
Genetic Variability of HUPRA Syndrome—A Case Report
by
 , ,
, ,
Edita Petrosyan
1,2,*,
Maria Molchanova
1,
Berta Kushnir
3,†,
Patritsia Povilaitite
4,†,
Polina Tsygankova
5,‡,
Ekaterina Zakharova
5,‡ and
Maria Proskura
2 1
Department of Hospital Pediatrics Named after Academician V.A. Tabolin, Pirogov Russian National Research Medical University, ul. Ostrovitjanova 1, Moscow 117997, Russia
2
Nephrology Department, Russian Children’s Clinical Hospital of Pirogov Russian National Research Medical University, Leninsky Prospect, 117, Moscow 119571, Russia
3
The Department of Pathology, Russian Children’s Clinical Hospital of Pirogov Russian National Research Medical University, Leninsky Prospect, 117, Moscow 119571, Russia
4
State Institution of Health Care, Rostov Region “Pathological Bureau”, Blagodatnaya, 170a, Rostov-on-Don 344015, Russia
5
Research Centre for Medical Genetics, Moskvorech’e, 1, Moscow 115552, Russia
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
‡
These authors contributed equally to this work.
Kidney Dial. 2023, 3(2), 196-203; https://doi.org/10.3390/kidneydial3020018
Submission received: 23 February 2023
/
Revised: 15 April 2023
/
Accepted: 18 April 2023
/
Published: 26 April 2023
(This article belongs to the Collection Teaching Cases in Nephrology, Dialysis and Transplantation)
Abstract
:HUPRA syndrome is a rare autosomal recessive mitochondrial disorder caused by a mutation in the SARS2 gene encoding mitochondrial seryl-tRNA synthetase (mtSerRS). It includes hyperuricemia, pulmonary hypertension, renal failure, and alkalosis. We present a case report of a boy aged 1 year 2 months with premature anemia, hyperuricemia, pulmonary hypertension, renal failure, and alkalosis and diagnosed with HUPRA syndrome. This disease is known to be progressive and fatal. A genetic test revealed a new previously undescribed heterozygous nucleotide variant in exons 14 and 1 of the SARS2 gene. The nucleotide substitution c.1295G > A (p.Arg432His) was detected in exon 14; according to the criteria of the American College of Medical Genetics (ACMG), this missense mutation is probably pathogenic. The nucleotide substitution c.227T > C (p.Leu76Pro) was detected in exon 1; according to the ACMG criteria, this missense mutation is a variant of unclear significance. We suggest that previously undescribed nucleotide substitutions in the SARS2 gene revealed in a patient with typical clinical presentation of the HUPRA syndrome should be considered as a pathogenic mutation.
1. Introduction
Hyperuricemia, pulmonary hypertension, renal failure, and alkalosis are symptoms of HUPRA syndrome (OMIM#613845), which is an orphan disease with an autosomal recessive inheritance, with a progressive course and leading to death at an early age. The disease was found to be caused by a mutation in the SARS2 nuclear gene encoding mitochondrial seryl-tRNA synthetase (mtSerRS) and has been known since 2011 [1]. In this article, we present a case report of a child with HUPRA syndrome with a new heterozygous mutation of the SARS2 gene.
2. A Case Report
A baby-boy patient (1 year 2 months old of non-consanguineous parents) was born at 36 weeks of gestation from the fifth pregnancy (the 1st and 2nd children in the family are healthy, the 3rd pregnancy ended with miscarriage at 7 weeks of gestation, and the 4th pregnancy miscarried at 22 weeks). The mother was diagnosed with thrombophilia. The boy was breast-fed until 5 months, when he presented with enterocolitis leading to poor weight gain and maldigestion and turned to bottle-feeding with a hydrolytic formula. Despite adequate food intake from the age of 7 months, the patient stopped gaining weight. At 7 months, he was also diagnosed with anemia (Hb 88 g/L [N, 110–140 g/L]), regarded as anemia of prematurity. Lab tests showed a normal serum iron level. Due to anemia, the child received erythropoietin, but this had an insufficient effect. Blood tests revealed a progressive increase in urea and creatinine, aspartate aminotransferase (111 U/L [N, <48 U/L]), and LDH levels (628 U/L [N, <320 U/L]), as well as an increase in parathyroid hormone (180 pg/mL [N, 5.7–34 pg/mL]), with normal levels of serum calcium, phosphorus, and alkaline phosphatase. Occasionally, the child demonstrated leukopenia (up to 2.0 × 109/L [N, 5.5–12.5 × 109/L]) and thrombocytopenia (up to 87 × 109/L [N, 180–400 × 109/L]). Urinary tests showed his transient proteinuria up to 400 mg/day [N, <250 mg/day] and hematuria.
Due to continued weight loss at the age of 1 year and 1 month, the child was admitted to a local hospital for the first time. Lab tests revealed signs of renal failure (creatinine level 172 µmol/L [N, 17–44 µmol/lL]), urea (54.7–74.6 mmol/L [N, 2.5–7.1 mmol/L]), hyperuricemia (989–1420 µmol/L [N, 120–320 µmol/L]), anemia (101 g/L [N, 110–140 g/L]), thrombocytopenia (90 × 109/L [N, 180–400 × 109/L]), hypokalemia (2.0 mmol/L [N, 3.6–5.6 mmol/L]), hyponatremia (117 mmol/L [N, 135–145 mmol/L]), and hypochloremic metabolic alkalosis (chloride 73 mmol/L [N, 96–111 mmol/L], pH 7.49 [N, 7.35–7.45]). The lactic acid level was not determined.
As the cause of kidney disease remained unknown, the boy was admitted to the Nephrology Department of the Russian Children’s Clinical Hospital. Upon admission, the child had a severe condition due to a low body weight (7.5 kg, <3 percentile), while height was 74 cm (25 percentile). Arterial blood pressure remained within the normal range of 97/63 mm Hg. The child showed polyuria 800 to 900 mL per day. Table 1 shows lab tests results.
Ultrasound examination revealed hyperechogeneous kidney parenchyma.
Initially, the correction of electrolyte disturbances was performed.
We conducted a differential diagnosis between the primary Bartter syndrome (hyponatremia, hypokalemia, and hypochloremic metabolic alkalosis), Gitelman syndrome (the presence of hypomagnesaemia), and pseudo-Bartter syndrome. We excluded the primary forms of Bartter and Gitelman syndrome due to the presence of cytopenic syndrome as well as severe hyperuricemia and early azotemia, while there were no signs of nephrocalcinosis in ultrasound examination.
According to echocardiography, the severe dilatation of the right side of the heart and signs of pulmonary hypertension were found. Cytopenic syndrome, hyperuricemia, and severe weight loss were considered as the course of an oncohematological disorder with a secondary kidney damage. To verify the diagnosis the child underwent a bone marrow puncture, which revealed evidence of dyserythropoiesis and dysmegakaryocytopoiesis. A renal biopsy was performed to determine the cause of kidney damage. After the biopsy, the child developed renal bleeding, which could not be controlled with hemostatic plasma therapy and required left-side nephrectomy. As a result of severe blood loss and secondary disseminated intravascular coagulation (DIC), the child developed multiple organ failure syndrome and died.
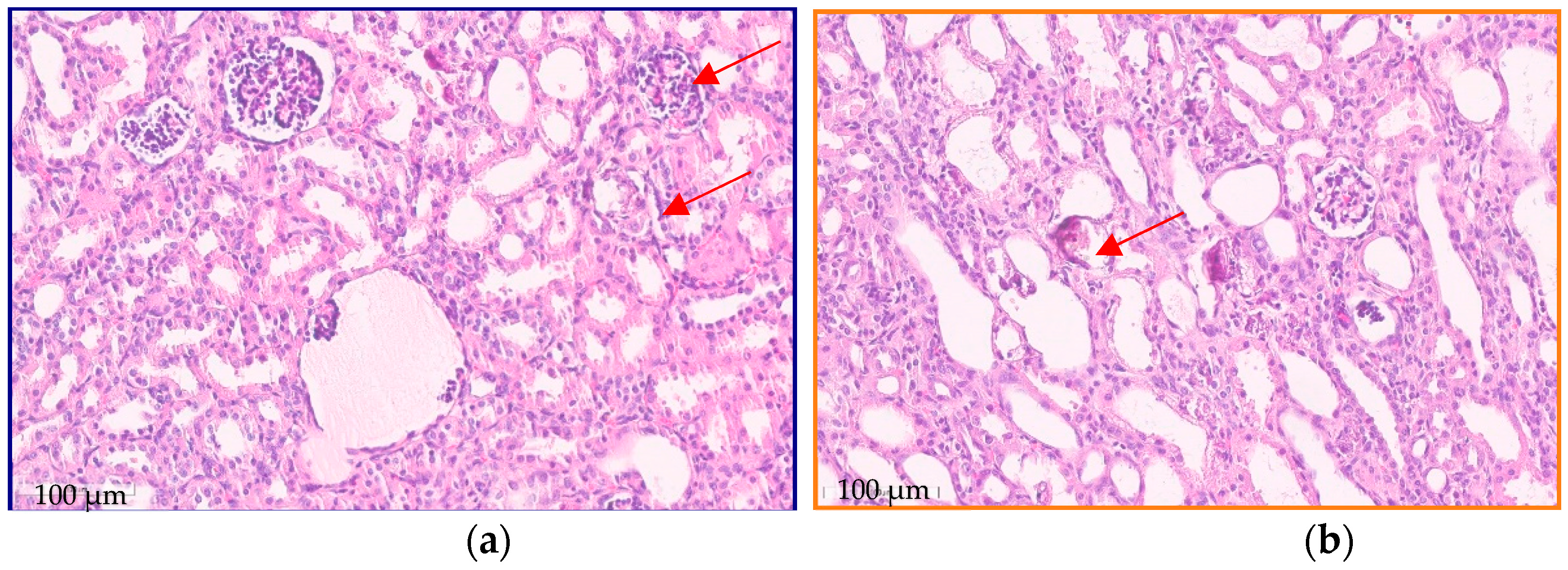
Renal tissue morphological examination revealed signs of focal segmental glomerulosclerosis; immature glomeruli, with large closely spaced podocytes; and dysmorphic glomeruli, including glomeruli with multiplication. Tubule changes such as foci of fibrosis and atrophy occupied about 20% of the microslide. There were signs of nephrocalcinosis (phosphate and calcium oxalate crystals) (Figure 1).
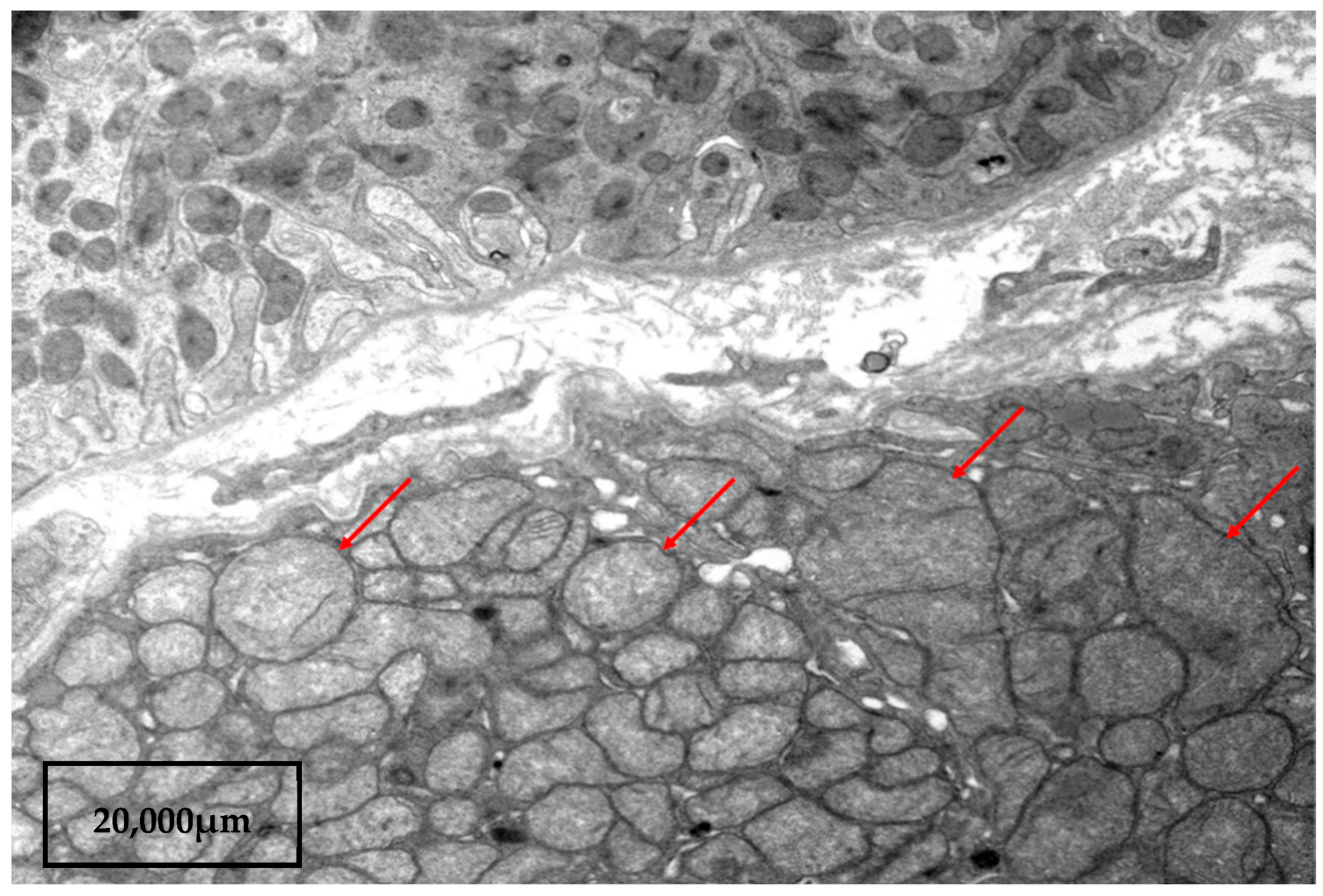
No arteriole changes were found. The immunofluorescent assay was negative. Electronic microscopic examination revealed flattening (fusion) of small processes of podocytes and accumulations of malformed mitochondria in tubulocytes. Mitochondria were found with an enlightened matrix and absent or single cristae (Figure 2).
Post mortem histological examination detected fibrosis, thickening of the walls of large arteries, and interstitial focal myocardial fibrosis.
Given that the child presented with such symptoms as hyperuricemia, metabolic hypochloremic alkalosis, early onset of renal failure, and signs of pulmonary hypertension, HUPRA syndrome was suspected.
To confirm the diagnosis, a genetic examination was carried out.
Genomic DNA was extracted from blood samples with the use of QiaAMP DNA-mini kit (Qiagen, Valencia, CA, USA), following the manufacturer’s protocol.
Whole-exome sequencing of the patient’s DNA was performed with the TruSeq DNA PCR-Free sample preparation kit on NovaSeq 6000 (Illumina, San Diego, CA, USA).
Regarding the bioinformatic pipeline, sequence readings were aligned to the human reference genome GRCh37 (hg19) using the Burrows–Wheeler Aligner (http://bio-bwa.sourceforge.net/, accessed on 1 November 2022). Single-nucleotide variants and small insertions and deletions (indels) were called with the Strelka2 Small Variant Caller (https://github.com/Illumina/strelka, accessed on 1 November 2022) and the Genome Analysis Toolkit v.4 (https://gatk.broadinstitute.org/, accessed on 1 November 2022). The reported variants were annotated with their genomic coordinates, allele frequency (gnomAD database, http://gnomad.broadinstitute.org/, accessed on 1 November 2022), functional consequence, and impact level on the gene product using SnpEff v5 (http://pcingola.github.io/SnpEff/, accessed on 1 November 2022). Variants were prioritized by the consensus score of the set of bioinformatic tools, which predict the pathogenicity of the variant and the deleterious effect on protein (SIFT, SIFT4G, Polyphen2, MutationAssessor, FATHMM, PROVEAN, DEOGEN2, LRT, PrimateAI, MetaSVM, MetaLR, SpliceAI, MMsplice, SPiP, Spidex, and ReMM). Data analysis was performed with an in-home “NGS-Data-Genome” interface.
The SARS2 variants, which were revealed by massive parallel sequencing, were validated by the Sanger sequencing in both parents, confirming the biallelic state.
Variants were named according to the HGVS nomenclature and validated using VariantValidator (https://variantvalidator.org/, accessed on 1 November 2022). The novel variants were classified according to ACMG recommendations and submitted to the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/, accessed on 1 November 2022).
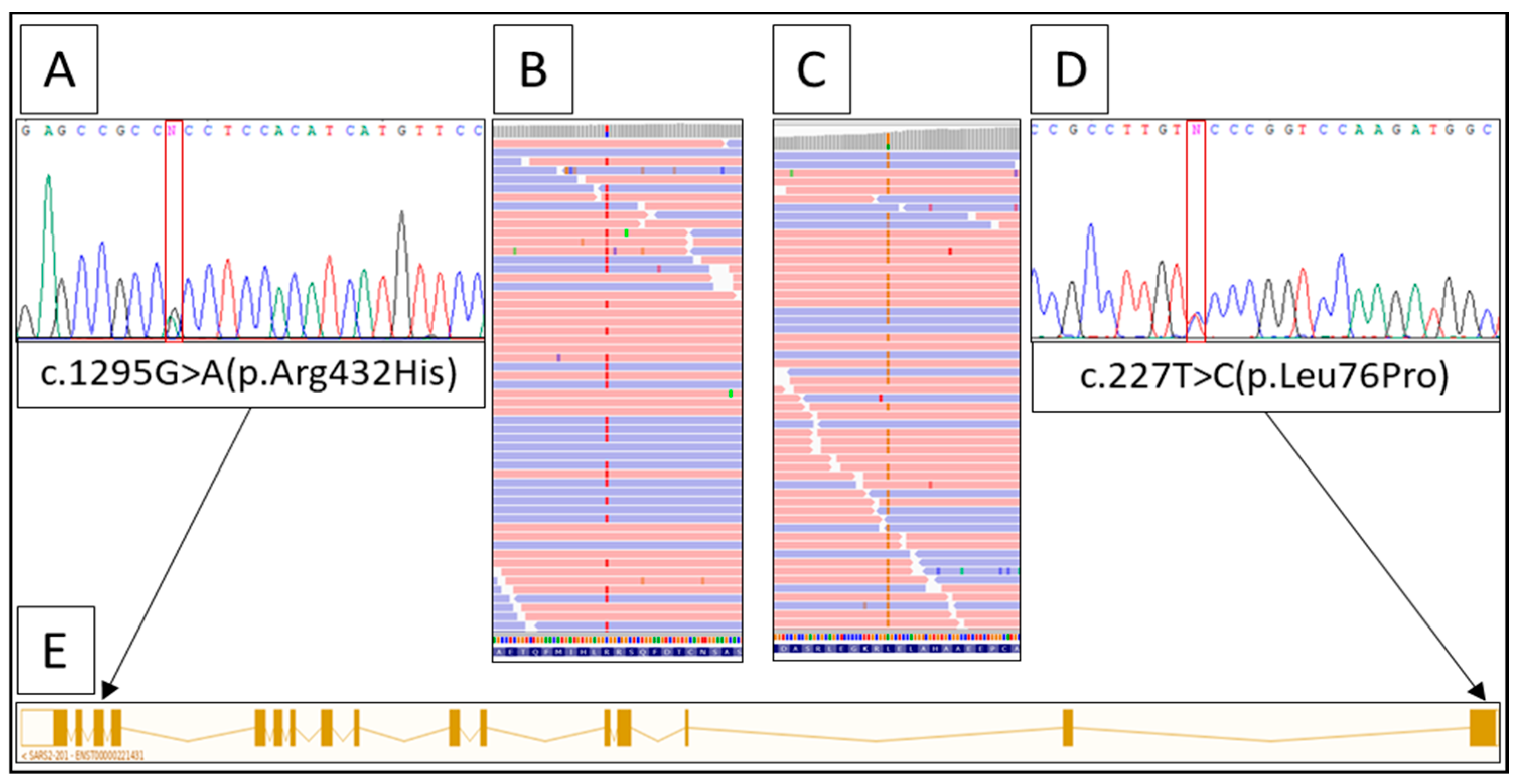
Whole-exome sequencing analysis revealed two heterozygous variants in the SARS2 gene (NM_017827.4:c.227T > C(p.Leu76Pro) in exon 1 and c.1295G > A (p.Arg432His) in exon 14 (Figure 3). Both variants were not described earlier in the literature or in the HGMD, ClinVar, or LOVD databases.
According to ACMG criteria, variant c.227T > C (p.Leu76Pro) is supposed to be a variant of uncertain significance. The variant is absent in the gnomAD database. ReMM (Regulatory Mendelian Mutation resource [2,3]) predicts a high pathogenicity score for this variant. The second mutant allele contains c.1295G > A (p.Arg432His), which has very low minor allele frequency in gnomAD (2 heterozygous carriers among 64,553 samples) and is likely pathogenic according to ACMG.
3. Discussion
Mitochondrial diseases are rare conditions caused by defects in mitochondria, leading to impaired cell respiration. Nervous system damage is present in most cases of diseases, and kidney damage is also diagnosed in some forms of this condition [4]. Both kidney parenchyma and neural tissue require high energy for adequate function. The proximal tubule, the ascending part of the Henle loop, is at risk of damage due to being rich in mitochondria requiring high adenosine triphosphate (ATP) levels for proper tubule transport system function [5]. This explains the fact that tubular dysfunctions of different varieties are quite often observed in mitochondrial disorders. Fanconi syndrome is the most common tubular dysfunction in children with mitochondrial diseases such as Kearns–Sayre syndrome, Pearson syndrome, Leigh encephalopathy, and CoQ10 deficiency [6,7,8,9,10,11,12,13]. Some patients with mitochondrial disorders were reported to have Barter-like phenotypes [14,15] or isolated hypermagnesuria [6].
The organelle function is controlled by nuclear and mitochondrial DNA, and mutations in nuclear genes account for 75–95% of all mitochondrial diseases [16]. A large number of nuclear-encoded factors, s.a. aminoacyl-tRNA synthetases (ARS), are required for the tRNA function to provide mitochondrial protein synthesis [17]. Each ARS is highly specific for amino acid matching and must match with the appropriate tRNA [18]. Belostotsky et al. (2011) revealed a mutation in the nuclear SARS2 gene encoding seryl-tRNA synthetase, which is charging tRNA with aminoacylated serine [1,19,20] in HUPRA syndrome. In translation, incoming tRNAs provide the serine required for mitochondrial protein synthesis. Mitochondrial proteins contribute to the formation of the mitochondrial respiratory chain complex [1]. Thus, due to the SARS2 gene mutation, this process is impaired. However, in most tissues, residual SARS2 activity is able to maintain a sufficient energy level, and the symptoms of the disease are observed only in the most “sensitive to lack of energy” tissues [1,21]. Various diseases are associated with mutations in different types of ARS genes, and most patients suffer from neurological disorders [22]. In contrast, initially missense mutations of the SARS2 gene leading to the HUPRA syndrome were described and the kidneys were affected, resulting in metabolic alkalosis and early onset of renal failure [1,23]. However, a new SARS2 gene homozygous mutation of the splicing leading to progressive spastic tetraparesis without kidney damage was described later [24]. This fact once again indicates that the clinical aspects of mitochondrial diseases can be quite diverse.
HUPRA syndrome is an extremely rare disease described in only 7 children [1,23,25,26]. The additional signs of the disease are premature birth (from 27 up to 37 weeks of gestation), developmental delay, anemia with insufficient response to erythropoietin treatment, hyponatremia, hypochloremia, hypomagnesemia, and a minimal increase in blood lactic acid. Despite progressive renal failure, metabolic alkalosis was detected. An uncommon imbalance between creatinine and urea levels was also present: urea and uric acid concentrations were significantly higher with a lower creatinine level. In this case, we observed similar metabolic disturbances. However, the noticeable difference was a persistent increase in LDH and AST serum levels that were not described in other papers. Thrombocytopenia and leukocytopenia in this clinical case were previously described in only two patients [1,26]. However, our patient did not develop diabetes mellitus, which was present in all three children in the Belostotsky et al. article (2011) [1].
An important difference between this particular clinical case and three patients described previously (underwent kidney biopsy) is the absence of the glomeruli changes [1,23] as this patient appeared to have signs of focal segmental glomerulosclerosis, as well as immature and dysmorphic glomeruli. However, tubular changes such as fibrosis and atrophy and the presence of enlarged mitochondria in tubule cells were observed both in this case and in Belostotsky’s paper. Additionally, in this case the patient showed no arteriole changes or positive immunofluorescent assay [1,23].
It should be noted that several studies reported the development of focal segmental glomerulosclerosis in patients with Bartter and Gitelman syndrome [27,28,29,30]. This allows us to assume that the glomerular disorders that we have identified may be the outcome of a general pathomorphological process, which is common to these syndromes. At the post mortem histological examination, fibrosis and thickening of the walls of large arteries and myocardial fibrosis were confirmed. There were also signs of pulmonary hypertension, apparently developed after renal bleeding resulting in DIC. Prior to that, there were no signs of pulmonary hypertension requiring any medications.
The cause of prolonged kidney hemorrhage after biopsy remained unclear. Despite procoagulant drug administration, plasma transfusion, nephrectomy, and surgical hemostasis bleeding were not controlled for 72 h. The kidney hemorrhage resulted in DIC. In comparison with the previously described patient histories, only Rivera et al. (2013) reported the pulmonary hemorrhage as the cause of death in one patient [23]. In other cases, there were no coagulation disorders.
Most children with HUPRA syndrome died; life expectancy ranged from 10 to 70 months [25]. Belostotsky et al. (2011) observed the most severe cases: children with homozygous pathogenic variant c.1169A > G (p.Asp390Gly), who had a life expectancy of approximately 12 months; their cause of death was pulmonary hypertension [1]. Rivera et al. (2013) reported children with homozygous mutation c.1205G > A (p. Arg402His), with a life expectancy of 21 to 26 months [23]. Zhou et al. (2021) described a significantly longer life expectancy of a girl with heterozygous nucleotide variants c.667G > A (p.Val223Met)/c.1205G > A (p Arg402His). Thus, this phenotype was regarded as less severe: the girl did not show pulmonary hypertension and metabolic alkalosis. The main symptom and leading cause of death was renal failure, and she died at the age of 70 months due to uremic intoxication [25]. Göknar et al. (2022) revealed the homozygous mutation c.515A > G [p.Asn172Ser] in a girl who was 4 years 4 months old and was alive at presentation. She showed all symptoms of HUPRA syndrome, except for metabolic alkalosis. Furthermore, the patient showed severe immunodeficiency, Chiari type 1 anomaly, and Graves’ disease. It is still unclear whether these conditions were associated with observed homozygous mutation or were random [26].
In this case, the child died at the age of 14 months, but the main cause of death was DIC due to kidney hemorrhage. At this age, he did not show severe pulmonary hypertension but reached end-stage kidney disease. In this child, heterozygous missense mutations c.1295G > A (p.Arg432His)/c.227T > C(p.Leu76Pro) still played an important role in the severity of the disease.
In HUPRA syndrome, the many causes of organ damage remain unclear. Pulmonary hypertension appears to be associated with the development of cardiomyopathy. Hyperuricemia is explained by the impaired fractional excretion of uric acid by the affected tubule cells or as a result of chronic kidney disease (CKD) [1], as well as increased uric acid production in ischemic cardiac tissue [31]. Our patient did not have severe cardiomyopathy, but there was a persistent increase in LDH and AST serum levels from infancy.
The main hypothesis is that the residual activity of the mutated SARS2 gene is the main cause of the symptoms. It is sufficient to maintain the function of the electron transport chain in most tissues, but the thick ascending part of the Henle loop tubule cells requires much more ATP to maintain active transport system functioning [1]. So, this tissue is affected more often, and the seryl-tRNA synthetase may have additional yet unexplored functions that affect certain types of cells at different stages of their differentiation [23].
In general, the clinical presentation of HUPRA syndrome is highly variable. It is debatable whether the severity of the disease depends on the homo- or heterozygous type of mutation. It is necessary to accumulate as much information about HUPRA syndrome as possible since this condition can be easily overlooked. Due to very few cases of the disease, each clinical case presentation may play an important role in diagnostics and treatment.
4. Conclusions
We presented a case report on an extremely rare HUPRA syndrome with new genetic nucleotide substitutions in the SARS2 gene, which demonstrates its genetic variability in different populations. We believe that most nephrologists and pediatricians should be familiar with HUPRA syndrome and consider it as one of the possible causes of renal failure in children.
Author Contributions
E.P. collected and analyzed the patient’s clinical data and compiled and edited the manuscript. M.M. reviewed the literature and wrote the manuscript. B.K. and P.P. conducted a morphological study. P.T. and E.Z. conducted a molecular genetic study and helped to write the manuscript. M.P. edited the article and carried out the translation. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the University Review Board, protocol code NO. 81 of 17/10/2022.
Informed Consent Statement
Informed consent was obtained the patient to publish this paper.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Belostotsky, R.; Ben-Shalom, E.; Rinat, C.; Becker-Cohen, R.; Feinstein, S.; Zeligson, S.; Segel, R.; Elpeleg, O.; Nassar, S.; Frishberg, Y. Mutations in the mitochondrial seryl-tRNA synthetase cause hyperuricemia, pulmonary hypertension, renal failure in infancy and alkalosis, HUPRA Syndrome. Am. J. Hum. Genet. 2011, 88, 193–200. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–423. [Google Scholar] [CrossRef]
- Kim, S.S.; Dey, K.K.; Weissbrod, O.; Márquez-Luna, C.; Gazal, S.; Price, A.L. Improving the informativeness of Mendelian disease-derived pathogenicity scores for common disease. Nat. Commun. 2020, 11, 6258. [Google Scholar] [CrossRef] [PubMed]
- Govers, L.P.; Toka, H.R.; Hariri, A.; Walsh, S.B.; Bockenhauer, D. Mitochondrial DNA mutations in renal disease: An overview. Pediatr. Nephrol. 2021, 36, 9–17. [Google Scholar] [CrossRef]
- Bhargava, P.; Schnellmann, R.G. Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 2017, 13, 629–646. [Google Scholar] [CrossRef]
- Emma, F.; Bertini, E.; Salviati, L.; Montini, G. Renal involvement in mitochondrial cytopathies. Pediatr. Nephrol. 2012, 27, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Niaudet, P.; Rotig, A. The kidney in mitochondrial cytopathies. Kidney Int. 1997, 51, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Ogier, H.; Lombes, A.; Scholte, H.R.; Poll-The, B.T.; Fardeau, M.; Alcardi, J.; Vignes, B.; Niaudet, P.; Saudubray, J.M. de Toni-Fanconi-Debré syndrome with Leigh syndrome revealing severe muscle cytochrome c oxidase deficiency. J. Pediatr. 1988, 112, 734–739. [Google Scholar] [CrossRef]
- Mochizuki, H.; Joh, K.; Kawame, H.; Imadachi, A.; Nozaki, H.; Ohashi, T.; Usui, N.; Eto, Y.; Kanetsuna, Y.; Aizawa, S. Mitochondrial encephalomyopathies preceded by de-Toni-Debré-Fanconi syndrome or focal segmental glomerulosclerosis. Clin. Nephrol. 1996, 46, 347–352. [Google Scholar]
- Mori, K.; Narahara, K.; Ninomiya, S.; Goto, Y.I.; Nonaka, I. Renal and skin involvement in a patient with complete Kearns–Sayre syndrome. Am. J. Med. Genet. 1991, 38, 583–587. [Google Scholar] [CrossRef]
- Topaloglu, R.; Lebre, A.S.; Demirkaya, E.; Kuskonmaz, B.; Coskun, T.; Derman, O.; Gurgey, A.; Gumruk, F. Two new cases with Pearson syndrome and review of Hacettepe experience. Turk. J. Pediatr. 2008, 50, 572–576. [Google Scholar] [PubMed]
- Lee, Y.S.; Yap, H.K.; Barshop, B.A.; Lee, Y.S.; Rajalingam, S.; Loke, K.Y. Mitochondrial tubulopathy: The many faces of mitochondrial disorders. Pediatr. Nephrol. 2001, 16, 710–712. [Google Scholar] [CrossRef]
- Martín-Hernández, E.; García-Silva, M.T.; Vara, J.; Campos, Y.; Cabello, A.; Muley, R.; Del Hoyo, P.; Martín, M.A.; Arenas, J. Renal pathology in children with mitochondrial diseases. Pediatr. Nephrol. 2005, 20, 1299–1305. [Google Scholar] [CrossRef]
- Emma, F.; Pizzini, C.; Tessa, A.; Di Giandomenico, S.; Onetti-Muda, A.; Santorelli, F.M.; Bertini, E.; Rizzoni, G. ‘Bartter-like’ phenotype in Kearns–Sayre syndrome. Pediatr. Nephrol. 2006, 21, 355–360. [Google Scholar] [CrossRef]
- Goto, Y.I.; Itami, N.; Kajii, N.; Tochimaru, H.; Endo, M.; Horai, S. Renal tubular involvement mimicking Bartter syndrome in a patient with Kearns–Sayre syndrome. J. Pediatr. 1990, 116, 904–910. [Google Scholar] [CrossRef]
- Graham, B.H. Diagnostic challenges of mitochondrial disorders: Complexities of two genomes. Mitochondrial Disord. Biochem. Mol. Anal. 2012, 837, 35–46. [Google Scholar]
- Hallberg, B.M.; Larsson, N.G. Making proteins in the powerhouse. Cell Metab. 2014, 20, 226–240. [Google Scholar] [CrossRef]
- Ling, J.; Reynolds, N.; Ibba, M. Aminoacyl-tRNA synthesis and translational quality control. Annu. Rev. Microbiol. 2009, 63, 61–78. [Google Scholar] [CrossRef]
- Antonellis, A.; Green, E.D. The role of aminoacyl-tRNA synthetases in genetic diseases. Annu. Rev. Genom. Hum. Genet. 2008, 9, 87–107. [Google Scholar] [CrossRef] [PubMed]
- Wellner, K.; Betat, H.; Morl, M. A tRNA’s fate is decided at its 3’ end: Collaborative actions of CCA-adding enzyme and RNases involved in tRNA processing and degradation. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2018, 1861, 433–441. [Google Scholar] [CrossRef] [PubMed]
- Scheper, G.C.; van der Klok, T.; van Andel, R.J.; van Berkel, C.G.; Sissler, M.; Smet, J.; Muravina, T.I.; Serkov, S.V.; Uziel, G.; Bugiani, M.; et al. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat. Genet. 2007, 39, 534–539. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Serrano, L.E.; Chihade, J.W.; Sissler, M. When a common biological role does not imply common disease outcomes: Disparate pathology linked to human mitochondrial aminoacyl-tRNA synthetases. J. Biol. Chem. 2019, 294, 5309–5320. [Google Scholar] [CrossRef] [PubMed]
- Rivera, H.; Martín-Hernández, E.; Delmiro, A.; García-Silva, M.T.; Quijada-Fraile, P.; Muley, R.; Arenas, J.; Martín, M.A.; Martínez-Azorín, F. A new mutation in the gene encoding mitochondrial seryl-tRNA synthetase as a cause of HUPRA syndrome. BMC Nephrol. 2013, 14, 195. [Google Scholar] [CrossRef] [PubMed]
- Linnankivi, T.; Neupane, N.; Richter, U.; Isohanni, P.; Tyynismaa, H. Splicing Defect in Mitochondrial Seryl-tRNA Synthetase Gene Causes Progressive Spastic Paresis Instead of HUPRA Syndrome. Hum. Mutat. 2016, 37, 884–888. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhong, C.; Yang, Q.; Zhang, G.; Yang, H.; Li, Q.; Wang, M. Novel SARS2 variants identified in a Chinese girl with HUPRA syndrome. Mol. Genet. Genom. Med. 2021, 9, 1650. [Google Scholar] [CrossRef]
- Göknar, N.; Keleşoğlu, E.; Kasap, N.; Üçkardeş, D.; Candan, C. A case of chronic kidney disease with pulmonary hypertension, hyperuricemia, immunodeficiency and other extrarenal findings: Questions. Pediatr. Nephrol. 2022, 37, 2615–2616. [Google Scholar] [CrossRef]
- Blethen, S.L.; Van Wyk, J.J.; Lorentz, W.B.; Jennette, J.C. Reversal of Bartter’s syndrome by renal transplantation in a child with focal segmental glomerular sclerosis. Am. J. Med. Sci. 1985, 289, 31–36. [Google Scholar] [CrossRef]
- Su, I.H.; Frank, R.; Gauthier, B.G.; Valderrama, E.; Simon, D.B.; Lifton, R.P.; Trachtman, H. Bartter syndrome and focal segmental glomerulosclerosis: A possible link between two diseases. Pediatr. Nephrol. 2000, 14, 970–972. [Google Scholar] [CrossRef]
- Bulucu, F.; Vural, A.; Yenicesu, M.; Caglar, K. Association of Gitelman’s syndrome and focal glomerulosclerosis. Nephron 1998, 79, 244. [Google Scholar] [CrossRef]
- Bettinelli, A.; Borsa, N.; Syren, M.L.; Mattiello, C.; Coviello, D.; Edefonti, A.; Giani, M.; Travi, M.; Tedeschi, S. Simultaneous mutations in the CLCNKB and SLC12A3 genes in two siblings with phenotypic heterogeneity in classic Bartter syndrome. Pediatr. Res. 2005, 58, 1269–1273. [Google Scholar] [CrossRef]
- Bendayan, D.; Shitrit, D.; Ygla, M.; Huerta, M.; Fink, G.; Kramer, M.R. Hyperuricemia as a prognostic factor in pulmonary arterial hypertension. Respir. Med. 2003, 97, 130–133. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Kidney biopsy: light microscopic features. (a) Dysmorphic glomeruli; some glomeruli are globally and segmentally sclerosed (red arrows). (b) Calcium oxalate crystals in the tubules (red arrows). Scale bar 100 µm.
Figure 1.
Kidney biopsy: light microscopic features. (a) Dysmorphic glomeruli; some glomeruli are globally and segmentally sclerosed (red arrows). (b) Calcium oxalate crystals in the tubules (red arrows). Scale bar 100 µm.

Figure 2.
Kidney biopsy: electron microscopy. Cluster of large malformed mitochondria in a tubule cells. Mitochondria with an enlightened matrix, absent or single cristae (red arrows). Scale bar 20,000 µm.
Figure 2.
Kidney biopsy: electron microscopy. Cluster of large malformed mitochondria in a tubule cells. Mitochondria with an enlightened matrix, absent or single cristae (red arrows). Scale bar 20,000 µm.

Figure 3.
Result of the molecular analysis. (A) Sanger sequencing of the SARS2 gene, exon 14 fragment. (B) IGV-browser with the fragment of exon 14, showing c.1295G > A (p.Arg432His) in heterozygous state. (C) IGV-browser with the fragment of exon 1, showing c.227T > C (p.Leu76Pro) in heterozygous state. (D) Sanger sequencing of the SARS2 gene, exon 1 fragment. (E) Scheme of the SARS2 transcript.
Figure 3.
Result of the molecular analysis. (A) Sanger sequencing of the SARS2 gene, exon 14 fragment. (B) IGV-browser with the fragment of exon 14, showing c.1295G > A (p.Arg432His) in heterozygous state. (C) IGV-browser with the fragment of exon 1, showing c.227T > C (p.Leu76Pro) in heterozygous state. (D) Sanger sequencing of the SARS2 gene, exon 1 fragment. (E) Scheme of the SARS2 transcript.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Laboratory analysis of the patient.
| Analysis | At Presentation 1 | Reference Value |
|---|---|---|
| Blood Hemoglobin, g/L | 100 | 110–140 |
| Red blood cell, × 1012/L | 4.5 | 3.9–4.5 |
| Platelet, ×109/L | 106 | 180–400 |
| White blood cell, ×109/L | 2.13 | 5.5–12.5 |
| Creatinine, µmol/L | 90.4 | 17–44 |
| Urea, mmol/L | 11 | 2.5–7.1 |
| Uric acid, µmol/L | 978 | 120–320 |
| LDH, U/L | 498 | <320 |
| ALT, U/L | 13 | <36 |
| AST, U/L | 120 | <48 |
| PTH, pg/mL | 233 | 25–65 |
| Sodium, mmol/L | 124 | 135–145 |
| Potassium, mmol/L | 3.1 | 3.6–5.6 |
| Chloride, mmol/L | 79 | 96–111 |
| Magnesium, mmol/L | 0.67 | 0.71–0.95 |
| Lactic acid (mmol/lL) | 3.0 | 0.27–2.2 |
| Coagulation test APPT, s | 47 | 24–34 |
| D-dimers, ng/mL | 218 | <243 |
| Blood gases (venous) | ||
| pH | 7.49 | 7.35–7.45 |
| HCO3−, mmol/L | 27.6 | 22–26 |
| Urine | ||
| Protein, g/L | abs | 0–0.14 |
1 Abnormal levels are shown in bold.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Petrosyan, E.; Molchanova, M.; Kushnir, B.; Povilaitite, P.; Tsygankova, P.; Zakharova, E.; Proskura, M. Genetic Variability of HUPRA Syndrome—A Case Report. Kidney Dial. 2023, 3, 196-203. https://doi.org/10.3390/kidneydial3020018
AMA Style
Petrosyan E, Molchanova M, Kushnir B, Povilaitite P, Tsygankova P, Zakharova E, Proskura M. Genetic Variability of HUPRA Syndrome—A Case Report. Kidney and Dialysis. 2023; 3(2):196-203. https://doi.org/10.3390/kidneydial3020018
Chicago/Turabian StylePetrosyan, Edita, Maria Molchanova, Berta Kushnir, Patritsia Povilaitite, Polina Tsygankova, Ekaterina Zakharova, and Maria Proskura. 2023. "Genetic Variability of HUPRA Syndrome—A Case Report" Kidney and Dialysis 3, no. 2: 196-203. https://doi.org/10.3390/kidneydial3020018