

Molecular Filters in Medicinal Chemistry

Definition
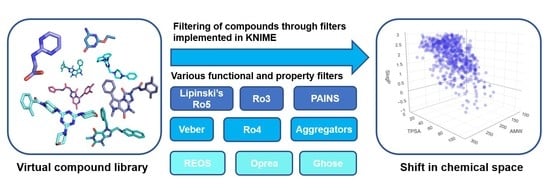
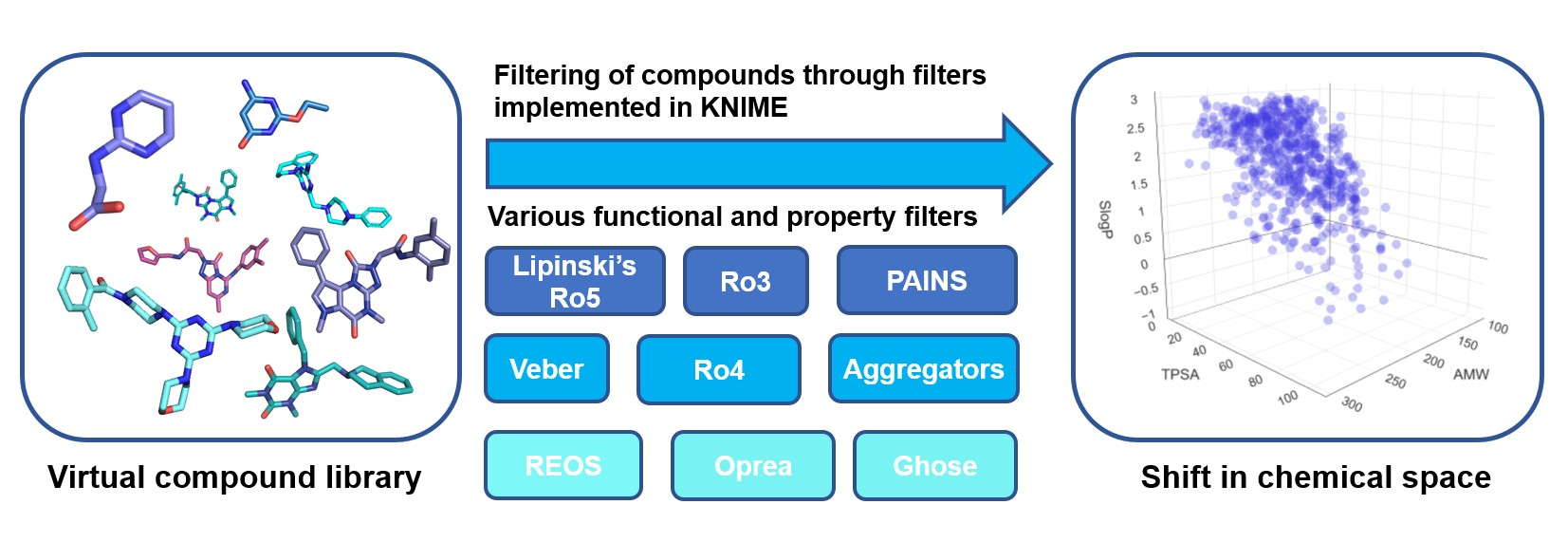
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Types of Filters
2.1. Functional Group Filters
2.2. Property Filters
2.2.1. Bioavailability
2.2.2. Drug-Likeness
2.2.3. Lead-Likeness
2.2.4. Central Nervous System Activity (Blood–Brain Barrier Permeability)
2.2.5. Protein–Protein Interaction Inhibitors
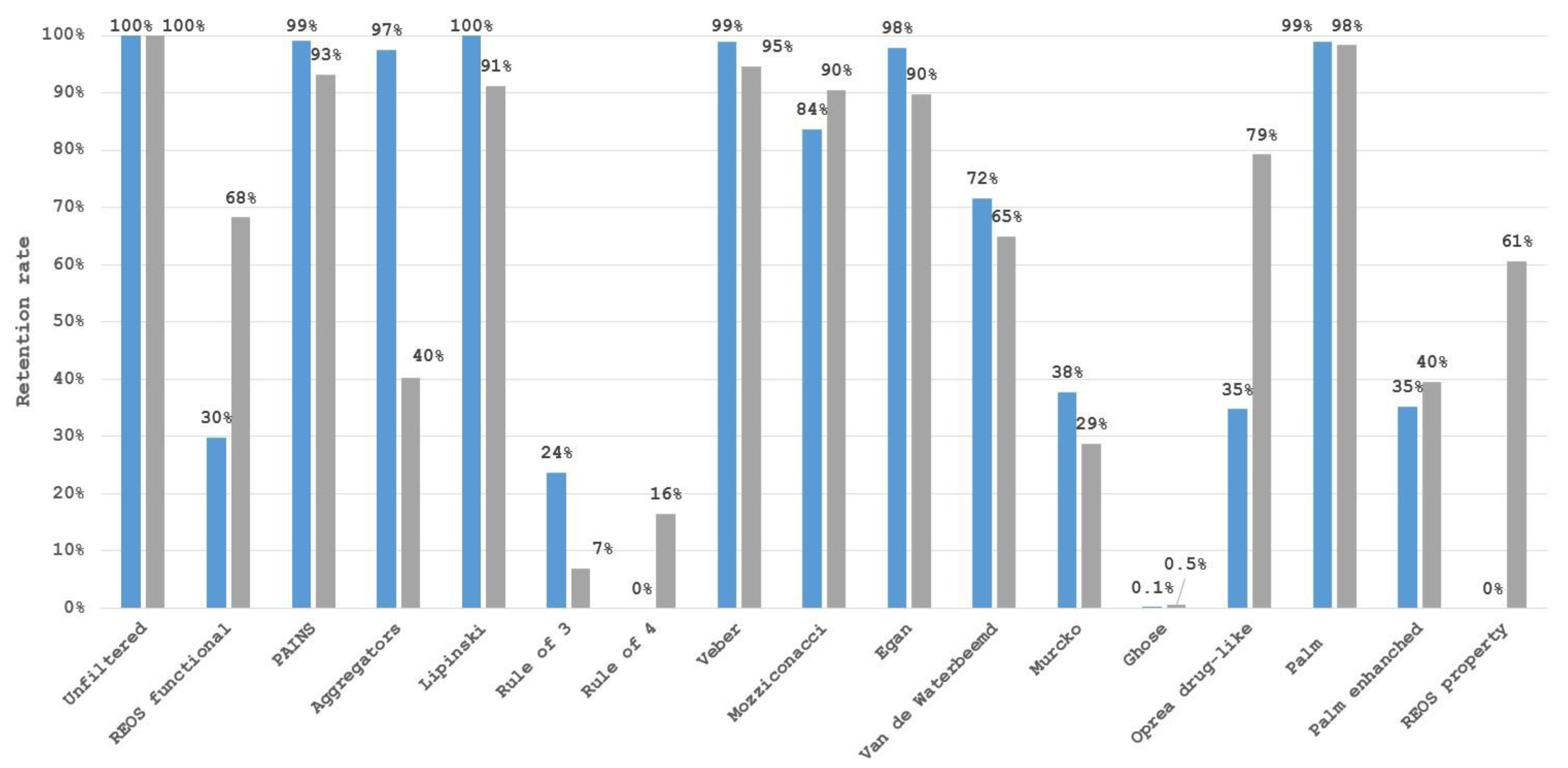
3. Limitations of Filter Use
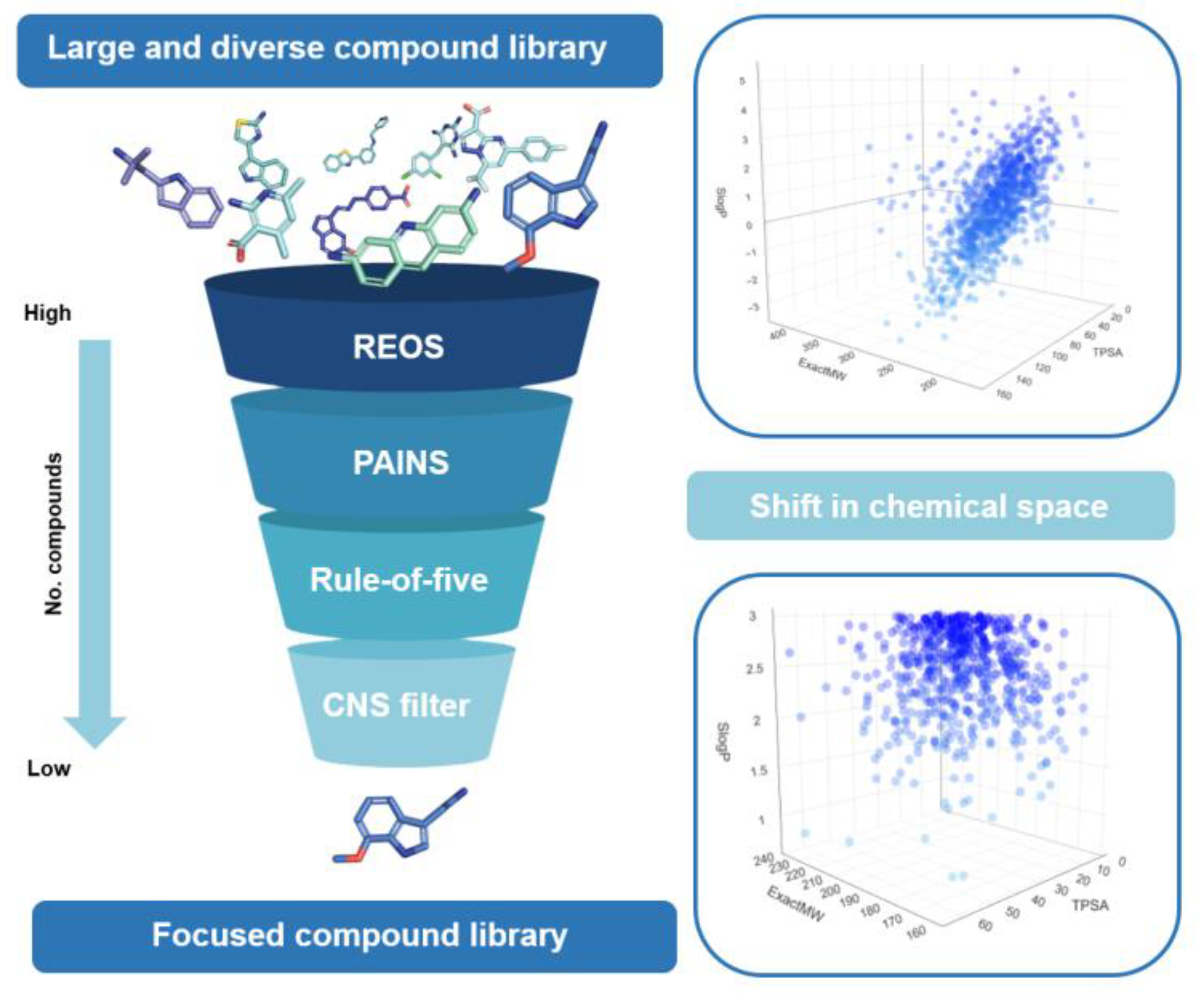
4. Impact on Chemical Space
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shoichet, B.K. Virtual Screening of Chemical Libraries. Nature 2004, 432, 862–865. [Google Scholar] [CrossRef] [PubMed]
- Doman, T.N.; McGovern, S.L.; Witherbee, B.J.; Kasten, T.P.; Kurumbail, R.; Stallings, W.C.; Connolly, D.T.; Shoichet, B.K. Molecular Docking and High-Throughput Screening for Novel Inhibitors of Protein Tyrosine Phosphatase-1B. J. Med. Chem. 2002, 45, 2213–2221. [Google Scholar] [CrossRef] [PubMed]
- Van Hilten, N.; Chevillard, F.; Kolb, P. Virtual Compound Libraries in Computer-Assisted Drug Discovery. J. Chem. Inf. Model. 2019, 59, 644–651. [Google Scholar] [CrossRef] [PubMed]
- Kralj, S.; Jukič, M.; Bren, U. Comparative Analyses of Medicinal Chemistry and Cheminformatics Filters with Accessible Implementation in Konstanz Information Miner (KNIME). Int. J. Mol. Sci. 2022, 23, 5727. [Google Scholar] [CrossRef] [PubMed]
- Blay, V.; Tolani, B.; Ho, S.P.; Arkin, M.R. High-Throughput Screening: Today’s Biochemical and Cell-Based Approaches. Drug Discov. Today 2020, 25, 1807–1821. [Google Scholar] [CrossRef]
- Bakken, G.A.; Bell, A.S.; Boehm, M.; Everett, J.R.; Gonzales, R.; Hepworth, D.; Klug-McLeod, J.L.; Lanfear, J.; Loesel, J.; Mathias, J.; et al. Shaping a Screening File for Maximal Lead Discovery Efficiency and Effectiveness: Elimination of Molecular Redundancy. J. Chem. Inf. Model. 2012, 52, 2937–2949. [Google Scholar] [CrossRef]
- Njoroge, M.; Njuguna, N.M.; Mutai, P.; Ongarora, D.S.B.; Smith, P.W.; Chibale, K. Recent Approaches to Chemical Discovery and Development against Malaria and the Neglected Tropical Diseases Human African Trypanosomiasis and Schistosomiasis. Chem. Rev. 2014, 114, 11138–11163. [Google Scholar] [CrossRef]
- Ruddigkeit, L.; van Deursen, R.; Blum, L.C.; Reymond, J.-L. Enumeration of 166 Billion Organic Small Molecules in the Chemical Universe Database GDB-17. J. Chem. Inf. Model. 2012, 52, 2864–2875. [Google Scholar] [CrossRef]
- Gorse, A.-D. Diversity in Medicinal Chemistry Space. Curr. Top. Med. Chem. 2006, 6, 3–18. [Google Scholar] [CrossRef]
- Jukič, M.; Janežič, D.; Bren, U. Ensemble Docking Coupled to Linear Interaction Energy Calculations for Identification of Coronavirus Main Protease (3CLpro) Non-Covalent Small-Molecule Inhibitors. Molecules 2020, 25, 5808. [Google Scholar] [CrossRef]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational Methods in Drug Discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Kralj, S.; Jukič, M.; Bren, U. Commercial SARS-CoV-2 Targeted, Protease Inhibitor Focused and Protein–Protein Interaction Inhibitor Focused Molecular Libraries for Virtual Screening and Drug Design. Int. J. Mol. Sci. 2021, 23, 393. [Google Scholar] [CrossRef] [PubMed]
- Thorpe, D.S.; Edith Chan, A.W.; Binnie, A.; Chen, L.C.; Robinson, A.; Spoonamore, J.; Rodwell, D.; Wade, S.; Wilson, S.; Ackerman-Berrier, M.; et al. Efficient Discovery of Inhibitory Ligands for Diverse Targets from a Small Combinatorial Chemical Library of Chimeric Molecules. Biochem. Biophys. Res. Commun. 1999, 266, 62–65. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like Properties and the Causes of Poor Solubility and Poor Permeability. J. Pharmacol. Toxicol. Methods 2000, 44, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T. Virtual Screening in Lead Discovery: A Viewpoint. Molecules 2002, 7, 51–62. [Google Scholar] [CrossRef]
- Muegge, I. Pharmacophore Features of Potential Drugs. Chem. Weinh. Bergstr. Ger. 2002, 8, 1976–1981. [Google Scholar] [CrossRef]
- Walters, W.P.; Murcko, A.A.; Murcko, M.A. Recognizing Molecules with Drug-like Properties. Curr. Opin. Chem. Biol. 1999, 3, 384–387. [Google Scholar] [CrossRef]
- Walters, W.P.; Murcko, M.A. Prediction of “Drug-Likeness”. Adv. Drug Deliv. Rev. 2002, 54, 255–271. [Google Scholar] [CrossRef]
- Lumley, J.A. Compound Selection and Filtering in Library Design. QSAR Comb. Sci. 2005, 24, 1066–1075. [Google Scholar] [CrossRef]
- Pascual, R.; Borrell, J.I.; Teixidó, J. Analysis of Selection Methodologies for Combinatorial Library Design. Mol. Divers. 2000, 6, 121–133. [Google Scholar] [CrossRef]
- Walters, W.P.; Namchuk, M. Designing Screens: How to Make Your Hits a Hit. Nat. Rev. Drug Discov. 2003, 2, 259–266. [Google Scholar] [CrossRef]
- Baell, J.B.; Holloway, G.A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. [Google Scholar] [CrossRef] [PubMed]
- Thorne, N.; Auld, D.S.; Inglese, J. Apparent Activity in High-Throughput Screening: Origins of Compound-Dependent Assay Interference. Curr. Opin. Chem. Biol. 2010, 14, 315–324. [Google Scholar] [CrossRef]
- Walters, W.P.; Stahl, M.T.; Murcko, M.A. Virtual Screening—An Overview. Drug Discov. Today 1998, 3, 160–178. [Google Scholar] [CrossRef]
- Rishton, G.M. Reactive Compounds and in Vitro False Positives in HTS. Drug Discov. Today 1997, 2, 382–384. [Google Scholar] [CrossRef]
- Yang, J.J.; Ursu, O.; Lipinski, C.A.; Sklar, L.A.; Oprea, T.I.; Bologa, C.G. Badapple: Promiscuity Patterns from Noisy Evidence. J. Cheminform. 2016, 8, 29. [Google Scholar] [CrossRef] [PubMed]
- Bruns, R.F.; Watson, I.A. Rules for Identifying Potentially Reactive or Promiscuous Compounds. J. Med. Chem. 2012, 55, 9763–9772. [Google Scholar] [CrossRef]
- Irwin, J.J.; Duan, D.; Torosyan, H.; Doak, A.K.; Ziebart, K.T.; Sterling, T.; Tumanian, G.; Shoichet, B.K. An Aggregation Advisor for Ligand Discovery. J. Med. Chem. 2015, 58, 7076–7087. [Google Scholar] [CrossRef]
- Huggins, D.J.; Venkitaraman, A.R.; Spring, D.R. Rational Methods for the Selection of Diverse Screening Compounds. ACS Chem. Biol. 2011, 6, 208–217. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- O′Hagan, S.; Swainston, N.; Handl, J.; Kell, D.B. A ‘Rule of 0.5’ for the Metabolite-Likeness of Approved Pharmaceutical Drugs. Metabolomics 2015, 11, 323–339. [Google Scholar] [CrossRef] [PubMed]
- Veber, D.F.; Johnson, S.R.; Cheng, H.-Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular Properties That Influence the Oral Bioavailability of Drug Candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef] [PubMed]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of Drug Absorption Using Multivariate Statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef] [PubMed]
- Dobson, P.D.; Kell, D.B. Carrier-Mediated Cellular Uptake of Pharmaceutical Drugs: An Exception or the Rule? Nat. Rev. Drug Discov. 2008, 7, 205–220. [Google Scholar] [CrossRef]
- Palm, K.; Luthman, K.; Unge, A.-L.; Strandlund, G.; Artursson, P. Correlation of Drug Absorption with Molecular Surface Properties. J. Pharm. Sci. 1996, 85, 32–39. [Google Scholar] [CrossRef]
- Palm, K.; Stenberg, P.; Luthman, K.; Artursson1, P. Polar Molecular Surface Properties Predict the Intestinal Absorption of Drugs in Humans. Pharm. Res. 1997, 14, 568–571. [Google Scholar] [CrossRef]
- Morin-Allory, L.; Mozziconacci, J.C.; Arnoult, E.; Baurin, N.; Marot, C. Preparation of a Molecular Database from a Set of 2 Million Compounds for Virtual Screening Applications: Gathering, Structural Analysis and Filtering; Institut de Chimie Organique et Analytique, Universite d’Orleans: Orleans, France, 2003. [Google Scholar]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.I. Property Distribution of Drug-Related Chemical Databases. J. Comput. Aided Mol. Des. 2000, 14, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Oprea, T.I.; Davis, A.M.; Teague, S.J.; Leeson, P.D. Is There a Difference between Leads and Drugs? A Historical Perspective. J. Chem. Inf. Comput. Sci. 2001, 41, 1308–1315. [Google Scholar] [CrossRef]
- Congreve, M.; Carr, R.; Murray, C.; Jhoti, H. A “rule of Three” for Fragment-Based Lead Discovery? Drug Discov. Today 2003, 8, 876–877. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H. (Eds.) Blood-Brain Barrier in Drug Discovery: Optimizing Brain Exposure of CNS Drugs and Minimizing Brain Side Effects for Peripheral Drugs; Wiley: Hoboken, NJ, USA, 2015; ISBN 978-1-118-78835-6. [Google Scholar]
- On behalf of the 2013 CINP Summit Group. Securing the Future of Drug Discovery for Central Nervous System Disorders. Nat. Rev. Drug Discov. 2014, 13, 871–872. [Google Scholar] [CrossRef] [PubMed]
- Van de Waterbeemd, H. Physicochemical Approaches to Drug Absorption. In Methods and Principles in Medicinal Chemistry; van de Waterbeemd, H., Testa, B., Eds.; Wiley: Hoboken, NJ, USA, 2008; Volume 40, ISBN 978-3-527-32051-6. [Google Scholar]
- van De Waterbeemd, H.; Camenisch, G.; Folkers, G.; Raevsky, O.A. Estimation of Caco-2 Cell Permeability Using Calculated Molecular Descriptors. Quant. Struct.-Act. Relatsh. 1996, 15, 480–490. [Google Scholar] [CrossRef]
- Ajay; Bemis, G.W.; Murcko, M.A. Designing Libraries with CNS Activity. J. Med. Chem. 1999, 42, 4942–4951. [Google Scholar] [CrossRef] [PubMed]
- Gupta, M.; Lee, H.J.; Barden, C.J.; Weaver, D.F. The Blood–Brain Barrier (BBB) Score. J. Med. Chem. 2019, 62, 9824–9836. [Google Scholar] [CrossRef]
- Morelli, X.; Bourgeas, R.; Roche, P. Chemical and Structural Lessons from Recent Successes in Protein–Protein Interaction Inhibition (2P2I). Curr. Opin. Chem. Biol. 2011, 15, 475–481. [Google Scholar] [CrossRef]
- Capuzzi, S.J.; Muratov, E.N.; Tropsha, A. Phantom PAINS: Problems with the Utility of Alerts for P an- A Ssay IN Terference Compound, S. J. Chem. Inf. Model. 2017, 57, 417–427. [Google Scholar] [CrossRef]
- Shultz, M.D. Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs: Miniperspective. J. Med. Chem. 2019, 62, 1701–1714. [Google Scholar] [CrossRef]
- Olah, M.M.; Bologa, C.G.; Oprea, T.I. Strategies for Compound Selection. Curr. Drug Discov. Technol. 2004, 1, 211–220. [Google Scholar] [CrossRef]
- Charifson, P.S.; Walters, W.P. Filtering Databases and Chemical Libraries. J. Comput. Aided Mol. Des. 2002, 16, 311–323. [Google Scholar] [CrossRef]
- Polishchuk, P.G.; Madzhidov, T.I.; Varnek, A. Estimation of the Size of Drug-like Chemical Space Based on GDB-17 Data. J. Comput. Aided Mol. Des. 2013, 27, 675–679. [Google Scholar] [CrossRef]
- Irwin, J.J.; Shoichet, B.K. ZINC—A Free Database of Commercially Available Compounds for Virtual Screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Lyu, J.; Irwin, J.J.; Shoichet, B.K. Modeling the expansion of virtual screening libraries. Nat. Chem. Biol. 2023, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Bender, B.J.; Gahbauer, S.; Luttens, A.; Lyu, J.; Webb, C.M.; Stein, R.M.; Fink, E.A.; Balius, T.E.; Carlsson, J.; Irwin, J.J. A practical guide to large-scale docking. Nat. Protoc. 2021, 16, 4799–4832. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kralj, S.; Jukič, M.; Bren, U. Molecular Filters in Medicinal Chemistry. Encyclopedia 2023, 3, 501-511. https://doi.org/10.3390/encyclopedia3020035
Kralj S, Jukič M, Bren U. Molecular Filters in Medicinal Chemistry. Encyclopedia. 2023; 3(2):501-511. https://doi.org/10.3390/encyclopedia3020035
Chicago/Turabian StyleKralj, Sebastjan, Marko Jukič, and Urban Bren. 2023. "Molecular Filters in Medicinal Chemistry" Encyclopedia 3, no. 2: 501-511. https://doi.org/10.3390/encyclopedia3020035