA damage tolerance mechanism, which is also the major focus of this review, is translesion synthesis (TLS), and TLS is mainly performed by a group of specific DNA polymerases called Y-family DNA polymerases [22,23] along with proliferating cell nuclear antigen (PCNA) and polζ, which consists of Rev3/Rev7, polθ, and polν. In humans, polymerase kappa (polκ), polymerase iota (polι), polymerase eta (polη), and Rev1 are members of the Y-family DNA polymerases, and they share common catalytic domains of the finger, thumb, palm, and little finger [24].
The major focus of this review is on one of the Y-family DNA polymerases, polη. Polη (UniProt ID: Q9Y253) was first recognized from the hypersensitivity of Xeroderma pigmentosum (XP) variant syndrome patients toward many skin diseases including skin cancer and its relationship with the defect in DNA synthesis and the defect in DNA repair for a thymidine dimer caused by UV [25,26,27,28]. Later, RAD30 gene of the yeast Saccharomyces cerevisiae was reported to encode Rad30, and Rad30 was shown to efficiently bypass a thymine–thymine dimer. Rad30 was called polη by the authors due to the fact that this is the seventh eukaryotic DNA polymerase [29]. A few months later it was also shown that XP variant syndrome patients carry mutations on the POLH gene, which is a human gene that encodes polη [30]. Even though polη is error-prone DNA polymerase without any proofreading functionality [31], polη was shown to be able to catalyze error-free bypass opposite a TT cyclobutene pyrimidine dimer (CPD) [32]. However, polη’s bypass across many other lesions is mostly still error-prone, and some examples of the error-prone lesion bypass by polη along with the structural insights for the mutagenicity are presented in the following sections.
Polη’s first crystal structure, though it was the catalytic domain not full-length, was published in 2010 [33], and polη was complexed with the CPD containing DNA and the incoming nucleotide. After this structure, a plethora of publications presented polη crystal structures complexed with DNA containing various DNA lesions including oxoG [34], oxoA [35,36], an abasic site [37], cisplatin GpG adduct [38], N7-alkylguanine [14,39,40], and hypoxanthine/xanthine [41], and so on. There have been quite a few reports regarding the facilitating factors for the promutagenic bypass of polη including syn–anti conformational equilibrium, catalytic metals, enol–keto tautomerism, and major or minor groove interaction through specific residues of polη. Each of these factors are discussed in the following sections in more detail. There are so many intriguing aspects for this TLS polymerase such as post-translational modification via ubiquitination and phosphorylation. There have been some reports revealing that polη’s translocation and UV tolerance activity are dependent on the phosphorylation on specific residues of polη including Ser587, Thr617, or Ser687 [42,43]. In addition, the interaction with monoubiquitinated proliferating cell nuclear antigen (PCNA) was reported to induce the polymerase switch from a replicative polymerase into polη [44], while the mono-ubiquitination of polη was reported to inhibit the interaction between polη and PCNA [45]. However, we focus on the recent investigations on the catalytic core of polη, especially polη’s lesion bypass, its promutagenicity, and the structural and biochemical experiments, that can give us clues for the promutagenic bypass of polη.
2. Overall and Catalytic Domain Structures of polη
Human polη is a 713-amino acid long protein with a catalytic N-terminus domain and a protein interacting C-terminus domain. Within the C-terminus domain, there are some important sequences such as a proliferating cell nuclear antigen (PCNA) interacting peptide (PIP), Rev1 interacting region (RIR), ubiquitin-binding zinc finger (UBZ) domain, and nuclear localization signal (NLS) (Figure 1A). There has not been any crystal structure published for the full-length polη, but there are some crystal structure or NMR structures of different C-terminal domains of polη available such as the UBZ domain (Figure 1B) [46] and Rev1 interacting region (Figure 1C) [47].
The crystal structure of the polη N-terminal catalytic domain (1-432) was first published in 2010 [33], and polη was complexed with CPD containing DNA in this structure. The crystal structure exhibited the conserved domains of Y-family DNA polymerases, and the finger, thumb, palm, and little finger domains (Figure 1D). In all the structures available ever since, the lesion containing DNA was bound between the thumb and little finger domains of polη. The catalytic domain of polη has been proven to be able to bypass a wide range of DNA lesions such as CPD [33], 8-oxoguanine [34], 8-oxoadenine [36], cisplatin adducts [15,38], oxaliplatin adduct [49], N7-methylguanine (N7mG) [39], N7-benzylguanine (N7BnG), N7-nitrogen half-mustard (NHMG) [14], O6-methylguanine [50], hypoxanthine, and xanthine [41], and the mutagenicity of the bypass of each of these lesions by polη is compiled in Table 1.
3. Catalytic Metal Cofactors—Effect of Displacement of Mg2+ by Mn2+
Most, if not all, of the published DNA polymerases, including polη, displayed two catalytic divalent metals in the active site [56]. A-metal is usually located between 3′-OH of the primer terminus and alpha-phosphate (Pα) of the incoming nucleotide. B-metal is located between the β- and γ-phosphate groups. For both metals, residues of polη give extra coordination, which is usually octahedral coordination, and the catalytic metals stabilize the transient negative charge formed during the nucleophilic addition of the incoming nucleotide and the departure of the pyrophosphate group (PPi).
The catalytic metal cofactor was found to be magnesium in many DNA polymerases including E. Coli DNA polymerase I (Klenow fragment) [57], T7 DNA polymerase [58], Sulfolobus solfataricus P2 DNA polymerase IV (Dpo4) [59,60], RB69 DNA polymerase [61], human DNA polymerase β (polβ) [35,62], human DNA polymerase ι (polι) [63], and many other polymerases along with polη. Calcium ion (Ca2+) is known to inhibit the nucleophilic attack of 3-OH’ of the primer terminus to Pα of the incoming nucleotide in many of the DNA polymerases including polη, and some of the early polη structures used Ca2+ so that the nucleotide insertion reaction could be blocked [34,64].
Manganese ion (Mn2+), compared with Mg2+, has been reported to play important roles in lesion bypass and in facilitating incorrect insertion, both of which often require the non-Watson–Crick base pairs, starting from the study of E. coli DNA polymerase I in 1970, which showed that Mn2+ increased the mutagenic replication [65,66,67]. Later, the similar increase in the mutagenic incorporation or catalytic activity of nucleotide upon the introduction of Mn2+ was shown in many other DNA polymerases including avian myeloblastosis virus (AMV) DNA polymerase [68], T4 DNA polymerase during the bypass across the abasic site [69], T7 DNA polymerase [70], human polβ [71], herpes simplex virus type-1 (HSV-1) polymerase [72], human DNA polymerase mu (polμ) [73], Sulfolobus solfataricus Dpo4 [74], human mitochondrial DNA polymerase gamma (polγ) for bypassing CPD [75], human DNA polymerase lambda (polλ) [76], human polι [77], and so on. In polη, Mn2+ was reported to facilitate the incorrect insertion across several lesions, especially medium-sized lesions. For example, the bypass of 5′S-8,5′-cyclo-2′-deoxyadenosine (cdA), which is one of the oxidative DNA lesions, by polη displayed that the substitution of Mg2+ by Mn2+ increased the reaction efficiency of the correct insertion of dTTP across cdA by 1400-fold (0.015 min−1 μM−1 vs. 21 min−1 μM−1) as shown in Table 2. [53]. The kcat values were similar to each other (8.6 min−1 vs. 10.1 min−1), but the Km value was drastically decreased upon the introduction of manganese (570 μM vs. 0.49 μM). The relative efficiency of the correct incorporation of dTTP between the undamaged dA and the damaged cdA was greatly increased when Mg2+ was replaced by Mn2+ (6.7 × 10−4 vs. 0.11). When the authors elucidated the crystal structures of polη complexed with cdA containing DNA in the presence of either Mg2+ or Mn2+ (Figure 2), the crystal structures were able to explain why cdA bypass was so inefficient when Mg2+ was present and how that inefficient bypass was rescued upon the substitution of Mg2+ by Mn2+. The introduction of Mn2+ does not alter the overall structure significantly as shown in Figure 2A. In the Mg2+-bound structure, there was only one Mg2+ found in the active site (Figure 2B), and this is one of the reasons why cdA bypass by polη was about 1300-fold less efficient compared to the undamaged dA (Table 2). Even though cdA in the template and the incoming non-hydrolyzable dTTP (dTTP*) form hydrogen bonds, they are not on the same plane and the hydrogen bonding interactions are not optimal. Furthermore, dA at the N+2 position of the template is positioned between the primer terminus and the incoming dTTP*, and this makes the distance between 3′-OH of the primer terminus and Pα of the incoming nucleotide 4.8 Å, which is quite far. This might be the reason why there was just one Mg2+ found in the active site (Figure 2B), which was also observed in some non-productive complexes in Ca2+-bound polη structures [37,55]. However, when Mn2+ was introduced instead of Mg2+, cdA and dTTP were found to be on the same plane with near-optimal geometry for the canonical Watson–Crick base pair along with the two catalytic Mn2+ metals in the incoming nucleotide binding site (Figure 2C). When the two crystal structures (with Mg2+ (gray) and Mn2+ (multi color)) were superposed, this striking difference is more clearly visible (Figure 2D). In particular, the dT (N+1) and dA (N+2) of the template DNA in the presence of Mg2+ showed disordered placement in the active site. However, the displacement of Mg2+ by Mn2+ and the presence of two catalytic Mn2+ instead of one, as in the case of Mg2+, in the active site stabilized the incoming dTTP across cdA, and this extra stabilization led to a more than 1000-fold increased reaction efficiency.
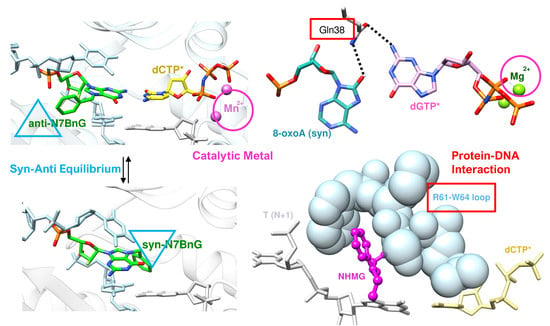
Another example that shows the effects of the catalytic metals in the lesion bypass by polη can be found in N7-alkylguanine (N7alkylG)-bound polη structures with Mg2+ or Mn2+ [14,39,40]. The N7-alkylguanine lesion including N7-methylguanine (N7mG or m7G) is one of the most common lesions for both DNA and RNA in the cells caused by methylation on the N7 position of guanine [78,79], and many of the N7alkylG lesions were reported to be bypassed by polη [14,39,40]. N7mG was readily bypassed by polη with slightly reduced efficiency compared to the undamaged dG (Table 3, Figure 3A,C,D) [39]. The crystal structure of polη complexed with Mg2+ and N7mG revealed that N7mG formed a canonical Watson–Crick base pair with the incoming dCTP* in the active site of polη and this base pair was well overlapped with the base pair between the undamaged dG and dCTP*. On the other hand, a bulky lesion, N7-nitrogen half-mustard guanine (NHMG), was bypassed inefficiently by polη, unlike the small lesion N7mG (Table 3, Figure 3B–D) [14], and the crystal structure was only elucidated in the presence of Mn2+. The crystal structure of polη bypassing NHMG complexed with the incoming dCTP* and Mn2+ revealed that NHMG and the incoming dCTP* can form a canonical Watson–Crick base pair when Mn2+, instead of Mg2+, is in the active site, though the base-pairing has about 20° propeller distortion between the two rings of NHMG and dCTP* (Figure 3B). Another important feature in the crystal structure is that only one Mn2+ ion was found at the B-site where β- and γ-phosphate groups are located. One metal in the active site is a crucial characteristic for the inactive form in many polymerases including polη, and this suggests that NHMG bypass by polη is inefficient and close to an inactive state. The exchange between Mg2+ and Mn2+ is closely related to another important factor that governs the mutagenic bypass of DNA lesions by polη, syn–anti conformational equilibrium, and it is discussed in detail in the next section.
4. Syn–Anti Conformational Change
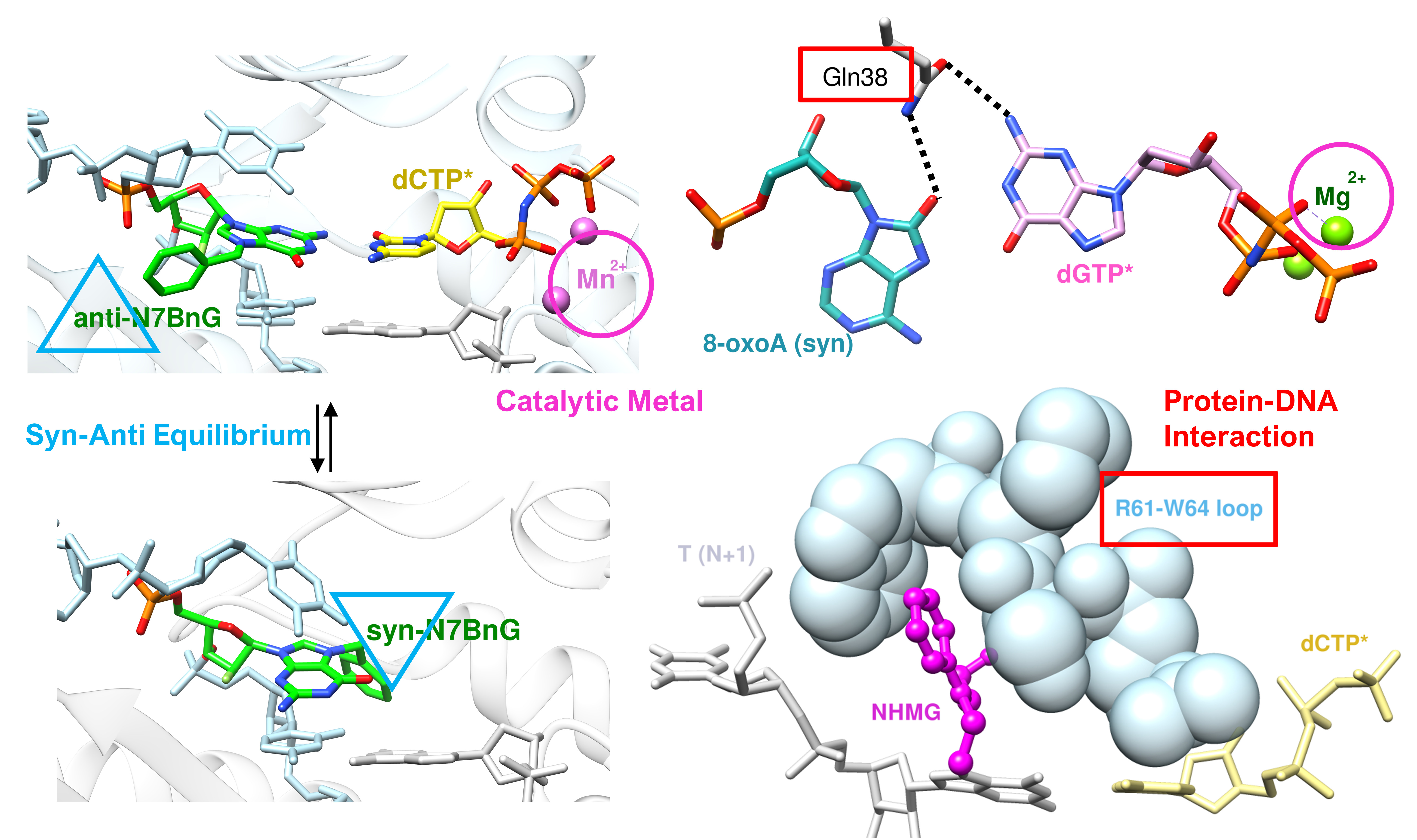
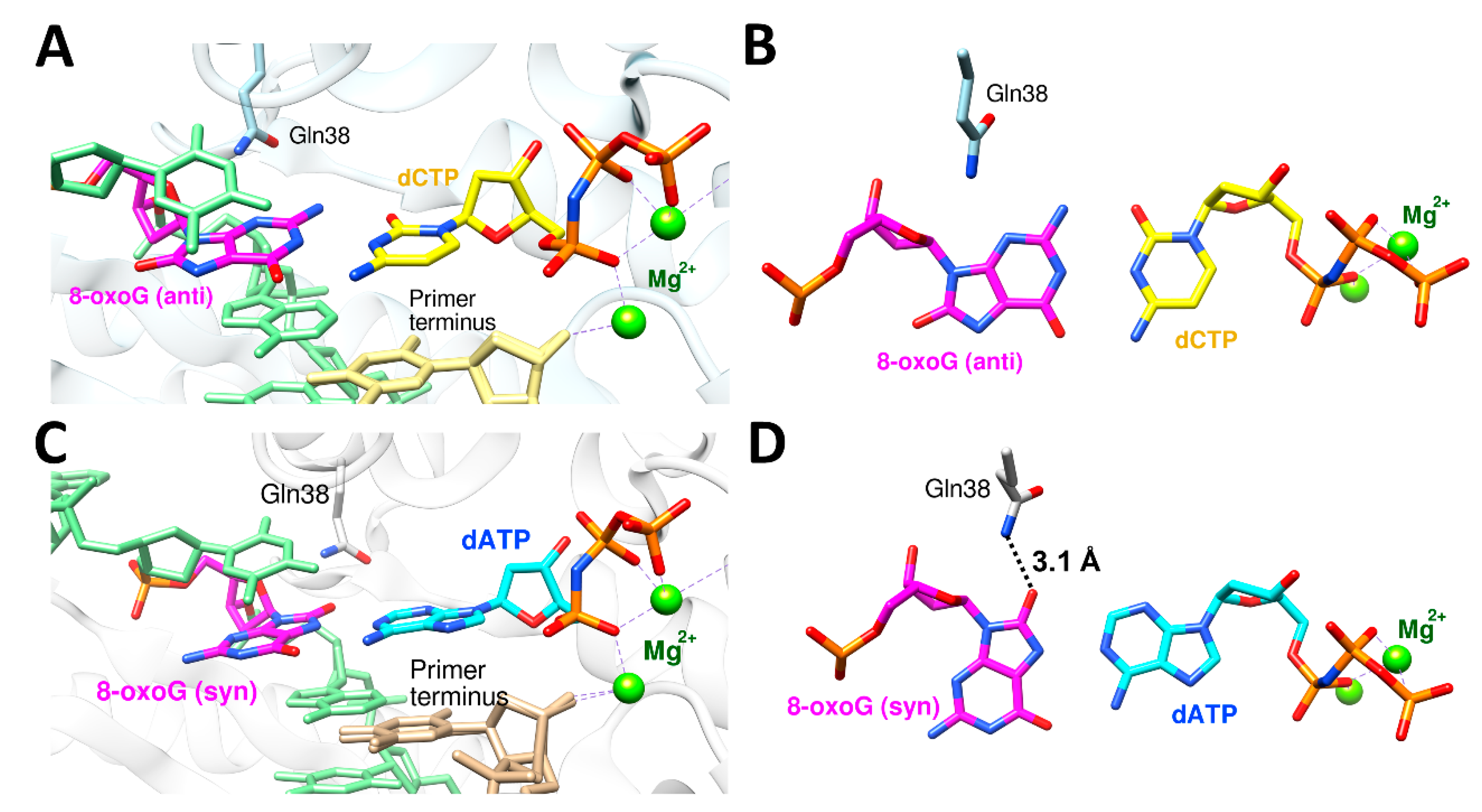
The nucleotides, both ribonucleotide and deoxynucleotide, are susceptible to syn–anti equilibrium both in theoretical and physiological conditions, and purine rings, especially guanine, have higher propensity for syn conformation than pyrimidine rings [80,81,82,83]. Some of the DNA damages including 8-oxoguanine (8-oxoG) and 8-oxoadenine (8-oxoA) and their mutagenic bypasses by polη, which cause C to A mutation (8-oxoG) or T to G mutation (8-oxoA), were reported to be affected by syn–anti conformational changes. [35,36,52]. To be able to accommodate bulky purine nucleotide (dATP or dGTP), 8-oxoG or 8-oxoA adopts a syn conformation in the active site of polη. The ratio between the correct and incorrect insertion by polη across 8-oxoG (dCTP vs. dATP) was 3.5:1 (0.52 vs. 0.15 μM−1 s−1), and the ratio between the correct and incorrect insertion across 8-oxoA (dTTP vs. dGTP) was 2:1 (10.5 vs. 5.1 10−3 μM−1 s−1) (Table 4) [35,52]. The crystal structure of polη complexed with 8-oxoG and the correct incoming nucleotide, dCTP, showed that 8-oxoG adopted an anti conformation in the active site for forming a Watson–Crick base pair with dCTP (Figure 4A,B). On the other hand, the crystal structure of polη complexed with 8-oxoG and the incorrect incoming purine nucleotide, dATP, revealed that 8-oxoG adopted a syn conformation across dATP to accommodate a bulky purine nucleotide (Figure 4C,D). Gln38 of polη is about 3.9 Å away from 8-oxoG in the insertion of the correct nucleotide, dCTP. However, in the insertion of the incorrect nucleotide, dATP, Gln38 of polη formed a hydrogen bond with the distance of 3.1 Å (Figure 4D). For 8-oxoA bypass, the crystal structure of polη complexed with 8-oxoA and the correct incoming nucleotide, dTTP, displayed that 8-oxoA adopted an anti conformation in the active site to form a Watson–Crick base pair with dTTP (Figure 5A,B). On the other hand, the crystal structure of polη complexed with 8-oxoA and the incorrect incoming purine nucleotide, dGTP, exhibited that 8-oxoA adopted a syn conformation across dGTP to accommodate a bulky dGTP. (Figure 5C,D). Gln38 of polη has no hydrogen bonding partner in the insertion of the correct nucleotide, dTTP, across 8-oxoA, but it forms two hydrogen bonds in the insertion of the incorrect nucleotide, dGTP, the first one between O8 of 8-oxoA and the amide group of Gln38 and the second one between N2 of dGTP and the carboxyl group of Gln38 with the distances of 3.5 Å and 3.3 Å, respectively. (Figure 5D). In both bypasses, syn conformation of 8-oxoG and 8-oxoA played crucial roles in accommodating purine incoming nucleotides, dATP and dGTP, across the lesions. The syn conformations of 8-oxoG and 8-oxoA and their hydrogen bonding interactions with their purine counterparts dATP and dGTP were further stabilized by the minor groove interaction via Gln38 of polη, and this extra stabilization was greater in 8-oxoA resulting in a higher mutagenicity of 8-oxoA bypass compared to 8-oxoG. The role of polη residues on the mutagenic bypass is further discussed in the following chapter.
Another lesion whose bypass is affected by syn–anti conformational change is N7-alkylG, especially N7BnG. As shown in the previous section, the mutagenicity of N7alkylG is greater in bulkier lesions. The smallest N7alkylG lesion, N7mG, shows almost no preference toward a syn conformation and behaves similarly to undamaged dG. On the other hand, the bulkier lesion such as NHMG depends more on syn–anti conformation for its mutagenic bypass by polη. The anti conformation of NHMG was supposed to be unfavorable due to the bulkiness of the nitrogen half-mustard moiety. However, the anti conformation was stabilized by the accommodation of the nitrogen half-mustard moiety in the R61-W64 loop of polη [14]. For the undamaged dG, N+1 position base in the template (1 base after the insertion) occupies the space right next to the R61-W64 loop (Figure 3A), while the bulky nitrogen half-mustard moiety snugly fits in the R61-W64 loop of polη in the bypass of NHMG (Figure 3B). When the NHMG-bound polη structure was superposed with the undamaged dG structure (Figure 3C) or the N7mG-bound structure (Figure 3D), dT at N+1 position was pushed away from the active site near the R61-W64 loop, while dT (N+1) was in the original position near the loop in both the dG- and N7mG-bound structures. If there had been no extra support from the R61-W64 loop, a syn conformation of NHMG might have been a dominant conformer in NHMG bypass. A little bit less bulky N7alkylG lesion, N7BnG, showed a balanced conformation depending on the environment such as the catalytic metals to help stabilize either conformation for the base pair with the incoming nucleotide. When Mg2+ was present, N7BnG adopted a syn conformation and there was missing density for the incoming dCTP (Figure 6A). However, when the catalytic magnesium was displaced by Mn2+, N7BnG was able to adopt an anti conformation and had a canonical base pair with the incoming dCTP (Figure 6B) [40].
5. Enol–Keto Tautomerization and polη Residues
Enol tautomer of the lesion containing nucleotide has been shown to play a crucial role in accommodating an incoming nucleotide across the lesion such as 8-oxoG, 8-oxoA, or N7mG, especially for the incorrect insertions in various DNA polymerases [35,39,84,85]. The kinetic studies and the structural studies via NMR recently exhibited that dTTP misincorporation across dG by DNA polymerases including polβ or polε employed a significant ratio of the enol tautomeric form of dG or the incoming dT to form a wobble G:T mismatch base pair [86]. It was also reported that N7-methylation of dG decreased the pKa of the N7-position and boosted the tautomerization of the N1 position of dG, which can lead to N7mG:dT or N7mG:dA mismatches [87,88]. In polη, several DNA lesions, including N7mG and xanthine (XT), have been shown to utilize tautomerization for the lesion bypass. For the bypass of bulky N7alkylG lesions such as N7BnG or NHMG, the enol–keto tautomerization does not have much effect on the mutagenicity due to the more prominent effect by syn–anti conformational change [14,40]. However, in the case of N7mG, which is the smallest lesion in the N7alkylG series, keto tautomer is the dominant form resulting in the facilitation of a correct insertion of dCTP across N7mG in polη [39]. Without protein contact, N7mG was shown to form a Watson–Crick-like base pair with the incoming dTTP leading to the incorrect insertion, which indicates that an enol tautomer of N7mG is involved in this N7mG:dTTP base pair in a protein contact-free environment [84]. The ratio between the correct (dCTP) and incorrect (dTTP) insertion across N7mG by polη is 14:1, and this is quite an increase from the ratio of 100:1 across the undamaged dG (Table 3). Similar to the protein contact-free environment, N7mG might form an enol tautomer in forming the base pair with the incoming dTTP, which leads to the increase in mutagenicity.
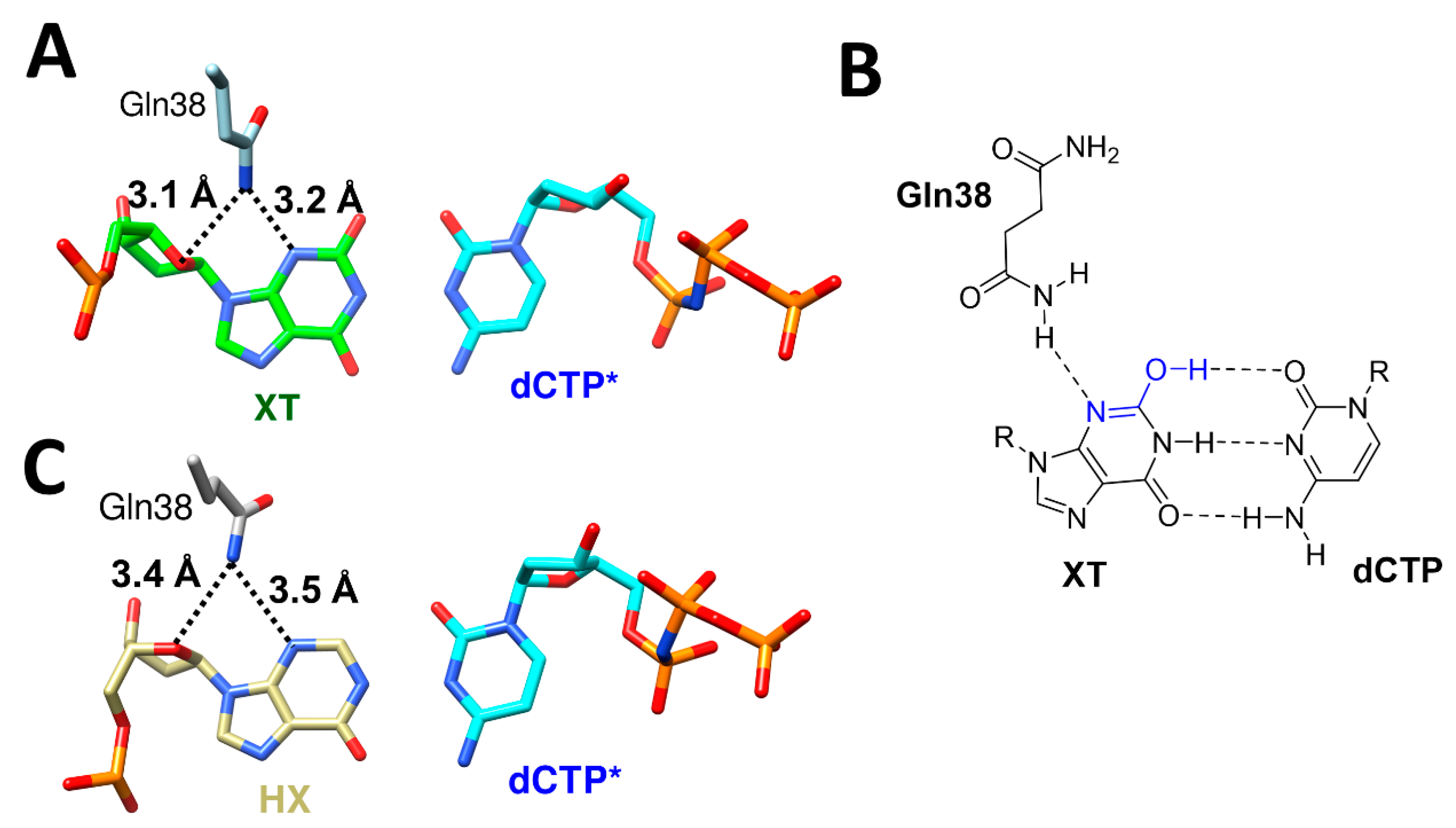
Another example for the involvement of tautomerization in DNA lesion bypass by polη can be seen in the bypass of xanthine (XT) and hypoxanthine (HX) [41]. XT is mainly formed through spontaneous or induced deamination from guanine, and this deamination changes hydrogen bonding donor–acceptor properties for the functional group at 2-position from the N2 (donor) to O2 (acceptor). This deamination increased mutagenicity more than 30-fold (100:1 kcat/Km ratio of correct (dCTP) to incorrect (dTTP) insertion in dG vs. 3:1 in XT, Table 5). Similarly, HX is formed from adenine through deamination, and it changes hydrogen bonding donor–acceptor properties for the functional group at 6-position and C1 of adenine, N6 (donor) to O6 (acceptor) and N1 (acceptor) to N1H (donor). This deamination completely inversed the insertion property (90:1 ratio of correct (dTTP) to incorrect (dCTP) insertion in dA vs. 1:70 in HX, Table 5). Without any protein contact, O2 keto tautomer is the dominant form of XT and leads to a repulsion with O2 of the incoming dCTP to form a canonical Watson–Crick base pair, which is a correct insertion (Figure 7A). Much to our surprise, the crystal structure of polη complexed with XT containing DNA and dCTP revealed that XT and dCTP formed a canonical Watson–Crick base pair with three hydrogen bonds between them, with Gln38 providing extra stabilization via N3 of XT (3.2 Å) and the ribose oxygen (3.1 Å) (Figure 7A,B). The structure also clearly displayed that O2 enol tautomer of XT and O2 of dCTP formed hydrogen bonding interaction. For the bypass of HX, O6 group adopted a keto tautomeric form to have a canonical Watson–Crick hydrogen bonding interaction with N4 of dCTP, with Gln38 providing extra stabilization via N3 of HX (3.5 Å) and the ribose oxygen (3.4 Å) (Figure 7C).
There have been a lot of reports regarding the specific residues of polη that are involved in the mutagenic bypass of DNA lesions. For example, the Arg61-Trp64 loop, which also involves Ser62 and Met63, offers crucial accommodation for the bypass of one of the bulky N7alkylG lesions, NHMG (Figure 3B) [14], and cisplatin adducts [38]. Moreover, the highly conserved Arg61 was reported to be involved in the alignment of the incoming nucleotide via the interaction with a base ring and the phosphate [34,51]. The enol–keto tautomerization was shown to be further stabilized by polη residues, especially by the minor groove interaction via Gln38. In the bypass of XT by polη, Gln38 gives extra stabilization to the enol tautomer of XT and stabilizes the canonical Watson–Crick base pair between XT and dCTP as a result (Figure 7A). This extra stabilization was given through the hydrogen bonding between the amino group of the side chain of Gln38 and N3 of XT, and this hydrogen bonding also stabilized the C2-N3 double bond and the O2 enol tautomer of XT (Figure 7B). The distance between the amino group of Gln38 and N3 of HX is about 3.5 Å, and there is no tautomer that can be stabilized by this interaction (Figure 7C).
6. Perspectives
DNA lesion repair and bypass are some of the most important areas of research in medical science. DNA lesion bypass is an important process that cells can use to keep themselves from replication halt, which can lead to cell death. TLS polymerases, including polη, have different specificity and efficiency toward different DNA lesions, and more research must be conducted to figure out a more accurate relationship between DNA lesion bypass and repair and the detailed mechanism of DNA lesion bypass so that DNA lesion bypass and its participant enzymes can be used or targeted by other fields of research including drug discovery. In this review, we briefly looked over the important factors that govern the DNA lesion bypass by polη, especially the mutagenic bypasses. The catalytic metals in the active site of polη that are coordinated by the phosphate groups of the incoming nucleotide, syn–anti conformational changes that enable purine–purine base pairs in the active site of polη, enol–keto tautomerization that gives some DNA lesions such as XT the ability to form Watson–Crick-like base pairs with an incoming nucleotide, and some of the residues of polη that offer extra stabilization for the base pair between DNA lesions and the incoming nucleotides were presented along with the case studies. Further investigations on the bypass of other DNA lesions and the comparison studies with other TLS and replicative polymerases will expand our knowledge and understanding on this crucial process that keeps cells from cell death caused by the forced halt of replication. In addition, it will be necessary and beneficial for polη’s versatile role in DNA lesion repair and bypass to be investigated in more detail.
Although DNA lesion bypass, even a promutagenic one, plays an important role in replication by preventing replication halt, the mutations caused by the promutagenic bypass via TLS polymerases including polη pose another threat to genome integrity. That is why there are layers of cellular systems that monitor and fix various DNA lesions and misincorporations along with a wide range of DNA repair processes that deal with those DNA lesions and mutations. A lot more research must be conducted to elucidate in more depth these highly complex processes, and the information coming through from future investigations will be extremely valuable for a wide range of biological science fields.
Funding
This review received no external funding.
Acknowledgments
I thank my colleagues in the School of Pharmacy at the University of Connecticut and the College of Pharmacy at the University of Texas at Austin.
Conflicts of Interest
The author declares no conflict of interest.
References
- Drablos, F.; Feyzi, E.; Aas, P.A.; Vaagbo, C.B.; Kavli, B.; Bratlie, M.S.; Pena-Diaz, J.; Otterlei, M.; Slupphaug, G.; Krokan, H.E. Alkylation damage in DNA and RNA—Repair mechanisms and medical significance. DNA Repair 2004, 3, 1389–1407. [Google Scholar] [CrossRef] [PubMed]
- Eadie, J.S.; Conrad, M.; Toorchen, D.; Topal, M.D. Mechanism of mutagenesis by O6-methylguanine. Nature 1984, 308, 201–203. [Google Scholar] [CrossRef] [PubMed]
- Monti, P.; Traverso, I.; Casolari, L.; Menichini, P.; Inga, A.; Ottaggio, L.; Russo, D.; Iyer, P.; Gold, B.; Fronza, G. Mutagenicity of N3-methyladenine: A multi-translesion polymerase affair. Mutat. Res. 2010, 683, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Gates, K.S.; Nooner, T.; Dutta, S. Biologically relevant chemical reactions of N7-alkylguanine residues in DNA. Chem. Res. Toxicol. 2004, 17, 839–856. [Google Scholar] [CrossRef]
- Hemnani, T.; Parihar, M.S. Reactive oxygen species and oxidative DNA damage. Indian J. Physiol. Pharmacol. 1998, 42, 440–452. [Google Scholar] [PubMed]
- Ames, B.N. Endogenous oxidative DNA damage, aging, and cancer. Free Radic. Res. Commun. 1989, 7, 121–128. [Google Scholar] [CrossRef] [PubMed]
- Talhaoui, I.; Couve, S.; Ishchenko, A.A.; Kunz, C.; Schar, P.; Saparbaev, M. 7,8-Dihydro-8-oxoadenine, a highly mutagenic adduct, is repaired by Escherichia coli and human mismatch-specific uracil/thymine-DNA glycosylases. Nucleic Acids Res. 2013, 41, 912–923. [Google Scholar] [CrossRef]
- Chen, W.; Balakrishnan, K.; Kuang, Y.; Han, Y.; Fu, M.; Gandhi, V.; Peng, X. Reactive oxygen species (ROS) inducible DNA cross-linking agents and their effect on cancer cells and normal lymphocytes. J. Med. Chem. 2014, 57, 4498–4510. [Google Scholar] [CrossRef]
- Kow, Y.W. Repair of deaminated bases in DNA. Free Radic. Biol. Med. 2002, 33, 886–893. [Google Scholar] [CrossRef]
- Caulfield, J.L.; Wishnok, J.S.; Tannenbaum, S.R. Nitric oxide-induced deamination of cytosine and guanine in deoxynucleosides and oligonucleotides. J. Biol. Chem. 1998, 273, 12689–12695. [Google Scholar] [CrossRef] [Green Version]
- Wink, D.A.; Kasprzak, K.S.; Maragos, C.M.; Elespuru, R.K.; Misra, M.; Dunams, T.M.; Cebula, T.A.; Koch, W.H.; Andrews, A.W.; Allen, J.S.; et al. DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science 1991, 254, 1001–1003. [Google Scholar] [CrossRef] [PubMed]
- Surova, O.; Zhivotovsky, B. Various modes of cell death induced by DNA damage. Oncogene 2013, 32, 3789–3797. [Google Scholar] [CrossRef] [PubMed]
- Norbury, C.J.; Zhivotovsky, B. DNA damage-induced apoptosis. Oncogene 2004, 23, 2797–2808. [Google Scholar] [CrossRef] [PubMed]
- Jung, H.; Rayala, N.K.; Lee, S. Translesion synthesis of the major nitrogen mustard-induced DNA lesion by human DNA polymerase eta. Biochem. J. 2020, 477, 4543–4558. [Google Scholar] [CrossRef]
- Gregory, M.T.; Park, G.Y.; Johnstone, T.C.; Lee, Y.S.; Yang, W.; Lippard, S.J. Structural and mechanistic studies of polymerase eta bypass of phenanthriplatin DNA damage. Proc. Natl. Acad. Sci. USA 2014, 111, 9133–9138. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008, 18, 27–47. [Google Scholar] [CrossRef]
- Kusakabe, M.; Onishi, Y.; Tada, H.; Kurihara, F.; Kusao, K.; Furukawa, M.; Iwai, S.; Yokoi, M.; Sakai, W.; Sugasawa, K. Mechanism and regulation of DNA damage recognition in nucleotide excision repair. Genes Environ. 2019, 41, 2. [Google Scholar] [CrossRef]
- Pecina-Slaus, N.; Kafka, A.; Salamon, I.; Bukovac, A. Mismatch Repair Pathway, Genome Stability and Cancer. Front. Mol. Biosci. 2020, 7, 122. [Google Scholar] [CrossRef]
- Wright, W.D.; Shah, S.S.; Heyer, W.D. Homologous recombination and the repair of DNA double-strand breaks. J. Biol. Chem. 2018, 293, 10524–10535. [Google Scholar] [CrossRef]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Knobel, P.A.; Marti, T.M. Translesion DNA synthesis in the context of cancer research. Cancer Cell Int. 2011, 11, 39. [Google Scholar] [CrossRef] [PubMed]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef]
- Yang, W. An overview of Y-Family DNA polymerases and a case study of human DNA polymerase eta. Biochemistry 2014, 53, 2793–2803. [Google Scholar] [CrossRef]
- Kraemer, K.H.; Slor, H. Xeroderma pigmentosum. Clin. Dermatol. 1985, 3, 33–69. [Google Scholar] [CrossRef]
- Setlow, R.B.; Regan, J.D.; German, J.; Carrier, W.L. Evidence that xeroderma pigmentosum cells do not perform the first step in the repair of ultraviolet damage to their DNA. Proc. Natl. Acad. Sci. USA 1969, 64, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, J.E. Defective repair replication of DNA in xeroderma pigmentosum. Nature 1968, 218, 652–656. [Google Scholar] [CrossRef]
- Epstein, J.H.; Fukuyama, K.; Reed, W.B.; Epstein, W.L. Defect in DNA synthesis in skin of patients with xeroderma pigmentosum demonstrated in vivo. Science 1970, 168, 1477–1478. [Google Scholar] [CrossRef]
- Johnson, R.E.; Prakash, S.; Prakash, L. Efficient bypass of a thymine-thymine dimer by yeast DNA polymerase, Poleta. Science 1999, 283, 1001–1004. [Google Scholar] [CrossRef]
- Masutani, C.; Kusumoto, R.; Yamada, A.; Dohmae, N.; Yokoi, M.; Yuasa, M.; Araki, M.; Iwai, S.; Takio, K.; Hanaoka, F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 1999, 399, 700–704. [Google Scholar] [CrossRef]
- Matsuda, T.; Bebenek, K.; Masutani, C.; Hanaoka, F.; Kunkel, T.A. Low fidelity DNA synthesis by human DNA polymerase-eta. Nature 2000, 404, 1011–1013. [Google Scholar] [CrossRef] [PubMed]
- Masutani, C.; Araki, M.; Yamada, A.; Kusumoto, R.; Nogimori, T.; Maekawa, T.; Iwai, S.; Hanaoka, F. Xeroderma pigmentosum variant (XP-V) correcting protein from HeLa cells has a thymine dimer bypass DNA polymerase activity. EMBO J. 1999, 18, 3491–3501. [Google Scholar] [CrossRef] [PubMed]
- Biertumpfel, C.; Zhao, Y.; Kondo, Y.; Ramon-Maiques, S.; Gregory, M.; Lee, J.Y.; Masutani, C.; Lehmann, A.R.; Hanaoka, F.; Yang, W. Structure and mechanism of human DNA polymerase eta. Nature 2010, 465, 1044–1048. [Google Scholar] [CrossRef]
- Su, Y.; Patra, A.; Harp, J.M.; Egli, M.; Guengerich, F.P. Roles of Residues Arg-61 and Gln-38 of Human DNA Polymerase eta in Bypass of Deoxyguanosine and 7,8-Dihydro-8-oxo-2′-deoxyguanosine. J. Biol. Chem. 2015, 290, 15921–15933. [Google Scholar] [CrossRef]
- Koag, M.C.; Jung, H.; Lee, S. Mutagenic Replication of the Major Oxidative Adenine Lesion 7,8-Dihydro-8-oxoadenine by Human DNA Polymerases. J. Am. Chem. Soc. 2019, 141, 4584–4596. [Google Scholar] [CrossRef] [PubMed]
- Koag, M.C.; Jung, H.; Lee, S. Mutagenesis mechanism of the major oxidative adenine lesion 7,8-dihydro-8-oxoadenine. Nucleic Acids Res. 2020, 48, 5119–5134. [Google Scholar] [CrossRef]
- Patra, A.; Zhang, Q.; Lei, L.; Su, Y.; Egli, M.; Guengerich, F.P. Structural and kinetic analysis of nucleoside triphosphate incorporation opposite an abasic site by human translesion DNA polymerase eta. J. Biol. Chem. 2015, 290, 8028–8038. [Google Scholar] [CrossRef]
- Zhao, Y.; Biertumpfel, C.; Gregory, M.T.; Hua, Y.J.; Hanaoka, F.; Yang, W. Structural basis of human DNA polymerase eta-mediated chemoresistance to cisplatin. Proc. Natl. Acad. Sci. USA 2012, 109, 7269–7274. [Google Scholar] [CrossRef]
- Koag, M.C.; Jung, H.; Kou, Y.; Lee, S. Bypass of the Major Alkylative DNA Lesion by Human DNA Polymerase eta. Molecules 2019, 24, 3928. [Google Scholar] [CrossRef]
- Jung, H.; Rayala, N.K.; Lee, S. Effects of N7-Alkylguanine Conformation and Metal Cofactors on the Translesion Synthesis by Human DNA Polymerase eta. Chem. Res. Toxicol. 2022, 35, 512–521. [Google Scholar] [CrossRef]
- Jung, H.; Hawkins, M.; Lee, S. Structural insights into the bypass of the major deaminated purines by translesion synthesis DNA polymerase. Biochem. J. 2020, 477, 4797–4810. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Cleaver, J.E.; Hatahet, Z.; Honkanen, R.E.; Chang, J.Y.; Yen, Y.; Chou, K.M. Human DNA polymerase eta activity and translocation is regulated by phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 16578–16583. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; You, C.; Wang, Y. The Functions of Serine 687 Phosphorylation of Human DNA Polymerase eta in UV Damage Tolerance. Mol. Cell. Proteom. 2016, 15, 1913–1920. [Google Scholar] [CrossRef]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: A possible mechanism for the polymerase switch in response to DNA damage. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Bienko, M.; Green, C.M.; Sabbioneda, S.; Crosetto, N.; Matic, I.; Hibbert, R.G.; Begovic, T.; Niimi, A.; Mann, M.; Lehmann, A.R.; et al. Regulation of translesion synthesis DNA polymerase eta by monoubiquitination. Mol. Cell 2010, 37, 396–407. [Google Scholar] [CrossRef]
- Bomar, M.G.; Pai, M.T.; Tzeng, S.R.; Li, S.S.; Zhou, P. Structure of the ubiquitin-binding zinc finger domain of human DNA Y-polymerase eta. EMBO Rep. 2007, 8, 247–251. [Google Scholar] [CrossRef]
- Pozhidaeva, A.; Pustovalova, Y.; D’Souza, S.; Bezsonova, I.; Walker, G.C.; Korzhnev, D.M. NMR structure and dynamics of the C-terminal domain from human Rev1 and its complex with Rev1 interacting region of DNA polymerase eta. Biochemistry 2012, 51, 5506–5520. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Ouzon-Shubeita, H.; Baker, M.; Koag, M.C.; Lee, S. Structural basis for the bypass of the major oxaliplatin-DNA adducts by human DNA polymerase eta. Biochem. J. 2019, 476, 747–758. [Google Scholar] [CrossRef]
- Patra, A.; Zhang, Q.; Guengerich, F.P.; Egli, M. Mechanisms of Insertion of dCTP and dTTP Opposite the DNA Lesion O6-Methyl-2′-deoxyguanosine by Human DNA Polymerase eta. J. Biol. Chem. 2016, 291, 24304–24313. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Gregory, M.T.; Biertumpfel, C.; Hua, Y.J.; Hanaoka, F.; Yang, W. Mechanism of somatic hypermutation at the WA motif by human DNA polymerase eta. Proc. Natl. Acad. Sci. USA 2013, 110, 8146–8151. [Google Scholar] [CrossRef] [PubMed]
- Patra, A.; Nagy, L.D.; Zhang, Q.; Su, Y.; Muller, L.; Guengerich, F.P.; Egli, M. Kinetics, structure, and mechanism of 8-Oxo-7,8-dihydro-2′-deoxyguanosine bypass by human DNA polymerase eta. J. Biol. Chem. 2014, 289, 16867–16882. [Google Scholar] [CrossRef] [PubMed]
- Weng, P.J.; Gao, Y.; Gregory, M.T.; Wang, P.; Wang, Y.; Yang, W. Bypassing a 8,5′-cyclo-2′-deoxyadenosine lesion by human DNA polymerase eta at atomic resolution. Proc. Natl. Acad. Sci. USA 2018, 115, 10660–10665. [Google Scholar] [CrossRef]
- Patra, A.; Banerjee, S.; Johnson Salyard, T.L.; Malik, C.K.; Christov, P.P.; Rizzo, C.J.; Stone, M.P.; Egli, M. Structural Basis for Error-Free Bypass of the 5-N-Methylformamidopyrimidine-dG Lesion by Human DNA Polymerase eta and Sulfolobus solfataricus P2 Polymerase IV. J. Am. Chem. Soc. 2015, 137, 7011–7014. [Google Scholar] [CrossRef]
- Su, Y.; Egli, M.; Guengerich, F.P. Mechanism of Ribonucleotide Incorporation by Human DNA Polymerase eta. J. Biol. Chem. 2016, 291, 3747–3756. [Google Scholar] [CrossRef] [PubMed]
- Beese, L.S.; Steitz, T.A. Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: A two metal ion mechanism. EMBO J. 1991, 10, 25–33. [Google Scholar] [CrossRef]
- Kaushik, N.; Pandey, V.N.; Modak, M.J. Significance of the O-helix residues of Escherichia coli DNA polymerase I in DNA synthesis: Dynamics of the dNTP binding pocket. Biochemistry 1996, 35, 7256–7266. [Google Scholar] [CrossRef]
- Doublie, S.; Tabor, S.; Long, A.M.; Richardson, C.C.; Ellenberger, T. Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 A resolution. Nature 1998, 391, 251–258. [Google Scholar] [CrossRef]
- Ling, H.; Boudsocq, F.; Woodgate, R.; Yang, W. Crystal structure of a Y-family DNA polymerase in action: A mechanism for error-prone and lesion-bypass replication. Cell 2001, 107, 91–102. [Google Scholar] [CrossRef]
- Jung, H.; Lee, S. Promutagenic bypass of 7,8-dihydro-8-oxoadenine by translesion synthesis DNA polymerase Dpo4. Biochem. J. 2020, 477, 2859–2871. [Google Scholar] [CrossRef]
- Franklin, M.C.; Wang, J.; Steitz, T.A. Structure of the replicating complex of a pol alpha family DNA polymerase. Cell 2001, 105, 657–667. [Google Scholar] [CrossRef]
- Sawaya, M.R.; Prasad, R.; Wilson, S.H.; Kraut, J.; Pelletier, H. Crystal structures of human DNA polymerase beta complexed with gapped and nicked DNA: Evidence for an induced fit mechanism. Biochemistry 1997, 36, 11205–11215. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Pence, M.G.; Christov, P.P.; Wawrzak, Z.; Choi, J.Y.; Rizzo, C.J.; Egli, M.; Guengerich, F.P. Basis of miscoding of the DNA adduct N2,3-ethenoguanine by human Y-family DNA polymerases. J. Biol. Chem. 2012, 287, 35516–35526. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Zhao, Y.; Yamagata, Y.; Hua, Y.J.; Yang, W. Watching DNA polymerase eta make a phosphodiester bond. Nature 2012, 487, 196–201. [Google Scholar] [CrossRef] [PubMed]
- El-Deiry, W.S.; Downey, K.M.; So, A.G. Molecular mechanisms of manganese mutagenesis. Proc. Natl. Acad. Sci. USA 1984, 81, 7378–7382. [Google Scholar] [CrossRef]
- Beckman, R.A.; Mildvan, A.S.; Loeb, L.A. On the fidelity of DNA replication: Manganese mutagenesis in vitro. Biochemistry 1985, 24, 5810–5817. [Google Scholar] [CrossRef]
- Miyaki, M.; Murata, I.; Osabe, M.; Ono, T. Effect of metal cations on misincorporation by E. coli DNA polymerases. Biochem. Biophys. Res. Commun. 1977, 77, 854–860. [Google Scholar] [CrossRef]
- Sirover, M.A.; Loeb, L.A. On the fidelity of DNA replication. Effect of metal activators during synthesis with avian myeloblastosis virus DNA polymerase. J. Biol. Chem. 1977, 252, 3605–3610. [Google Scholar] [CrossRef]
- Hays, H.; Berdis, A.J. Manganese substantially alters the dynamics of translesion DNA synthesis. Biochemistry 2002, 41, 4771–4778. [Google Scholar] [CrossRef]
- Tabor, S.; Richardson, C.C. Effect of manganese ions on the incorporation of dideoxynucleotides by bacteriophage T7 DNA polymerase and Escherichia coli DNA polymerase I. Proc. Natl. Acad. Sci. USA 1989, 86, 4076–4080. [Google Scholar] [CrossRef] [Green Version]
- Pelletier, H.; Sawaya, M.R.; Wolfle, W.; Wilson, S.H.; Kraut, J. A structural basis for metal ion mutagenicity and nucleotide selectivity in human DNA polymerase beta. Biochemistry 1996, 35, 12762–12777. [Google Scholar] [CrossRef]
- Villani, G.; Tanguy Le Gac, N.; Wasungu, L.; Burnouf, D.; Fuchs, R.P.; Boehmer, P.E. Effect of manganese on in vitro replication of damaged DNA catalyzed by the herpes simplex virus type-1 DNA polymerase. Nucleic Acids Res. 2002, 30, 3323–3332. [Google Scholar] [CrossRef] [PubMed]
- Dominguez, O.; Ruiz, J.F.; Lain de Lera, T.; Garcia-Diaz, M.; Gonzalez, M.A.; Kirchhoff, T.; Martinez, A.C.; Bernad, A.; Blanco, L. DNA polymerase mu (Pol mu), homologous to TdT, could act as a DNA mutator in eukaryotic cells. EMBO J. 2000, 19, 1731–1742. [Google Scholar] [CrossRef] [PubMed]
- Vaisman, A.; Ling, H.; Woodgate, R.; Yang, W. Fidelity of Dpo4: Effect of metal ions, nucleotide selection and pyrophosphorolysis. EMBO J. 2005, 24, 2957–2967. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Baruch-Torres, N.; Iwai, S.; Herrmann, G.K.; Brieba, L.G.; Yin, Y.W. Human Mitochondrial DNA Polymerase Metal Dependent UV Lesion Bypassing Ability. Front. Mol. Biosci. 2022, 9, 808036. [Google Scholar] [CrossRef]
- Garcia-Diaz, M.; Bebenek, K.; Krahn, J.M.; Pedersen, L.C.; Kunkel, T.A. Role of the catalytic metal during polymerization by DNA polymerase lambda. DNA Repair 2007, 6, 1333–1340. [Google Scholar] [CrossRef]
- Frank, E.G.; Woodgate, R. Increased catalytic activity and altered fidelity of human DNA polymerase iota in the presence of manganese. J. Biol. Chem. 2007, 282, 24689–24696. [Google Scholar] [CrossRef]
- Park, J.W.; Ames, B.N. 7-Methylguanine adducts in DNA are normally present at high levels and increase on aging: Analysis by HPLC with electrochemical detection. Proc. Natl. Acad. Sci. USA 1988, 85, 7467–7470. [Google Scholar] [CrossRef]
- Hu, G.; Tsai, A.L.; Quiocho, F.A. Insertion of an N7-methylguanine mRNA cap between two coplanar aromatic residues of a cap-binding protein is fast and selective for a positively charged cap. J. Biol. Chem. 2003, 278, 51515–51520. [Google Scholar] [CrossRef]
- Haschemeyer, A.E.; Rich, A. Nucleoside conformations: An analysis of steric barriers to rotation about the glycosidic bond. J. Mol. Biol. 1967, 27, 369–384. [Google Scholar] [CrossRef]
- Wilson, H.R.; Rahman, A. Nucleoside conformation and non-bonded interactions. J. Mol. Biol. 1971, 56, 129–142. [Google Scholar] [CrossRef]
- Son, T.D.; Guschlbauer, W.; Gueron, M. Flexibility and conformations of guanosine monophosphates by the Overhauser effect. J. Am. Chem. Soc. 1972, 94, 7903–7911. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, G.; Vojtechovsky, J.; Clowney, L.; Brunger, A.T.; Berman, H.M. New parameters for the refinement of nucleic acid-containing structures. Acta Crystallogr. D Biol. Crystallogr. 1996, 52, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Koag, M.C.; Kou, Y.; Ouzon-Shubeita, H.; Lee, S. Transition-state destabilization reveals how human DNA polymerase beta proceeds across the chemically unstable lesion N7-methylguanine. Nucleic Acids Res. 2014, 42, 8755–8766. [Google Scholar] [CrossRef] [PubMed]
- Reed, A.J.; Suo, Z. Time-Dependent Extension from an 8-Oxoguanine Lesion by Human DNA Polymerase Beta. J. Am. Chem. Soc. 2017, 139, 9684–9690. [Google Scholar] [CrossRef]
- Kimsey, I.J.; Szymanski, E.S.; Zahurancik, W.J.; Shakya, A.; Xue, Y.; Chu, C.C.; Sathyamoorthy, B.; Suo, Z.; Al-Hashimi, H.M. Dynamic basis for dG*dT misincorporation via tautomerization and ionization. Nature 2018, 554, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Lawley, P.D.; Brookes, P. Acidic dissociation of 7:9-dialkylguanines and its possible relation to mutagenic properties of alkylating agents. Nature 1961, 192, 1081–1082. [Google Scholar] [CrossRef]
- Kou, Y.; Koag, M.C.; Lee, S. N7 methylation alters hydrogen-bonding patterns of guanine in duplex DNA. J. Am. Chem. Soc. 2015, 137, 14067–14070. [Google Scholar] [CrossRef]
- Yasui, M.; Suzuki, N.; Miller, H.; Matsuda, T.; Matsui, S.; Shibutani, S. Translesion synthesis past 2′-deoxyxanthosine, a nitric oxide-derived DNA adduct, by mammalian DNA polymerases. J. Mol. Biol. 2004, 344, 665–674. [Google Scholar] [CrossRef]
- Yasui, M.; Suenaga, E.; Koyama, N.; Masutani, C.; Hanaoka, F.; Gruz, P.; Shibutani, S.; Nohmi, T.; Hayashi, M.; Honma, M. Miscoding properties of 2′-deoxyinosine, a nitric oxide-derived DNA Adduct, during translesion synthesis catalyzed by human DNA polymerases. J. Mol. Biol. 2008, 377, 1015–1023. [Google Scholar] [CrossRef]
Figure 1.
Structure of human polη. (A) The schematic diagram of the overall structure of polη. The N-terminal catalytic core including little finger domain and the C-terminal binding/regulatory domains including RIR, UBZ, PIP, and NLS are shown. (B) The crystal structure of UBZ domain of polη (PDB ID: 3WUP). Two catalytic histidine residues and two cysteine residues coordinating Zn2+ ion are shown. (C) The NMR structure of Rev1 interacting region (RIR) of polη complexed with Rev1 C-terminus region (Rev1-CT) (PDB ID: 2LSK). The two crucial phenylalanine residues of polη and aspartate residue of Rev1 are shown. (D) The overall structure of polη catalytic domain complexed with xanthine (XT) and the incoming dCTP* (PDB ID: 6WK6). All the conserved domains, finger, palm, thumb, and little finger, are shown. Figures in this review were prepared using Chimera [48].
Figure 1.
Structure of human polη. (A) The schematic diagram of the overall structure of polη. The N-terminal catalytic core including little finger domain and the C-terminal binding/regulatory domains including RIR, UBZ, PIP, and NLS are shown. (B) The crystal structure of UBZ domain of polη (PDB ID: 3WUP). Two catalytic histidine residues and two cysteine residues coordinating Zn2+ ion are shown. (C) The NMR structure of Rev1 interacting region (RIR) of polη complexed with Rev1 C-terminus region (Rev1-CT) (PDB ID: 2LSK). The two crucial phenylalanine residues of polη and aspartate residue of Rev1 are shown. (D) The overall structure of polη catalytic domain complexed with xanthine (XT) and the incoming dCTP* (PDB ID: 6WK6). All the conserved domains, finger, palm, thumb, and little finger, are shown. Figures in this review were prepared using Chimera [48].

Figure 2.
The overall structure and the structural comparison of cdA bypass by polη with Mg2+ (PDB ID: 6M7U) and Mn2+ (PDB ID: 6M7O). (A) The overall structure of polη with cdA containing DNA and the incoming dTTP* in the presence of Mn2+ (plum ribbon) superposed with Mg2+ containing polη-cdA structure (gray). (B) The non-optimal interaction between cdA and the incoming dTTP* in the presence of Mg2+. There is just one Mg2+ catalytic metal with long distance between the primer terminus and Pα of dTTP*. (C) The near-optimal interaction between cdA and the incoming dTTP* in the presence of Mn2+. There are two catalytic Mn2+ metals with close proximity between the primer terminus and Pα of dTTP*. (D) The superposed close-up structures of polη-cdA:Mn2+ (multi color) and polη-cdA:Mg2+ (gray).
Figure 2.
The overall structure and the structural comparison of cdA bypass by polη with Mg2+ (PDB ID: 6M7U) and Mn2+ (PDB ID: 6M7O). (A) The overall structure of polη with cdA containing DNA and the incoming dTTP* in the presence of Mn2+ (plum ribbon) superposed with Mg2+ containing polη-cdA structure (gray). (B) The non-optimal interaction between cdA and the incoming dTTP* in the presence of Mg2+. There is just one Mg2+ catalytic metal with long distance between the primer terminus and Pα of dTTP*. (C) The near-optimal interaction between cdA and the incoming dTTP* in the presence of Mn2+. There are two catalytic Mn2+ metals with close proximity between the primer terminus and Pα of dTTP*. (D) The superposed close-up structures of polη-cdA:Mn2+ (multi color) and polη-cdA:Mg2+ (gray).

Figure 3.
The structure comparison between NHMG (PDB ID: 6V5K) and undamaged dG (PDB ID: 4O3N)/N7mG (PDB ID: 6UI2) bypass by polη. (A) The bypass of undamaged dG by polη showed T (N+1) positioned right at R61-W64 loop (presented in ball presentation). (B) The bypass of NHMG by polη showed the nitrogen half-mustard moiety positioned right in R61-W64 loop, and this loop utilization by NHMG kicked T (N+1) away from the active site. (C) When the two structures (NHMG and dG) were superposed, the movement of T (N+1) and S62 enabled NHMG to position in R61-W64 loop. (D) When NHMG- and N7mG-bound structures were superposed, similar things were observed as in dG bypass.
Figure 3.
The structure comparison between NHMG (PDB ID: 6V5K) and undamaged dG (PDB ID: 4O3N)/N7mG (PDB ID: 6UI2) bypass by polη. (A) The bypass of undamaged dG by polη showed T (N+1) positioned right at R61-W64 loop (presented in ball presentation). (B) The bypass of NHMG by polη showed the nitrogen half-mustard moiety positioned right in R61-W64 loop, and this loop utilization by NHMG kicked T (N+1) away from the active site. (C) When the two structures (NHMG and dG) were superposed, the movement of T (N+1) and S62 enabled NHMG to position in R61-W64 loop. (D) When NHMG- and N7mG-bound structures were superposed, similar things were observed as in dG bypass.

Figure 4.
The active site structures of polη complexed with 8-oxoG and the incoming nucleotides, dCTP (PDB ID: 4O3P) and dATP (PDB ID: 4O3O). (A) 8-oxoG (anti) and the incoming dCTP* in the active site of polη with catalytic Mg2+, primer, template, and Gln38 shown. (B) The canonical base pair between 8-oxoG (anti) and dCTP* with no additional interaction with Gln38. (C) 8-oxoG (syn) and the incoming dATP* in the active site of polη with catalytic Mg2+, primer, template, and Gln38 shown. The primer terminus shows two alternative conformations. (D) Non-canonical base pair between 8-oxoG (syn) and dATP*, and this base pair is further stabilized by the hydrogen bonding interaction with Gln38.
Figure 4.
The active site structures of polη complexed with 8-oxoG and the incoming nucleotides, dCTP (PDB ID: 4O3P) and dATP (PDB ID: 4O3O). (A) 8-oxoG (anti) and the incoming dCTP* in the active site of polη with catalytic Mg2+, primer, template, and Gln38 shown. (B) The canonical base pair between 8-oxoG (anti) and dCTP* with no additional interaction with Gln38. (C) 8-oxoG (syn) and the incoming dATP* in the active site of polη with catalytic Mg2+, primer, template, and Gln38 shown. The primer terminus shows two alternative conformations. (D) Non-canonical base pair between 8-oxoG (syn) and dATP*, and this base pair is further stabilized by the hydrogen bonding interaction with Gln38.

Figure 5.
The active site structures of polη complexed with 8-oxoA and the incoming nucleotides, dTTP (PDB ID: 6PL8) and dGTP (PDB ID: 6PLC). (A) 8-oxoA (anti) and the incoming dTTP* in the active site of polη with catalytic Mg2+, primer, template, and Gln38 shown. (B) The canonical base pair between 8-oxoA(anti) and dTTP* with no additional interaction with Gln38. (C) 8-oxoA (syn) and the incoming dGTP* in the active site of polη with catalytic Mg2+, primer, template, and Gln38 shown. The primer terminus shows two alternative conformations. (D) Non-canonical base pair between 8-oxoA (syn) and dGTP*, and this base pair is further stabilized by the hydrogen bonding interaction with Gln38.
Figure 5.
The active site structures of polη complexed with 8-oxoA and the incoming nucleotides, dTTP (PDB ID: 6PL8) and dGTP (PDB ID: 6PLC). (A) 8-oxoA (anti) and the incoming dTTP* in the active site of polη with catalytic Mg2+, primer, template, and Gln38 shown. (B) The canonical base pair between 8-oxoA(anti) and dTTP* with no additional interaction with Gln38. (C) 8-oxoA (syn) and the incoming dGTP* in the active site of polη with catalytic Mg2+, primer, template, and Gln38 shown. The primer terminus shows two alternative conformations. (D) Non-canonical base pair between 8-oxoA (syn) and dGTP*, and this base pair is further stabilized by the hydrogen bonding interaction with Gln38.

Figure 6.
The bypass of N7BnG by polη and the role of syn–anti equilibrium and catalytic metals in the bypass. (A) In the presence of Mg2+, N7BnG displayed syn conformation, and this conformation hindered the base pair with the incoming dCTP* (PDB ID: 7L69). (B) In the presence of Mn2+, N7BnG showed anti conformation, and this conformation enabled the base pair with the incoming dCTP* (PDB ID: 6W5X).
Figure 6.
The bypass of N7BnG by polη and the role of syn–anti equilibrium and catalytic metals in the bypass. (A) In the presence of Mg2+, N7BnG displayed syn conformation, and this conformation hindered the base pair with the incoming dCTP* (PDB ID: 7L69). (B) In the presence of Mn2+, N7BnG showed anti conformation, and this conformation enabled the base pair with the incoming dCTP* (PDB ID: 6W5X).

Figure 7.
The bypass of XT (PDB ID: 6WK6) and HX (PDB ID: 6MQ8) by polη and the role of Gln38 in the bypass. (A) The canonical Watson–Crick base pair between XT and the incoming dCTP* and the extra stabilization from Gln38. (B) The Watson–Crick base pair between XT and dCTP* utilized the enol tautomer of XT, and this enol tautomer was stabilized by the hydrogen bonding with Gln38. (C) The canonical Watson–Crick base pair between HX and the incoming dCTP, and Gln38 is in close proximity with HX.
Figure 7.
The bypass of XT (PDB ID: 6WK6) and HX (PDB ID: 6MQ8) by polη and the role of Gln38 in the bypass. (A) The canonical Watson–Crick base pair between XT and the incoming dCTP* and the extra stabilization from Gln38. (B) The Watson–Crick base pair between XT and dCTP* utilized the enol tautomer of XT, and this enol tautomer was stabilized by the hydrogen bonding with Gln38. (C) The canonical Watson–Crick base pair between HX and the incoming dCTP, and Gln38 is in close proximity with HX.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Representative DNA lesions bypassed by polη.
| DNA Lesion. | Mutagenic Property | References |
|---|---|---|
| Cyclobutane pyrimidine dimer (CPD) | Error-free insertion of dATP across TT | [33] |
| Cisplatin intrastrand crosslink GpG | Error-free insertion of dCTP | [38] |
| T:G Mismatch | Error-prone insertion of dTTP | [51] |
| 8-Oxoguanine (8-oxoG) | Error-prone insertion with 3.5:1 ratio of dCTP:dATP | [52] |
| 8-Oxoadenine (8-oxoA) | Error-prone insertion with 2:1 ratio of dTTP:dGTP | [35] |
| 8,5′-cyclo-2′-deoxyadenosine (cdA) | Error-free insertion of dTTP across cdA (Mg2+) | [53] |
| Abasic (AP) site | Purine nucleotide insertion across AP next to dT/dC | [37] |
| 5-N-methylformamidopyrimidine dG | Error-free insertion of dCTP across FapydG | [54] |
| Ribonucleotide insertion | dCTP:rCTP ratio is 1:0.005 across dG | [55] |
| N7-methylguanine (N7mG) | Error-free insertion with 14:1 ratio of dCTP:dTTP | [39] |
| N7-benzylguanine (N7BnG) | Error-free insertion with 10:1 ratio of dCTP:dTTP | [40] |
| O6-methylguanine (O6mG) | Error-prone insertion with 1:1 ratio of dTTP:dCTP | [50] |
| Xanthine (XT) | Error-prone insertion with 3:1 ratio of dCTP:dTTP | [41] |
| Hypoxanthine (HX) | Exclusive error-prone insertion of dCTP | [41] |
| N7-nitrogen half-mustard (NHMG) | Error-free insertion with 10:1 ratio of dCTP:dTTP | [14] |
Table 2.
Kinetic parameters for nucleotide incorporation opposite cdA and dA by polη [53].
Table 2.
Kinetic parameters for nucleotide incorporation opposite cdA and dA by polη [53].
| Template:dNTP | Km (μM) | kcat (min−1) | kcat/Km (min−1 μM−1) | f a |
|---|---|---|---|---|
| dA:dTTP (Mg2+) | 5.4 ± 0.7 | 109 ± 13 | 20 | 1 |
| cdA:dTTP (Mg2+) | 570 ± 70 | 8.6 ± 0.5 | 0.015 | 6.7 × 10−4 |
| dA:dTTP (Mn2+) | 0.44 ± 0.04 | 82 ± 5 | 186 | 1 |
| cdA:dTTP (Mn2+) | 0.49 ± 0.07 | 10.1 ± 0.2 | 21 | 0.11 |
a Relative efficiency: (kcat/Km)[undamaged insertion]/(kcat/Km)[damaged insertion].
| Template:dNTP | Km (μM) | kcat (10−3 s−1) | kcat/Km (10−3 s−1 μM−1) | f a |
|---|---|---|---|---|
| dG:dCTP | 2.7 ± 0.3 | 120.6 ± 6.1 | 46 | 1 |
| dG:dTTP | 159.3 ± 2.7 | 74.8 ± 0.9 | 0.5 | 0.01 |
| N7mG:dCTP | 4.3 ± 0.4 | 56.4 ± 2.7 | 13 | 1 |
| N7mG:dTTP | 52.5 ± 1.7 | 49.3 ± 0.1 | 0.9 | 0.07 |
| N7BnG:dCTP | 10.2 ± 2.4 | 20.6 ± 3.6 | 2.1 | 1 |
| N7BnG:dTTP | 51.7 ± 5.3 | 11.5 ± 0.3 | 0.2 | 0.1 |
| N7BnG:dCTP (Mn2+) | 5.6 ± 0.9 | 38.7 ± 4.4 | 6.9 | 1 |
| N7BnG:dTTP (Mn2+) | 18.6 ± 1.9 | 17.8 ± 2.1 | 1.0 | 0.14 |
| NHMG:dCTP | 113.4 ± 1.4 | 40.9 ± 1.3 | 0.36 | 1 |
| NHMG:dTTP | 146.3 ± 5.1 | 5.5 ± 0.1 | 0.037 | 0.1 |
a Misincorporation frequency: (kcat/Km)[incorrect insertion]/(kcat/Km)[correct insertion].
| Template:dNTP | Km (μM) | kcat (10−3 s−1) | kcat/Km (10−3 s−1 μM−1) | f a |
|---|---|---|---|---|
| dG:dCTP | 1.3 ± 0.2 | 1330 ± 50 | 1000 | 1 |
| dG:dATP | 92 ± 23 | 100 ± 10 | 1.1 | 0.001 |
| oxoG:dCTP | 2.3 ± 0.2 | 1200 ± 30 | 520 | 1 |
| oxoG:dATP | 5.4 ± 0.6 | 780 ± 30 | 150 | 0.28 |
| dA:dTTP | 5.4 ± 0.2 | 90.9 ± 5.8 | 17 | 1 |
| dA:dGTP | 76.3 ± 4.8 | 6.3 ± 0.5 | 0.08 | 0.005 |
| oxoA:dTTP | 3.6 ± 0.3 | 37.3 ± 2.3 | 11 | 1 |
| oxoA:dGTP | 4.9 ± 0.3 | 24.8 ± 1.3 | 5.1 | 0.46 |
| oxoA:dTTP (Q38A) | 71.3 ± 4.4 | 172.6 ± 5.6 | 2.4 | 1 |
| oxoA:dGTP (Q38A) | 113.8 ± 2.8 | 10.5 ± 0.1 | 0.092 | 0.037 |
a Misincorporation frequency: (kcat/Km)[incorrect insertion]/(kcat/Km)[correct insertion].
| Template:dNTP | Km (μM) | kcat (10−3 s−1) | kcat/Km (10−3 s−1 μM−1) | f a |
|---|---|---|---|---|
| dG:dCTP | 2.7 ± 0.3 | 120.6 ± 6.1 | 46 | 1 |
| dG:dTTP | 159.3 ± 2.7 | 74.8 ± 0.9 | 0.5 | 0.01 |
| XT:dCTP | 10.7 ± 0.9 | 123.3 ± 3.6 | 11.5 | 1 |
| XT:dTTP | 20.6 ± 0.9 | 82.2 ± 4.2 | 4.0 | 0.34 |
| dA:dTTP | 5.4 ± 0.2 | 90.9 ± 5.8 | 17 | 1 |
| dA:dCTP | 80.3 ± 3.2 | 15.2 ± 2.5 | 0.19 | 0.011 |
| HX:dTTP | 21.9 ± 1.4 | 11.7 ± 0.2 | 0.54 | 1 |
| HX:dCTP | 4.6 ± 0.4 | 170.5 ± 4.1 | 37.4 | 69 |
a Misincorporation frequency: (kcat/Km)[incorrect insertion]/(kcat/Km)[correct insertion].
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Jung, H. Contributing Factors for Mutagenic DNA Lesion Bypass by DNA Polymerase Eta (polη). DNA 2022, 2, 205-220. https://doi.org/10.3390/dna2040015
AMA Style
Jung H. Contributing Factors for Mutagenic DNA Lesion Bypass by DNA Polymerase Eta (polη). DNA. 2022; 2(4):205-220. https://doi.org/10.3390/dna2040015
Chicago/Turabian StyleJung, Hunmin. 2022. "Contributing Factors for Mutagenic DNA Lesion Bypass by DNA Polymerase Eta (polη)" DNA 2, no. 4: 205-220. https://doi.org/10.3390/dna2040015