
Comparative Analysis and Classification of SARS-CoV-2 Spike Protein Structures in PDB


Abstract
:1. Introduction
2. Methods
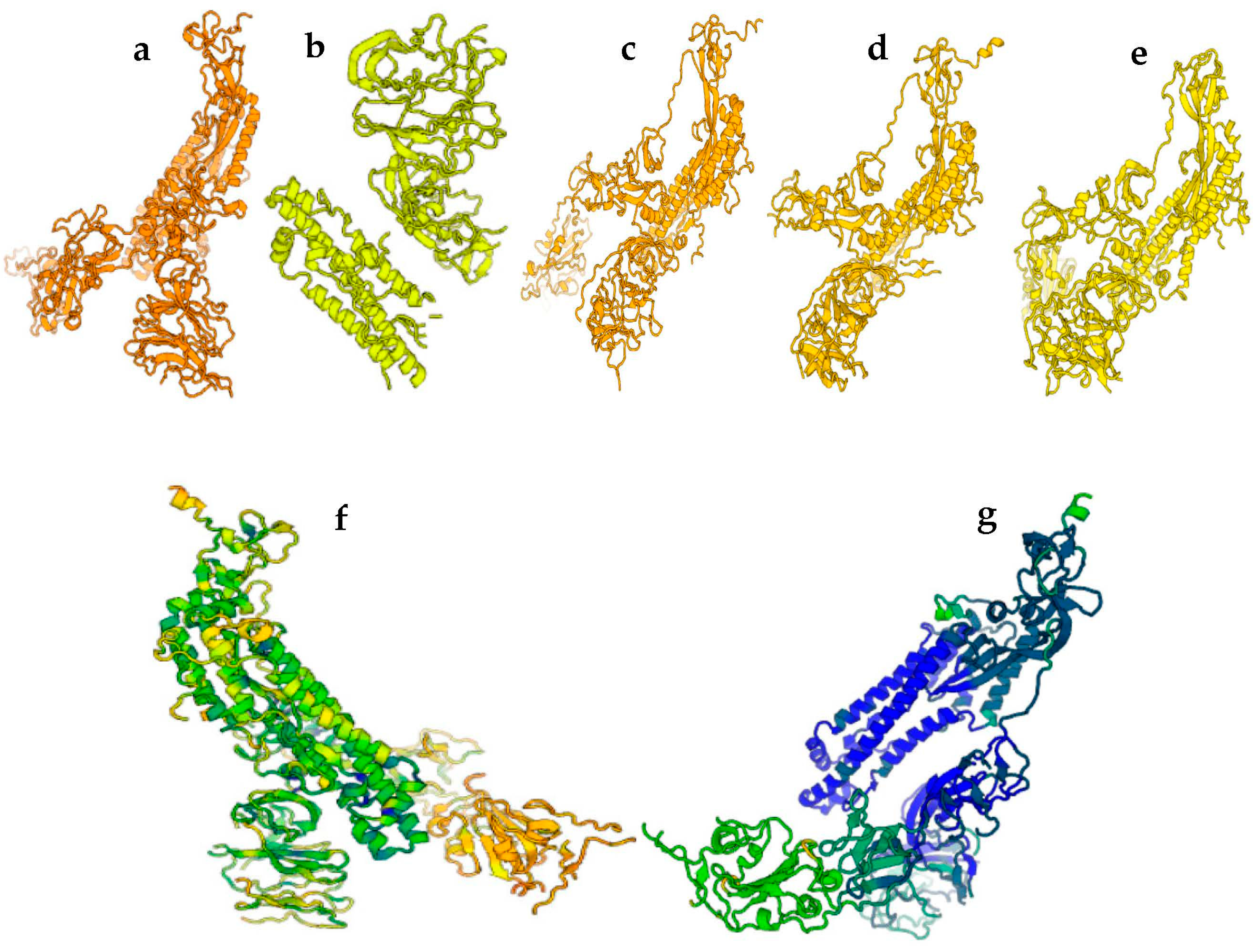
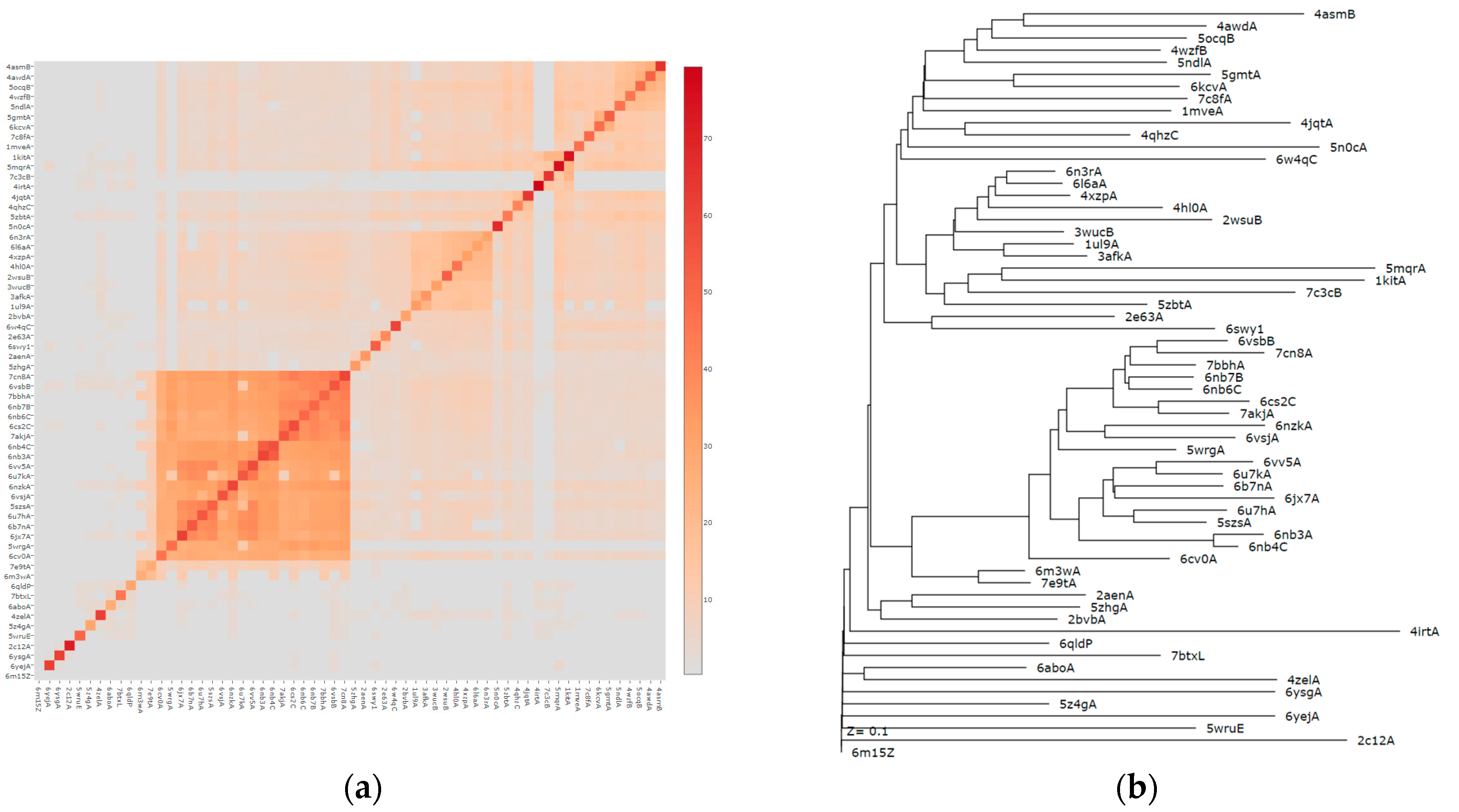
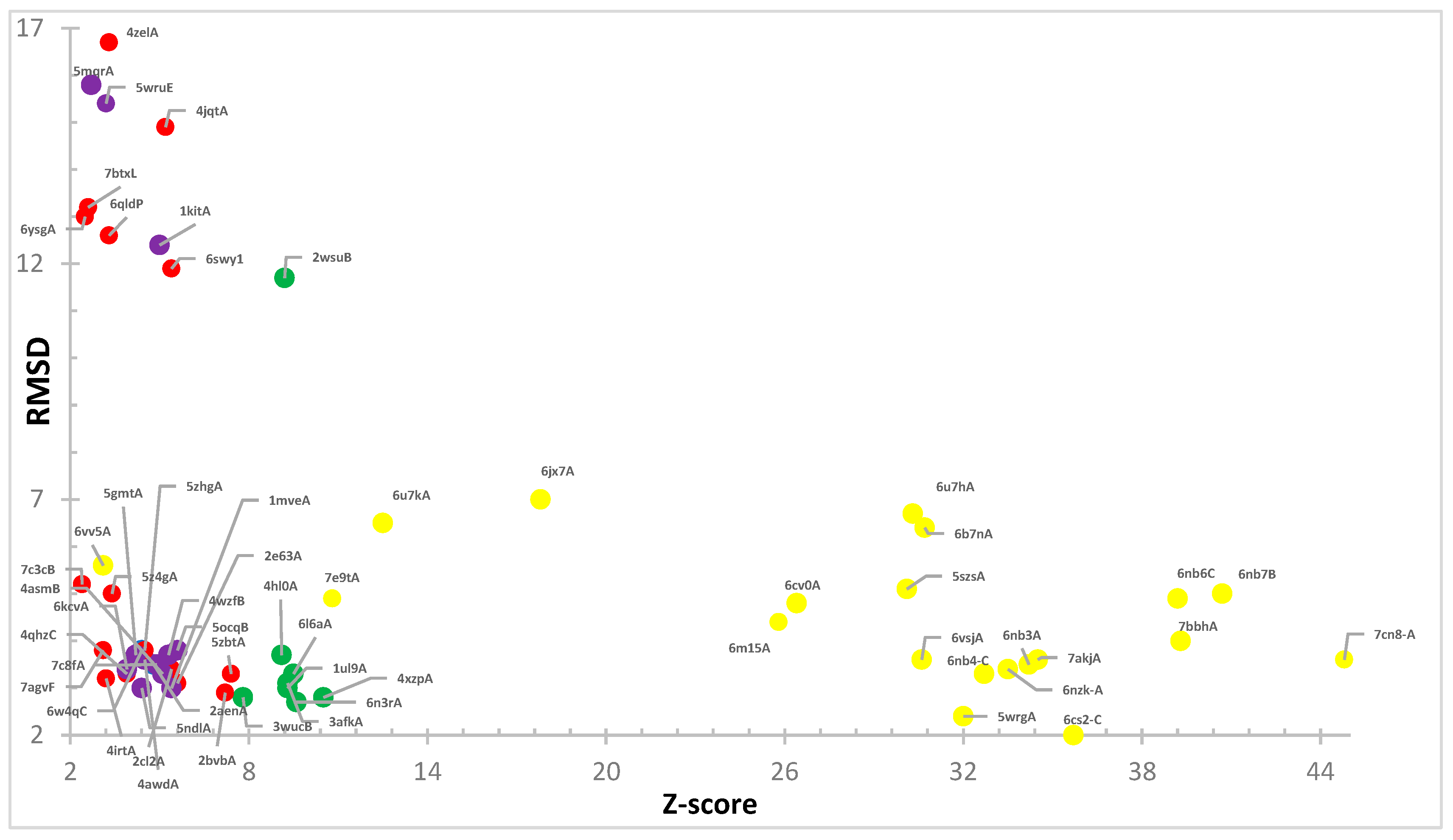
3. Results
4. Structures Classification and Their Description
4.1. Drug Development
4.1.1. Antibodies Based on S Protein of SARS-CoV-2 Variants
4.1.2. Small Protein Inhibitors
5. Discussion
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.-M.; Wang, W.; Song, Z.-G.; Hu, Y.; Tao, Z.-W.; Tian, J.-H.; Pei, Y.-Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lata, S.; Akif, M. Comparative protein structure network analysis on 3CLpro from SARS-CoV-1 and SARS-CoV-2. Proteins 2021, 89, 1216–1225. [Google Scholar] [CrossRef] [PubMed]
- Greasley, S.E.; Noell, S.; Plotnikova, O.; Ferre, R.; Liu, W.; Bolanos, B.; Fennell, K.; Nicki, J.; Craig, T.; Zhu, Y.; et al. Structural basis for the in vitro efficacy of nirmatrelvir against SARS-CoV-2 variants. J. Biol. Chem. 2022, 298, 101972. [Google Scholar] [CrossRef]
- Fu, L.; Ye, F.; Feng, Y.; Yu, F.; Wang, Q.; Wu, Y.; Zhao, C.; Sun, H.; Huang, B.; Niu, P.; et al. Both Boceprevir and GC376 efficaciously inhibit SARS-CoV-2 by targeting its main protease. Nat. Commun. 2020, 11, 4417. [Google Scholar] [CrossRef] [PubMed]
- Nawaz, M.S.; Fournier-Viger, P.; He, Y. S-PDB: Analysis and classification of SARS-CoV-2 Spike protein structures. In Proceedings of the 2022 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Las Vegas, NV, USA, 6–8 December 2022; pp. 2259–2265. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.-H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef] [PubMed]
- Rawat, P.; Jemimah, S.; Ponnuswamy, P.K.; Gromiha, M.M. Why are ACE2 binding coronavirus strains SARS-CoV/SARS-CoV-2 wild and NL63 mild? Proteins 2021, 89, 389–398. [Google Scholar] [CrossRef]
- Nawaz, M.S.; Fournier-Viger, P.; Shojaee, A.; Fujita, H. Using artificial intelligence techniques for COVID-19 genome analysis. Appl. Intell. 2021, 51, 3086–3103. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef] [Green Version]
- Burley, S.K.; Berman, H.M.; Kleywegt, G.J.; Markley, J.L.; Nakamura, H.; Valenkar, S. Protein Data Bank (PDB): The Single Global Macromolecular Structure Archive. Protein Crystallogr. Methods Mol. Biol. 2017, 1607, 627–641. [Google Scholar] [CrossRef] [Green Version]
- Lawson, C.L.; Baker, M.L.; Best, C.; Bi, C.; Dougherty, M.; Feng, P.; Ginkel, G.V.; Devkota, B.; Lagerstedt, I.; Ludtke, S.J.; et al. EMDataBank.org: Unified data resource for CryoEM. Nucleic Acids Res. 2011, 39 (Suppl. 1), D456–D464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holm, L. Dali server: Structural unification of protein families. Nucleic Acids Res. 2022, 50, W210–W215. [Google Scholar] [CrossRef] [PubMed]
- Holm, L. Using DALI for Protein Structure Comparison. In: Structural Bioinformatics. Methods Mol. Biol. 2020, 2112, 29–42. [Google Scholar] [CrossRef] [PubMed]
- Walls, C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuier, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 Spike glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef] [Green Version]
- Pinto, D.; Park, Y.-J.; Beltramello, M.; Walls, A.C.; Tortorici, M.A.; Bianchi, S.; Jaconi, S.; Culap, K.; Zatta, F.; Marco, A.D.; et al. Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature 2020, 583, 290–295. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Sun, D.; Sang, Z.; Kim, Y.J.; Xiang, Y.; Cohen, T.; Belford, A.K.; Huet, A.; Conway, J.F.; Sun, J.; Taylor, D.J.; et al. Potent neutralizing nanobodies resist convergent circulating variants of SARS-CoV-2 by targeting diverse and conserved epitopes. Nat. Commun. 2021, 12, 4676. [Google Scholar] [CrossRef]
- Tai, L.; Zhu, G.; Yang, M.; Cao, L.; Xing, X.; Yin, G.; Chan, C.; Qin, C.; Rao, C.; Wang, X.; et al. Nanometer-resolution in situ structure of the SARS-CoV-2 postfusion spike protein. Proc. Natl. Acad. Sci. USA 2020, 118, e2112703118. [Google Scholar] [CrossRef] [PubMed]
- Tortorici, M.A.; Czudnochowski, N.; Starr, T.N.; Marzi, R.; Walls, A.C.; Zatta, F.; Bowen, J.E.; Jaconi, S.; Di’lulio, D.; Wang, A.; et al. Broad sarbecovirus neutralization by a human monoclonal antibody. Nature 2021, 597, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Xiong, X.; Park, Y.-J.; Tortorici, M.A.; Snijder, J.; Quispre, J.; Cameoni, E.; Gopal, R.; Dai, M.; Lanzavecchia, A.; et al. Unexpected receptor functional mimicry elucidates activation of coronavirus fusion. Cell 2020, 183, 1732. [Google Scholar] [CrossRef] [PubMed]
- Ni, D.; Lau, K.; Turelli, P.; Raclot, C.; Beckert, B.; Nazarov, S.; Pojer, F.; Myasnikov, A.; Stahlberg, H.; Trono, D. Structural analysis of the Spike of the Omicron SARS-COV-2 variant by cryo-EM and implications for immune evasion. BioRxiv 2021. [Google Scholar] [CrossRef]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Qiao, S.; Yu, J.; Zeng, J.; Shan, S.; Tian, L.; Lan, J.; Zhang, L.; Wang, W. Bat and pangolin coronavirus spike glycoprotein structures provide insights into SARS-CoV-2 evolution. Nat. Commun. 2021, 12, 1607. [Google Scholar] [CrossRef]
- Yang, T.J.; Yu, P.-Y.; Chang, Y.-C.; Liang, K.-H.; Tso, H.-C.; Ho, M.-R.; Chen, W.-Y.; Lin, H.-T.; Wu, H.-C.; Hsu, S.-T.D. Effect of SARS-CoV-2 B.1.1.7 mutations on spike protein structure and function. Nat. Struct. Mol. Biol. 2021, 28, 731–739. [Google Scholar] [CrossRef]
- Xu, S.; Wang, Y.; Wang, Y.; Zhang, C.; Hong, Q.; Gu, C.; Xu, R.; Wang, T.; Yang, Y.; Zang, J.; et al. Mapping cross-variant neutralizing sites on the SARS-CoV-2 spike protein. Emerg. Microbes. Infect. 2022, 11, 351–367. [Google Scholar] [CrossRef]
- Liu, C.; Zhou, D.; Nutalai, R.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Ginn, H.M.; Dejnirattisai, W.; Supasa, P.; Mentzer, A.J.; Wang, B.; et al. The antibody response to SARS-CoV-2 Beta underscores the antigenic distance to other variants. Cell Host Microbe 2021, 30, 53–68.e12. [Google Scholar] [CrossRef]
- Liang, Q.; Wang, Y.; Zhang, S.; Sun, J.; Sun, W.; Li, J.; Liu, Y.; Li, M.; Cheng, L.; Jiang, Y.; et al. RBD trimer mRNA vaccine elicits broad and protective immune responses against SARS-CoV-2 variants. iScience 2022, 25, 104043. [Google Scholar] [CrossRef]
- Cao, Y.; Yisimayi, A.; Bai, Y.; Huang, W.; Li, X.; Zhang, Z.; Yuan, T.; An, R.; Wang, J.; Ziao, T.; et al. Humoral immune response to circulating SARS-CoV-2 variants elicited by inactivated and RBD-subunit vaccines. Cell Res. 2021, 31, 732–741. [Google Scholar] [CrossRef]
- Zhang, Q.; Ju, B.; Ge, J.; Chan, J.F.-W.; Cheng, L.; Wang, R.; Huang, W.; Fang, M.; Chen, R.; Zhou, B.; et al. Potent and protective IGHV3-53/3-66 public antibodies and their shared escape mutant on the spike of SARS-CoV-2. Nat. Commun. 2021, 12, 4210. [Google Scholar] [CrossRef]
- Du, S.; Liu, P.; Zhang, Z.; Xiao, T.; Yasimayi, A.; Huang, W.; Wang, Y.; Cao, Y.; Xie, X.S.; Xiao, J. Structures of SARS-CoV-2 B.1.351 neutralizing antibodies provide insights into cocktail design against concerning variants. Cell Res. 2021, 31, 1130–1133. [Google Scholar] [CrossRef] [PubMed]
- Dejnirattisai, W.; Zhou, D.; Supasa, P.; Liu, C.; Mentzer, A.J.; Ginn, H.M.; Zhao, Y.; Duyvesteyn, H.M.E.; Tuekprakhon, A.; Nutalai, R.; et al. Antibody evasion by the P.1 strain of SARS-CoV-2. Cell 2021, 184, 2939–2954.e9. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.C.; Liu, C.; Zhang, C.; Wang, Y.; Hong, Q.; Xu, S.; Li, Z.; Yang, Y.; Huang, Z.; Cong, Y. Structural basis for SARS-CoV-2 Delta variant recognition of ACE2 receptor and broadly neutralizing antibodies. Nat. Commun. 2022, 13, 871. [Google Scholar] [CrossRef]
- Liu, C.; Ginn, H.M.; Dejnirattisai, W.; Supasa, P.; Wang, B.; Tuekprakhon, A.; Nutalai, R.; Zhou, D.; Mentzer, A.J.; Zhao, Y.; et al. Reduced neutralization of SARS-CoV-2 B.1.617 by vaccine and convalescent serum. Cell 2021, 184, 4220–4236.e13. [Google Scholar] [CrossRef] [PubMed]
- Chi, X.; Guo, Y.; Zhang, G.; Sun, H.; Zhang, J.; Li, M.; Chen, Z.; Han, J.; Zhang, Y.; Zhang, X.; et al. Broadly neutralizing antibodies against Omicron-included SARS-CoV-2 variants induced by vaccination. Signal Transduct. Target Ther. 2022, 7, 139. [Google Scholar] [CrossRef]
- Yin, W.; Xu, Y.; Xu, P.; Cao, X.; Wu, C.; Gu, C.; He, X.; Wang, X.; Huang, S.; Yuan, Q.; et al. Structures of the Omicron spike trimer with ACE2 and an anti-Omicron antibody. Science 2022, 375, 1048–1053. [Google Scholar] [CrossRef]
- Wang, Z.; Muecksh, F.; Cho, A.; Gaebler, C.; Hoffmann, H.-H.; Ramos, V.; Zong, S.; Cipolla, M.; Johnson, B.; Schmidt, F.; et al. Analysis of memory B cells identifies conserved neutralizing epitopes on the N-terminal domain of variant SARS-Cov-2 spike proteins. Immunity 2022, 55, 998–1012.e8. [Google Scholar] [CrossRef]
- Zhou, T.; Wang, L.; Misasi, J.; Pegu, A.; Zhang, Y.; Harris, D.R.; Olia, A.S.; Talana, C.A.; Yang, E.S.; Chen, M.; et al. Structural basis for potent antibody neutralization of SARS-CoV-2 variants including B.1.1.529. Science 2022, 376, eabn8897. [Google Scholar] [CrossRef]
- Wang, K.; Jia, Z.; Bao, L.; Wang, L.; Cao, L.; Chi, H.; Hu, Y.; Li, Q.; Zhou, Y.; Zhu, Q.; et al. Memory B cell repertoire from triple vaccinees against diverse SARS-CoV-2 variants. Nature 2022, 603, 919–925. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Zhan, W.; Yang, Z.; Tu, C.; Hu, G.; Zhang, X.; Song, W.; Du, S.; Zhu, Y.; Huang, K.; et al. Broad neutralization of SARS-CoV-2 variants by an inhalable bispecific single-domain antibody. Cell 2022, 185, 1389–1401. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Gao, Y.; Li, T.; Li, T.; Lu, Y.; Zheng, L.; Liu, Y.; Yang, T.; Luo, F.; Song, S.; et al. Structures of Omicron spike complexes and implications for neutralizing antibody development. Cell Rep. 2022, 39, 110770. [Google Scholar] [CrossRef] [PubMed]
- Nutalai, R.; Zhou, D.; Tuekprakhon, A.; Ginn, H.M.; Supasa, P.; Liu, C.; Huo, J.; Mentzer, A.J.; Duyvesteyn, H.M.E.; Dijokaite-Guraliuc, A.; et al. Potent cross-reactive antibodies following Omicron breakthrough in vaccinees. Cell 2022, 185, 2116–2131. [Google Scholar] [CrossRef] [PubMed]
- McCallum, M.; Bassi, J.; Marco, A.D.; Chen, A.; Walls, A.C.; Di’lulio, J.; Tortorici, M.A.; Navarro, M.-J.; Sillaci-Frengi, C.; Saliba, C.; et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science 2021, 373, 648–654. [Google Scholar] [CrossRef]
- Nguyen, A.T.; Szeto, C.; Jayasinghe, D.; Lobos, C.A.; Halim, H.; Chatzileontiadou, D.S.M.; Grant, E.J.; Gras, S. SARS-CoV-2 Spike-Derived peptides presented by HLA molecules. Biophysica 2021, 1, 194–203. [Google Scholar] [CrossRef]
- Wu, D.; Kolesnikov, A.; Yin, R.; Guest, J.D.; Gowathaman, R.; Shmelev, A.; Serdyuk, Y.; Dianov, D.V.; Efimov, G.A.; Pierce, B.G.; et al. Structural assessment of HLA-A2-restricted SARS-CoV-2 spike epitopes recognized by public and private T-cell receptors. Nat. Commun. 2022, 3, 19. [Google Scholar] [CrossRef]
- Cao, L.; Goreshnik, I.; Coventry, B.; Case, J.B.; Miller, L.; Kozodoy, L.; Chen, R.E.; Garter, L.; Walls, A.C.; Park, Y.-J.; et al. De novo design of picomolar SARS-CoV-2 miniprotein inhibitors. Science 2020, 370, 426–431. [Google Scholar] [CrossRef]
- Rosa, A.; Pye, V.E.; Graham, C.; Muir, L.; Seow, J.; Ng, K.W.; Cook, N.J.; Rees-Spear, C.; Parker, E.; Santos, M.S.D.; et al. SARS-CoV-2 can recruit a heme metabolite to evade antibody immunity. Sci. Adv. 2021, 7, eabg7607. [Google Scholar] [CrossRef]
- Ma, C.; Xia, Z.; Sacco, M.D.; Hu, Y.; Townsend, J.A.; Meng, X.; Choza, J.; Tan, H.; Jang, J.; Gongora, M.V.; et al. Discovery of Di- and Trihaloacetamides as covalent SARS-CoV-2 main protease inhibitors with high target specificity. J. Am. Chem. Soc. 2021, 143, 20697–20709. [Google Scholar] [CrossRef]
- Quan, B.X.; Shuai, H.; Xia, A.-J.; Hou, Y.; Zeng, R.; Liu, X.-L.; Lin, G.-F.; Qiao, J.-X.; Li, W.-P.; Wang, F.-L.; et al. An orally available Mpro inhibitor is effective against wild-type SARS-CoV-2 and variants including Omicron. Nat. Microbio. 2022, 7, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Mao, C.; Luan, X.; Shen, D.-D.; Shen, Q.; Su, H.; Wang, X.; Zhou, F.; Zhao, W.; Gao, M.; et al. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by Remdesivir. Science 2020, 368, 1499–1504. [Google Scholar] [CrossRef]
- Peng, Q.; Peng, R.; Yuan, B.; Wang, M.; Zhao, J.; Fu, L.; Qi, J.; Shi, Y. Structural Basis of SARS-CoV-2 Polymerase Inhibition by Favipiravir. Innovation 2021, 2, 100080. [Google Scholar] [CrossRef]
- Neydenova, K.; Muir, K.W.; Wu, L.-F.; Zhang, Z.; Coscia, F.; Peet, M.J.; Castro-Hartmann, P.; Qian, P.; Sader, K.; Dent, K. Structure of the SARS-CoV-2 RNA-dependent RNA polymerase in the presence of favipiravir-RTP. Proc. Natl. Acad. Sci. USA 2021, 118, e2021946118. [Google Scholar] [CrossRef]
- Narayanan, A.; Narwal, M.; Majowicz, S.A.; Varricchio, C.; Toner, S.A.; Ballatore, C.; Brancale, A.; Murakami, K.; Jose, J. Identification of SARS-CoV-2 inhibitors targeting Mpro and PLpro using in-cell-protease assay. Commun. Biol. 2022, 5, 169. [Google Scholar] [CrossRef]
- Rosas-Lemus, M.; Minasov, G.; Shuvalova, L.; Inniss, N.L.; Kiryukhina, O.; Brunzelle, J.; Satchell, K.J.F. High-resolution structures of the SARS-CoV-2 2'-O-methyltransferase reveal strategies for structure-based inhibitor design. Sci. Signal. 2020, 3, 202. [Google Scholar] [CrossRef]
- Ren, P.X.; Shang, W.-J.; Yin, W.-C.; Ge, H.; Wang, L.; Zhang, X.-L.; Li, B.-Q.; Li, H.-L.; Xu, Y.-C.; Xu, E.H.; et al. A multi-targeting drug design strategy for identifying potent anti-SARS-CoV-2 inhibitors. Acta Pharmacol. Sin. 2022, 43, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Wrobel, A.G.; Benton, D.J.; Roustan, C.; Borg, A.; Hussain, S.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.L.; Gamblin, S.J. Evolution of the SARS-CoV-2 spike protein in the human host. Nat. Commun. 2022, 13, 1178. [Google Scholar] [CrossRef] [PubMed]
- Browuer, P.J.M.; Caniels, T.G.; van der Straten, K.; Snitselaar, J.L.; Aldon, Y.; Bangaru, S.; Torres, J.L.; Okba, N.M.A.; Claireaux, M.; Kerster, G.; et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science 2020, 369, 643–650. [Google Scholar] [CrossRef]
- Cao, Y.; Wang, J.; Jian, F.; Xiao, T.; Song, W.; Yisimayi, A.; Huang, W.; Li, Q.; Wang, P.; An, R.; et al. Omicron escapes the majority of existing SARS-CoV-2 neutralizing antibodies. Nature 2022, 602, 657–663. [Google Scholar] [CrossRef]
- Malik, J.A.; Ahmed, S.; Mir, A.; Shinde, M.; Bender, O.; Alshammari, F.; Ansari, M.; Anwar, S. The SARS-CoV-2 mutations versus vaccine effectiveness: New opportunities to new challenges. J. Infect. Public Health 2022, 15, 228–240. [Google Scholar] [CrossRef] [PubMed]
- WHO Solidarity Trial Consortium. Repurposed antiviral drugs for COVID-19—Interim WHO Solidarity Trial Results. N. Engl. J. Med. 2021, 384, 497–511. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Beltran, W.F.; Denis, K.J.S.; Hoelzemer, A.; Lam, E.C.; Nitido, A.D.; Sheehan, M.L.; Berrios, C.; Ofoman, O.; Chang, C.C.; Hauser, B.M.; et al. mRNA-based COVID-19 vaccine boosters induce neutralizing immunity against SARS-CoV-2 Omicron variant. Cell 2022, 185, 457–466. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Year | Documents (%) |
|---|---|
| 2020 | 26 (18.2) |
| 2021 | 75 (52.4) |
| 2022 | 42 (29.4) |
| Parameters | PDB25 | PDB50 | PDB90 |
|---|---|---|---|
| SARS-CoV-2 S protein | 1 (7E9T (10.8, 78%)) | 3 (7N9E (42.9, 96%), 7QO9 (26.2, 73%), 7E9T (10.8, 78%) | 48 (7AD1 (48.8, 97%), 7N9C (48.1, 99%), 7RA8 (48.1, 97%), 7SN3 (47.8, 97%), 7KE8 (47.6), 98%), 7LAA (47.4, 99%), 7TPH (47.3, 96%), 7Q9P 47.2, 96%), 7NS6 (47, 98%), 7C2L (46.6, 98%), 6XKL (46.6, 99%), 7LCN (46.1, 99%), 7TM0 (46, 97%), 7VX7 (45.8, 98%), 7THK (45.3, 98%), 7NDB (45.3, 99%), 7MTC (44.8, 100%), 7MTD (44.4, 99%), 7MTE (44.3, 100%), 7SBT (44.1, 94%), 7N0G (43.9, 98%), 7FCE (43.2, 99%), 7TOU (43.1, 99%), 7VXF (43, 99%), 7CN9 (42.9, 96%), 7N9E (42.8, 96%), 7P7A (42.7, 98%), 7WVO (42.5, 97%), 7TLA (42.5, 99%), 7R15 (42.2, 96%), 7FCD (41.3, 98%), 7WP9 (41.3, 97%), 7SG4 (41.3, 79%), 7TGE (41.2, 97%), 5X5B (40.1, 73%), 7WEV (39.6, 99%), 7N9T (39.5, 96%), 7L2D (38.3, 99%), 6ZGH (37.5, 99%), 7T9J (35.1, 98%), 7SBR (35.1, 98%), 7KRQ (33.9, 99%), 7TL1 (33.6), 96%, 6ZOZ (32.9, 99%), 7QO9 (26.2 73%), 7E7X (22.1, 88%), 7S0E (20.6, 92%), 7E9T (10.8, 78%) |
| S protein of other viruses | 8 | 10 | 22 |
| Other | 37 | 78 | 108 |
| Total | 46 | 91 | 178 |
| Search Strategy | PID | Z-Score, % Identity | Blast % Identity | Spike Protein |
|---|---|---|---|---|
| PDB25 | 7CN8 | 44.8, 94 | 92.25 | Pangolin (PCoV_GX) |
| 6CS2 | 35.7, 78 | 76.51 | SARS-CoV | |
| 6NZK | 33.5, 31 | 35.82 | Coronavirus OC43 | |
| 6NB4 | 32.7, 32 | 34.73 | MERS-CoV | |
| 5I08 | 27.4, 31 | 37.86 | Prefusion human coronavirus HKU1 | |
| 6CV0 | 26.2, 25 | 35.69 | Avian infectious bronchitis coronavirus | |
| 6M15 | 25.8, 26 | 32.69 | Rhinolophus bat coronavirus HKU2 | |
| 6JX7 | 17.8, 20 | 32.86 | Feline infectious peritonitis (FIP) virus | |
| PDB50 | 6NB3 | 34.2, 30 | 35.82 | MERS-CoV |
| 6VV5 | 31.0, 28 | 33.89 | Porcine epidemic diarrhea virus (PEDV) | |
| 6B7N | 30.5, 27 | 31.01 | Prefusion porcine delta coronavirus | |
| 5SZS | 30.1, 28 | 29.86 | Human coronavirus NL63 spike trimer | |
| PDB90 | 7C2L | 46.6, 98 | 99.75 | Duvenhage virus phosphoprotein |
| 6NB7 | 40.7, 77 | 75.63 | SARS-CoV | |
| 7BBH | 39.3, 92 | 89.27 | Smuggled Guangdong pangolin | |
| 6NB6 | 39.2, 33 | 75.63 | SARS-CoV | |
| 7AKJ | 34.5, 79 | 73.15 | SARS-CoV | |
| 5WRG | 32.6, 77 | 74.75 | SARS-CoV | |
| 6VSJ | 30.6, 29 | 36.14 | Mouse coronavirus | |
| 6U7H | 30.3, 27 | 55 | Human coronavirus 229E | |
| 6U7K | 12.5, 12 | 59 | PEDV | |
| 6M3W | 10.9, 71 | 89.82 | SARS-CoV | |
| PDBall | 7CN4(C) | 44, 95 | 94.48 | Bat coronavirus RaTG13 |
| 6CRW(C) | 43.7, 78 | 76.61 | Escherichia virus T4 | |
| 6ZGF(B) | 43, 97 | 95.66 | Bat coronavirus RaTG13 | |
| 6CRX(B) | 43, 76 | 76.61 | Escherichia virus T4 | |
| 6CS1(C) | 42.4, 78 | 76.61 | Escherichia virus T4 | |
| 6CRZ(B) | 42.3, 76 | 76.61 | Escherichia virus T4 | |
| 6CS0(B) | 42.3, 76 | 76.61 | Escherichia virus T4 |
| Variant | PDB IDs | % |
|---|---|---|
| Alpha | 7R0Z, 7R10, 7R13, 7R14, 7R15, 7R1A, 7SXX, 7SXY, 7SY1, 7SY2, 7SXR, 7FEM, 7FET, 7EDF, 7EDG, 7EDH, 7EDI, 7EDJ, 7EH5, 7EKF, 7EAZ, 7EB0, 7EB3, 7EB4, 7EB5, 7LWV, 7LWU, 7LWT, 7LX5, 7LWS, 7DX1, 7DX2, 7KRQ, 7KRR, 7KRS, 6XS6, 7KDI, 7KDH, 7KDL, 7KDJ, 7KDK, 7KDG, 7KEC, 7KEB, 7KEA, 7KE9, 7KE8, 7KE7, 7KE6, 7KE4, 7CM4, 7BNM, 7BNN | 12.44 |
| Beta | 7S5P, 7S5Q, 7S5R, 7R11, 7R16. 7R17, 7WCR, 7WCZ, 7WD0, 7WD7, 7WD8, 7WD9, 7WDF, 7RNJ, 7VX1, 7VX4, 7VXD, 7VXF, 7VXK, 7VXM, 7WEV, 7Q9P, 7Q9M, 7Q9K, 7Q9J, 7Q9I, 7Q9G, 7Q9F, 7Q6E, 7Q0I, 7Q0H, 7Q0G, 7PS7, 7PS6, 7PS5, 7PS4, 7PS2, 7PS1, 7PS0, 7PRZ, 7PRY, 7SXT, 7SXV, 7SXW, 7SY3, 7SY4, 7FJS, 7FJO, 7FJN, 7V8C, 7V7Z, 7V80, 7EKG, 7V2A, 7V26, 7RAQ, 7V76, 7V77, 7E3L, 7E3K, 7LYK, 7LYL, 7LYN, 7LYO, 7LYP, 7LYQ, 7LYM, 7MY3, 7MY2, 7N1Y, 7N1X, 7N1W, 7N1V, 7N1U, 7N1T, 7N1Q, 7RA8, 7RAL, 7LWW, 7E7X, 7E7Y, 7E86, 7E88, 7E8C, 7E8F, 7CHO, 7CHP, 7CHS, 7EY0, 7EY4, 7EY5, 7EYA, 7EZV, 7ZFC, 7ZR7, 7ZR8, 7ZR9, 7ZRC | 24.41 |
| Gamma | 7SXU, 7SY7, 8SY9. 7SBS, 7SBT, 7V79, 7V78, 7V7A, 7V81, 7V82, 7V83, 7V84, 7NXC, 7NX6, 7NX7, 7NX8, 7NX9, 7NXA, 7NXB, 7M8K, 7MJG, 7MJH, 7MJI, 7MJJ, 7MJK, 7MJL, 7MJN, 7MJM, 7MY3, 7MY2, 7EKC | 7.51 |
| Delta | 7TOZ, 7TOY, 7TOX, 7TOV, 7TOU, 7TPL, 7TPH, 7TPF, 7TPE, 7TPC, 7TPA, 7TP9, 7TP8, 7TP7, 7TPF2, 7TP1, 7TP0, 7WBQ, 7SXS, 7SXZ, 7SY0, 7V7N, 7V7O, 7W9I, 7W9F, 7W9E, 7W9C, 7W9B, 7W99, 7W98, 7W97, 7W92, 7V7P, 7V7Q, 7V7R, 7V7S, 7V88, 7V89, 7V8A, 7V8B, 7SBK, 7SBL, 7SBO, 7LQ7, 7Z0Y, 7Z0X, 7X7D, 7E9N, 7F46, 7E9O, 7E9P, 7E9Q, 7ENF, 7ENG, 7TEX, 7TEY, 7TEW, 7V7T, 7V7U, 7V7V, 7OR9, 7ORB, 7ORA, 7WWL, 7WWM | 15.25 |
| Omicron | 7WVQ, 7WVP, 7WVO, 7WVN, 7U0N, 7U0D, 7WUH, 7TGY, 7TGW, 7TGX, 7TGE, 7WP9, 7WPA, 7WPB, 7WPC, 7WPD, 7WPE, 7WPF, 7WRV, 7QO7, 7SY5, 7SY6, 7QNW, 7WBL, 7WBP, 7T9J, 7T9K, 7T9L, 7TB4, 7R40, 7THK, 7TOW, 7TF8, 7TEI, 7TLD, 7TLC, 7TLA, 7TL9, 7TL1, 7THT, 7THE, 7TNW, 7TO4, 7SO9, 7SOA, 7UAP, 7UAQ, 7UAR, 7TM0, 7TLY, 7TN0, 7TLZ, 7WK5, 7WK4, 7WK6, 7WK9, 7WK2, 7WK3, 7WK8, 7WKA, 7WK9, 7QO9, 7UB0, 7UB5, 7UB6, 7RZQ, 7RZR, 7RZS, 7RZT, 7RZU, 7RZV, 7TIK, 7TB8, 7T,C9, 7U,.0D, 7TCA, 7TBF, 7TCC, 7TB4, 7WEF, 7WEE, 7WED, 7WEC, 7WEB, 7WEA, 7WE9, 7WE8, 7WE7, 7WLC, 7WHI, 7WHJ, 7WHK, 7WG6, 7WG7, 7WG8, 7WG9, 7WGB, 7WGC, 7WS9, 7WS8, 7WS8, 7WS7, 7WS6, 7WS5, 7WS4, 7WS3, 7WS2, 7WS1, 7WS0, 7WHH, 7ZF3, 7ZFB, 7ZFD, 7ZFE, 7ZFF, 7ZF4, 7ZF5, 7ZF6, 7ZF7, 7ZF8, 7ZF9, 7ZFA | 28.63 |
| Kappa | 7VX5, 7VX9, 7VXA, 7VXB, 7VXC, 7VXE, 7VXI, 7V7D, 7V7E, 7V7F, 7V7G, 7V7H, 7V7I, 7V7J, 7V85, 7V86, 7V87, 7SBP, 7SBQ, 7SBR, 7SOB, 7SOC, 7SOD, 7SOE, 7SOF, 7TF5, 7TF4, 7TF3, 7TF2, 7TF1, 7TF0, 7TEZ | 7.74 |
| Mink | 7R12, 7R18, 7R19, 7R1B, 7LWQ, 7LWP, 7LWO, 7LWN, 7LWM, 7LWL, 7LWK, 7LWJ, 7LWI | 3 |
| Epsilon | 7N8H, 7N8I | 0.50 |
| Brisdelta | 7ODL, 70D3 | 0.50 |
| Ref | Domain | Antibodies | Variants |
|---|---|---|---|
| [28] | RBD | RBD-chAb15 and RBD-chAb 45 | Alpha |
| [29] | RBD | S5D2, S5G2, S3H3 | Alpha, Beta, Kappa Delta |
| [30] | RBD, NTD | Beta-6, -22, -24, -27, -29, -32, -38, -40, -43, -44, -47, -48, -49, -50, -53, -54, Covox-222, -45 | Beta |
| [31] | RBD | T6 | Wild-type, Beta, Gamma |
| [32] | RBD, NTD | N11, N9, BD-623, -508, -515, | Beta |
| [33] | RBD | P5A-1D2, P5A-3C8, P22A-1D1 | Beta |
| [34] | RBD | BD-813, -744, -667, -771 -821, -804, -812, -836 | Beta |
| [35] | RBD | 222 | Alpha, Beta, Gamma |
| [36] | RBD | 8D3 | Delta |
| [37] | RBD | 222, 278, 45, 253 75, | Kappa and Delta |
| [38] | RBD | ZWD12, ZWC6 | Alpha, Beta, Gamma, Kappa, Delta, Omicron |
| [39] | RBD | JMB2002 | Omicron, Alpha, Beta, Gamma |
| [40] | NTD | C1520, C1791 | Omicron |
| [40] | RBD | LY-CoV1404, S309, A19-46.1, S2E12, COV2-2196, B1-182.1, A23-58.1 | Omicron |
| [41] | RBD | XGv265, -282, -289, -347, | VOC particularly Omicron |
| [42] | RBD | BN03 | Omicron |
| [43] | RBD | Vir-S309 | Beta and Omicron |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aslam, M.; Nawaz, M.S.; Fournier-Viger, P.; Li, W. Comparative Analysis and Classification of SARS-CoV-2 Spike Protein Structures in PDB. COVID 2023, 3, 452-471. https://doi.org/10.3390/covid3040034
Aslam M, Nawaz MS, Fournier-Viger P, Li W. Comparative Analysis and Classification of SARS-CoV-2 Spike Protein Structures in PDB. COVID. 2023; 3(4):452-471. https://doi.org/10.3390/covid3040034
Chicago/Turabian StyleAslam, Memoona, M. Saqib Nawaz, Philippe Fournier-Viger, and Wenjin Li. 2023. "Comparative Analysis and Classification of SARS-CoV-2 Spike Protein Structures in PDB" COVID 3, no. 4: 452-471. https://doi.org/10.3390/covid3040034