
Intravascular Large B-Cell Lymphoma: Two Cases Observed at a Single Institution

 , and
, and
Abstract
:1. Introduction
2. Case Report
2.1. Case 1
2.2. Case 2
3. Discussion
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. Review Series the Updated who Classification of Hematological Malignancies the 2016 Revision of the World Health Organization Classification of Lymphoid Neoplasms. Available online: http://ashpublications.org/blood/article-pdf/127/20/2375/1393632/2375.pdf (accessed on 22 May 2022).
- Ponzoni, M.; Campo, E.; Nakamura, S. Intravascular Large B-Cell Lymphoma: A Chameleon with Multiple Faces and Many Masks. 2018. Available online: http://ashpublications.org/blood/article-pdf/132/15/1561/1406631/blood737445.pdf (accessed on 22 May 2022).
- Matsue, K.; Abe, Y.; Narita, K.; Kobayashi, H.; Kitadate, A.; Takeuchi, M.; Miura, D.; Takeuchi, K. Diagnosis of intravascular large B cell lymphoma: Novel insights into clinicopathological features from 42 patients at a single institution over 20 years. Br. J. Haematol. 2019, 187, 328–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, D.; Kapuria, D.; Nanua, S.; Gaur, R. A Case of De Novo CD5+ Disseminated Intravascular Large B-Cell Lymphoma Presenting as Multiorgan Failure. Case Rep. Hematol. 2016, 2016, 6239416. [Google Scholar]
- Ponzoni, M.; Ferreri, A.J.M.; Campo, E.; Facchetti, F.; Mazzucchelli, L.; Yoshino, T.; Murase, T.; Pileri, S.A.; Doglioni, C.; Zucca, E.; et al. Definition, diagnosis, and management of intravascular large B-cell lymphoma: Proposals and perspectives from an international consensus meeting. J. Clin. Oncol. 2007, 25, 3168–3173. [Google Scholar] [CrossRef] [PubMed]
- Charifa, A.; Paulson, N.; Levy, L.; Perincheri, S.; Lee, A.; Mcniff, J.M.; Ko, C. Intravascular Large B-Cell Lymphoma: Clinical and Histopathologic Findings. Yale J. Biol. Med. 2020, 93, 35–40. [Google Scholar]
- Murase, T. An Asian variant of intravascular large B-cell lymphoma: Clinical, pathological and cytogenetic approaches to diffuse large B-cell lymphoma associated with haemophagocytic syndrome. Br. J. Haematol. 2000, 111, 826–834. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, Y.; Zhu, Y.; Zhang, W. Prognosis of intravascular large b cell lymphoma (Ivlbcl): Analysis of 182 patients from global case series. Cancer Manag. Res. 2020, 12, 10531–10540. [Google Scholar] [CrossRef] [PubMed]
- Geyer, H.; Karlin, N.; Palen, B.; Mesa, R. Asian-Variant Intravascular Lymphoma in the African Race. Rare Tumors 2012, 4, 26–29. [Google Scholar] [CrossRef] [Green Version]
- Brunet, V.; Marouan, S.; Routy, J.P.; Hashem, M.A.; Bernier, V.; Simard, R.; Paterlla, T.; Lamarre, L.; Theoret, G.; Carrier, C.; et al. Retrospective study of intravascular large B-cell lymphoma cases diagnosed in Quebec. Medicine 2017, 96, e5985. [Google Scholar] [CrossRef]
- Su, D.W.; Pasch, W.; Costales, C.; Siddiqi, I.; Mohrbacher, A. Asian-Variant Intravascular Large B-Cell Lymphoma. Bayl. Univ. Med. Cent. Proc. 2017, 30, 186–189. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, S.; Pakbaz, Z. Intravascular large B-cell lymphoma in Hispanics: A case series and literature review. J. Community Hosp. Intern. Med. Perspect. 2020, 10, 562–566. [Google Scholar] [CrossRef]
- Morigi, A.; Stefoni, V.; Argnani, L.; Broccoli, A.; Zinzani, P.L. Intravascular Large B-cell Lymphoma Successfully Treated with Autologous Transplantation. Clin. Lymphoma Myeloma Leuk. 2021, 21, e678–e679. [Google Scholar] [CrossRef] [PubMed]
- Asif, A.A.; Tharoor, M.; Fischer, J.L. Intravascular Large B Cell Lymphoma—Still a Diagnostic Dilemma. J. Community Hosp. Intern. Med. Perspect. 2022, 12, 43–47. [Google Scholar] [CrossRef]
- Kama, K.; la Rosée, P.; Czock, D.; Bosch-Schips, J.; Illerhaus, G. Hemophagocytic Syndrome-Associated Intravascular Large B-cell Lymphoma With Dialysis-Dependent End-Stage Renal Disease Treated With Autologous Stem Cell Transplantation Using a Modified TEAM Regimen. Cureus 2022, 14, 25885. [Google Scholar] [CrossRef]
- Han, Y.; Li, Q.; Wang, D.; Peng, L.; Huang, T.; Ou, C.; Yang, K.; Wang, J. Case Report: Intravascular Large B-Cell Lymphoma: A Clinicopathologic Study of Four Cases With Review of Additional 331 Cases in the Literature. Front. Oncol. 2022, 12, 883141. Available online: https://www.frontiersin.org/articles/10.3389/fonc.2022.883141/full (accessed on 24 September 2022). [CrossRef]
- Sakaguchi, K.; Watari, T. Early Diagnosis of Intravascular Large B-Cell Lymphoma by Random Skin Biopsy. Eur. J. Case Rep. Intern. Med. 2022. Available online: https://www.ejcrim.com/index.php/EJCRIM/article/view/3497 (accessed on 24 September 2022).
- di Fonzo, H.; Contardo, D.; Carrozza, D.; Finocchietto, P.; Crisson, A.R.; Cabral, C.; Juarez, M.D.L.A. Intravascular large B cell lymphoma presenting as fever of unknown origin and diagnosed by random skin biopsies: A case report and literature review. Am. J. Case Rep. 2017, 18, 482–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boonsakan, P.; Iamsumang, W.; Chantrathammachart, P.; Chayavichitsilp, P.; Suchonwanit, P.; Rutnin, S. Prognostic Value of Concurrent Expression of C-MYC and BCL2 in Intravascular Large B-Cell Lymphoma: A 10-Year Retrospective Study. Biomed. Res. Int. 2020, 2020, 1350820. [Google Scholar] [CrossRef] [PubMed]
- García-Muñoz, R.; Rubio-Mediavilla, S.; Robles-de-Castro, D.; Muñoz, A.; Herrera-Pérez, P.; Rabasa, P. Intravascular large B cell lymphoma. Leuk. Res. Rep. 2014, 3, 21–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, K.; Kiyoi, H. Current progress and future perspectives of research on intravascular large B-cell lymphoma. Cancer Sci. 2021, 112, 3953–3961. [Google Scholar] [CrossRef]
- Miura, Y.; Ooi, J.; Matsuo, T.; Yamamoto, T.; Sumiyoshi, R.; Saito, S.; Matsumoto, K.; Tashiro, H.; Shirafuji, M. Successful myeloablative unrelated bone marrow transplantation for relapsed intravascular large B cell lymphoma after autologous peripheral blood stem cell transplantation. Ann. Hematol. 2021, 100, 3067–3069. [Google Scholar] [CrossRef]
- Sawamoto, A.; Narimatsu, H.; Suzuki, T.; Kurahashi, S.; Sugimoto, T.; Sugiura, I. Long-term remission after autologous peripheral blood stem cell transplantation for relapsed intravascular lymphoma. Bone Marrow Transpl. 2006, 37, 233–234. [Google Scholar] [CrossRef]
- Meissner, J.; Finel, H.; Dietrich, S.; Boumendil, A.; Kanfer, E.; Laboure, G.; Abecasis, M.; Cornelissen, J.; Delage, J.; Finke, J.; et al. Autologous hematopoietic stem cell transplantation for intravascular large B-cell lymphoma: The European Society for Blood and Marrow Transplantation experience. Bone Marrow Transpl. 2017, 52, 650–652. [Google Scholar] [CrossRef] [PubMed]
- Tucci, A.; Martelli, M.; Rigacci, L.; Riccomagno, P.; Cabras, M.G.; Salvi, F.; Stelitano, C.; Fabbri, A.; Storti, S.; Fogazzi, S.; et al. Comprehensive geriatric assessment is an essential tool to support treatment decisions in elderly patients with diffuse large B-cell lymphoma: A prospective multicenter evaluation in 173 patients by the Lymphoma Italian Foundation (FIL). Leuk. Lymphoma 2015, 56, 921–926. [Google Scholar] [CrossRef] [PubMed]



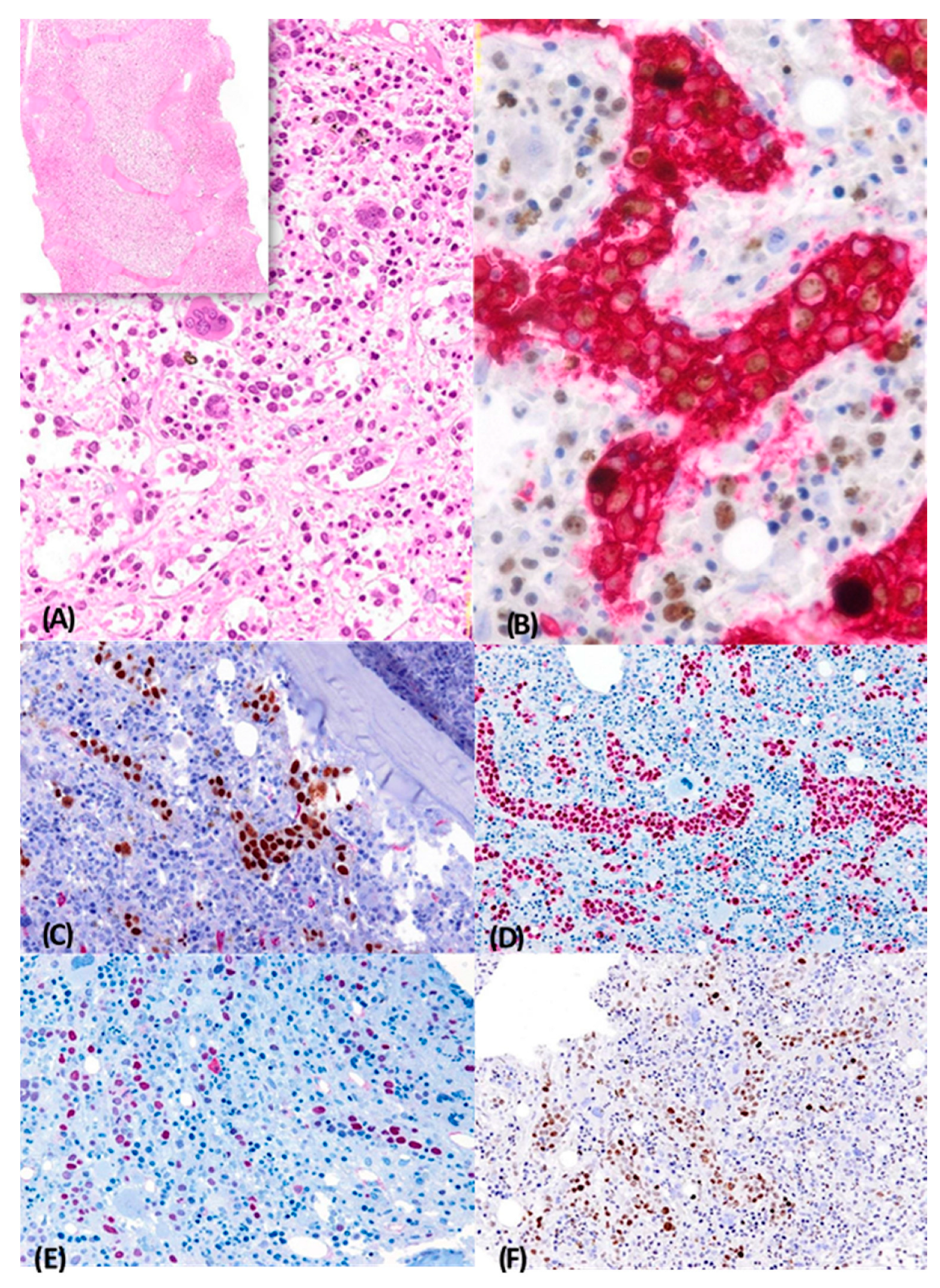{kind=link}
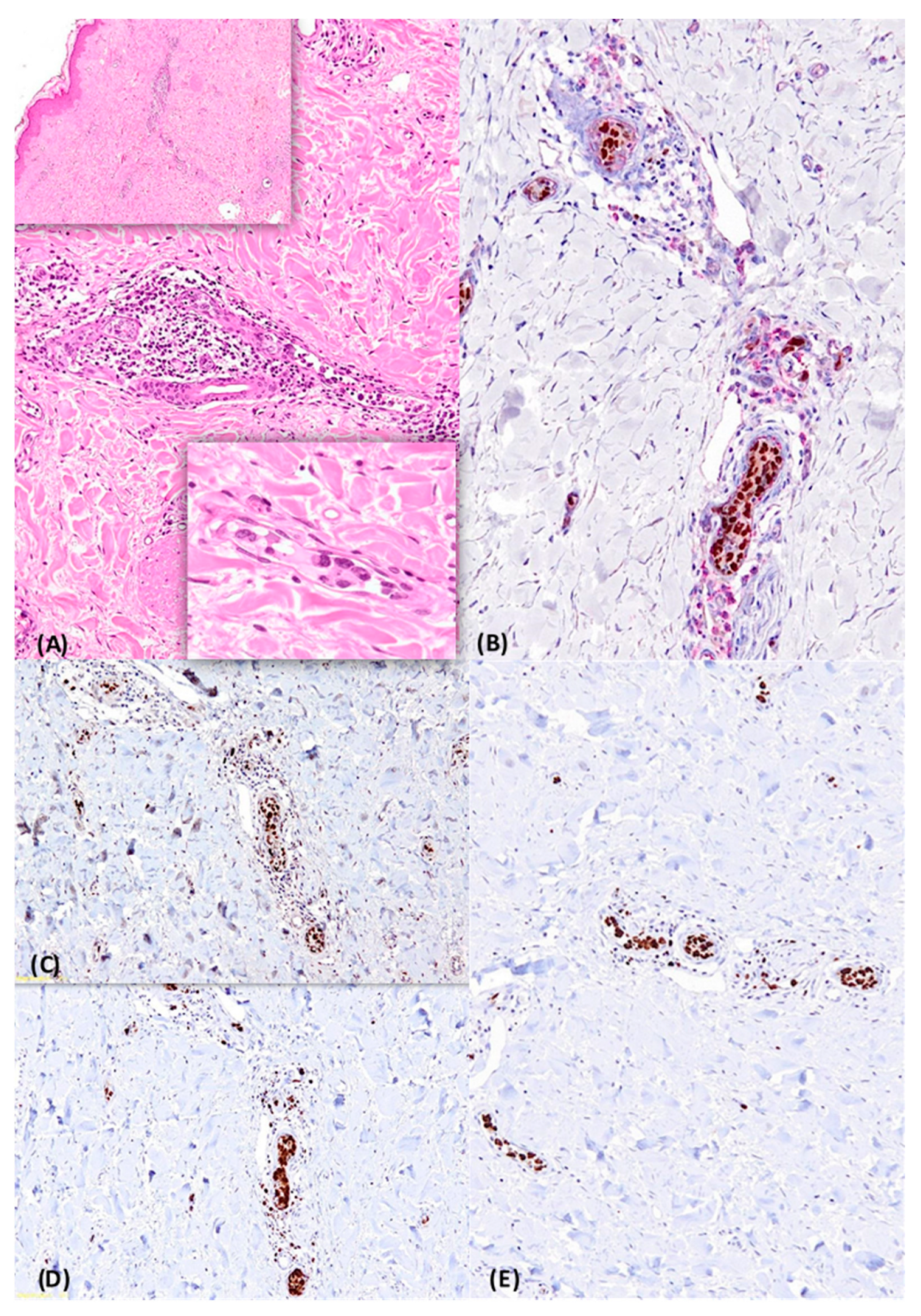{kind=link}
| Clinical Features | Case 1 | Case 2 |
|---|---|---|
| Sex/age (years) | M/43 | F/76 |
| Ethnicity | African | Caucasian |
| Primary location | BM | Skin |
| BM | Positive | Negative |
| Symptoms | Fever, fatigue, night sweats, abdominal pain, hypoxemia | Fever, fatigue |
| HB (g/mL) | 7.3 | 9.4 |
| WBC count | 10.7 × 109/L | 5.4 × 109/L |
| Platelets | 49 × 109/L | 180 × 109/L |
| LDH (U/L) | 5.852 | 552 |
| CRP (mg/L) | 20 | 7.9 |
| Albumin (g/L) | 1.9 | 3.8 |
| Ferritin (ng/mL) | 1120 | 450 |
| PTT (23.4–35.7 s) | 38 | 26 |
| PT (12–14.2 s) | 17 | 13 |
| CT | Hepatomegaly, splenomegaly, bilateral ground-glass opacities of lung and pleural effusion | Mild splenomegaly |
| PET-CT | Spleen FDG uptake | Spleen FDG uptake |
| Case 1 | Case 2 | |
|---|---|---|
| CD20 | + | + |
| CD79a | + | + |
| MUM 1 | + | + |
| BCL2 | + | + |
| BCL6 | + | + |
| CD3 | − | − |
| CD5 | +/− | + |
| Cyclin D1 | − | − |
| CD10 | − | − |
| c MYC | + | + |
| EBER-ISH | − | − |
| Case 1 | Asian-variant intravascular lymphoma in the African race [9] | Rare Tumors (2012) | Holly Geyer et al. |
| Cases 2 and 3 | Retrospective study of intravascular large B-cell lymphoma cases diagnosed in Quebec [10] | Medicine clinical case report (2017) | Vanessa Brunet et al. |
| Case 4 | Asian-variant intravascular large B-cell lymphoma [11] | Proc (Bayl Univ Med Cent) (2017) | Derrick W. Su et al. |
| Cases 5, 6, and 7 | Intravascular large B-cell lymphoma in Hispanics: a case series and literature review [12] | Journal of Community Hospital Internal Medicine Perspectives (2020) | Sonha Nguyena and Zahra Pakbaz |
| Case 8 | Intravascular Large B-cell Lymphoma Successfully Treated with Autologous Transplantation [13] | Clinical Lymphoma, Myeloma and Leukemia (2021) | Alice Morigi et al. |
| Case 9 | Intravascular Large B Cell Lymphoma Still a Diagnostic Dilemma [14] | Journal of Community Hospital Internal Medicine Perspectives, Volume 12, Issue 1, Article 9 (2022) | Abuzar A. Asif et al. |
| Case 10 | Hemophagocytic Syndrome-Associated Intravascular Large B-cell Lymphoma With Dialysis-Dependent End-Stage Renal Disease Treated With Autologous Stem Cell Transplantation Using a Modified TEAM Regimen [15] | Cureus (2022) | Kama K. et al. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miccolis, R.M.; De Santis, G.; Buquicchio, C.; Santeramo, T.M.; Leo, M.; Germano, C.R.; Lerario, G.; Carluccio, V.; Mallano, S.; Cardisciani, L.; et al. Intravascular Large B-Cell Lymphoma: Two Cases Observed at a Single Institution. BioMed 2023, 3, 50-58. https://doi.org/10.3390/biomed3010004
Miccolis RM, De Santis G, Buquicchio C, Santeramo TM, Leo M, Germano CR, Lerario G, Carluccio V, Mallano S, Cardisciani L, et al. Intravascular Large B-Cell Lymphoma: Two Cases Observed at a Single Institution. BioMed. 2023; 3(1):50-58. https://doi.org/10.3390/biomed3010004
Chicago/Turabian StyleMiccolis, Rosanna Maria, Gaetano De Santis, Caterina Buquicchio, Teresa Maria Santeramo, Mariangela Leo, Candida Rosaria Germano, Giovanna Lerario, Vera Carluccio, Sonia Mallano, Lina Cardisciani, and et al. 2023. "Intravascular Large B-Cell Lymphoma: Two Cases Observed at a Single Institution" BioMed 3, no. 1: 50-58. https://doi.org/10.3390/biomed3010004