
The Relevance of Cavity Creation for Several Phenomena Occurring in Water


Dipartimento di Scienze e Tecnologie, Università degli Studi del Sannio, Via Francesco de Sanctis, Snc, 82100 Benevento, Italy
Liquids 2023, 3(1), 57-65; https://doi.org/10.3390/liquids3010006
Submission received: 14 December 2022
/
Revised: 30 December 2022
/
Accepted: 1 January 2023
/
Published: 9 January 2023
(This article belongs to the Special Issue Modeling of Liquids Behavior: Experiments, Theory and Simulations)
Abstract
:The solvent-excluded volume effect is an under-appreciated general phenomenon occurring in liquids and playing a fundamental role in many cases. It is quantified and characterized by means of the theoretical concept of cavity creation and its Gibbs free energy cost. The magnitude of the reversible work of cavity creation proves to be particularly large in water, and this fact plays a key role for, among other things, the poor solubility of nonpolar species, the formation of host–guest complexes, and the folding of globular proteins. An analysis of some examples is provided in the present review.
1. Introduction
The starting point of any theory able to describe processes occurring in liquids (i.e., pure liquids or solutions) is the recognition that a suitable void space, a cavity, has to be created to allow solute insertion [1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18,19,20,21]. This is the simple consequence of the fact that liquids are a condensed state of matter and each molecule possess its own body. Cavity creation leads to a decrease in the number of configurations accessible to liquid molecules and thus leads to a solvent-excluded volume effect [22,23,24,25]. These words are right, but the matter has to be spelled out in more detail to reach a correct understanding (note that cavity creation is a theoretical concept and cannot be studied by performing experiments). Keeping a constant temperature and pressure, the creation of a cavity leads to an increase in liquid volume by the partial molar volume of the cavity itself. This volume increase does not cancel the solvent-excluded volume effect. If the cavity is to exist, the center of liquid molecules (assumed to be spherical) cannot go beyond the solvent-accessible surface area, SASA, and WASA in water [26], of the cavity itself. This means that the shell between the cavity van der Waals surface and the SASA is excluded to liquid molecules, causing a decrease in accessible configurations for basic geometric reasons. The latter constraint does affect the translational motion of all the liquid molecules, not solely the ones in the first solvation shell of the cavity (i.e., of the solute molecule to be hosted).
The solvent-excluded volume effect can be measured by calculating the reversible work of cavity creation, ΔGC, by means of analytical theories or computer simulations. Classic scaled particle theory, SPT [23,24,25,27,28,29], is a simple, geometry-based statistical mechanical model providing analytical formulas to calculate ΔGC for cavities of simple shape (i.e., a sphere, a prolate spherocylinder, and others) in liquids made up of hard particles. Its use in the case of water may appear strange, but it works well because the real liquid density is used as input in classic SPT calculations (i.e., density provides indirect information on the strength of the intermolecular attractions existing among liquid molecules; in addition, on the H-bonds between water molecules [30,31]). The classic SPT formulas to create a spherical cavity in a liquid (neglecting the pressure–volume term for its smallness at P = 1 atm) are:
where R is the gas constant, αP is the isobaric thermal expansion coefficient of the liquid, ξ is the volume packing density of the liquid, which is defined as the ratio of the physical volume of a mole of liquid molecules over the liquid molar volume, v1 (i.e., ξ = π · σ13 · NAv/6 · v1); x = σC/σ1, and σ1 is the hard sphere diameter of liquid molecules; σC is the cavity diameter, defined as the diameter of the spherical region from which any part of liquid molecules is excluded. The ∆GC(SPT) magnitude depends upon the volume packing density of the liquid, ξ, that is, the fraction of the total liquid volume really occupied by liquid molecules, and the effective hard sphere diameter, σ1, of liquid molecules [25,31]. On increasing ξ, the void volume decreases and ∆GC increases; on decreasing σ1, the void volume is partitioned into smaller pieces and ∆GC increases (i.e., a significant fraction of the liquid volume is void, but most of these voids are too small to host an atom or a molecule). This implies that the effective diameter of liquid molecules is a fundamental length-scale for the liquid itself. The validity of these arguments has been verified and confirmed in several cases over the years [31].
ΔGC = RT · {−ln(1 − ξ) + [3ξ/(1 − ξ)] · x + [3ξ/(1 − ξ)] · x2 + [9ξ2/2(1 − ξ)2] · x2}
ΔHC = [RT2⋅ξ⋅αP/(1 − ξ)3]⋅[(1 − ξ)2 + 3(1 − ξ)⋅x + 3(1 + 2ξ)⋅x2]
2. Solvation of Noble Gases
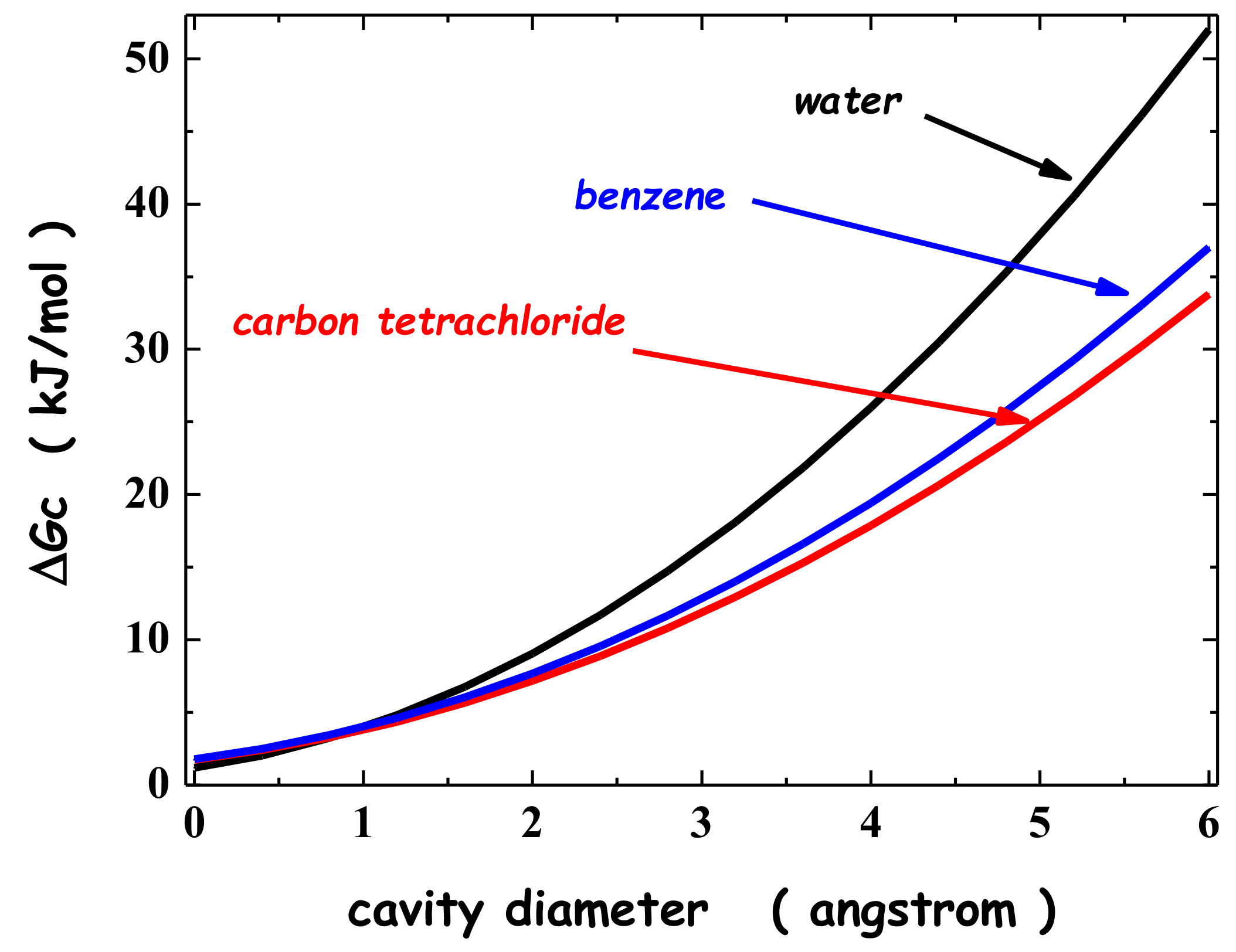
A further test is provided here, by analyzing the solvation (i.e., the transfer from a fixed position in the gas phase to a fixed position in the liquid phase) of noble gases in water, carbon tetrachloride, CCl4, and benzene, C6H6. Experimental thermodynamic data at 25 °C and 1 atm [32,33,34], reported in Table 1, emphasize that: (1) noble gases are poorly soluble in water, with them being characterized by large positive ΔG∙ values, caused by large negative entropy changes; (2) Ar is characterized by positive ΔG∙ values also in CCl4 and C6H6, while Xe is characterized by a negative ΔG∙ value in benzene. The ΔGC(SPT) values calculated for noble gases in the three liquids, whose molecules are assumed to be spherical, at 25 °C and 1 atm, are listed in the eighth column of Table 1. They prove to be largely positive in all cases. Actually, they are significantly larger in water with respect to the other two liquids (see the trends reported in Figure 1).
This holds because, even though the volume packing density of water is the smallest, ξ = 0.383 for water versus 0.503 for CCl4, and 0.513 for C6H6, the water molecules are the smallest, σ = 2.80 Å for water, 5.37 Å for CCl4, and 5.26 Å for C6H6 [35,36,37]. In this respect, it is important to underscore that the effective hard sphere diameter assigned to water molecules is physically reliable because it corresponds to the location of the first peak in the oxygen–oxygen radial distribution function of water [38], the distance between two H-bonded water molecules. The size effect prevails because it is the molecular cause of the markedly larger number density of water: at 25 °C and 1 atm, ρ(in moles per liter) = 55.3 for water, 10.3 for CCl4, and 11.2 for benzene [37]. The magnitude of the solvent-excluded volume effect associated with cavity creation depends strongly upon the liquid number density: the entropy loss is larger with the greater the number of affected molecules. This is why the size is so important. The reliability of using classic SPT for water has been further confirmed recently by the agreement between the ΔGC(SPT) values and those obtained by computer simulations in detailed water models [39].
Moreover, a simple formula devised by Pierotti [29] allows for the calculation of the interaction energy Ea between the noble gases and the three liquids. The Pierotti’s formula is:
where σ12 = (σ1 + σ2)/2 and ε12 = (ε1ε2)1/2, and where ε1 and ε2 are the Lennard–Jones parameters for the liquid and solute, respectively. The Ea estimates, listed in the ninth column of Table 1, are negative and, when added to the ΔGC(SPT) values, produce numbers that are close to the experimental ΔG∙ ones for all of the three liquids. The success is mainly because the solvent-excluded volume effect associated with solute insertion in a liquid is correctly accounted for by calculating the reversible work of cavity creation.
Ea = −(64/3) · ξ · ε12 · (σ12/σ1)3
Estimates of the enthalpy change associated with cavity creation, ΔHC(SPT), calculated by means of Equation (2) and listed in the third column of Table 2, are positive in all of the three liquids and are close to the values of the ΔH∙ − Ea difference, listed in the fourth column of Table 2. This suggests that the structural reorganization of liquid molecules upon noble gas insertion is an endothermic process at 25 °C and 1 atm (i.e., in the case of water, there is no indication of iceberg formation [40,41,42,43]). Actually, the ΔHC(SPT) values of water are significantly smaller than those of the other two liquids [37]; this is a consequence of the smaller isobaric thermal expansion coefficient αP of water with respect to those of the other two liquids [37] (look at the values reported in the notes of Table 1). The latter quantity, present in the classic SPT formula of ΔHC [29], is a measure of the ensemble correlation between fluctuations in volume and fluctuations in enthalpy, and so it can account for the liquid structural reorganization upon cavity creation. The smallness of the αP of water is due to the strength of water–water H-bonds, in comparison to the weakness of van-der-Waals-type interactions occurring among benzene and carbon tetrachloride molecules [37]. Therefore, the ΔHC(SPT) values indicate that cavity creation does not cause the breakage of water–water H-bonds [30,31], but a significant breakage of van der Waals interactions occurs in the other two liquids [37].
Estimates of the entropy change associated with cavity creation, ΔSC(SPT), listed in the fifth column of Table 2, are close to the total solvation entropy changes, listed in the last column of Table 2, in all of the three considered liquids. This finding indicates that the process of cavity creation is the main process responsible of the negative solvation entropy change [31,37]. In water, the ΔSC(SPT) values are largely negative, increasing in magnitude with the solute diameter [37]. This entropy loss cannot be due to an increase in water structural order [44,45], because it comes from a hard sphere approach. It is due to the decrease in the number of accessible configurations for water molecules because of cavity creation (i.e., the solvent-excluded volume effect). Such a decrease in the number of accessible configurations also occurs in the other two liquids, but it is masked by a largely positive entropy change due to the structural reorganization upon cavity creation [25,37]. The latter structural reorganization, however, has a markedly different magnitude in water and the two organic liquids; it is also characterized by a complete enthalpy-entropy compensation in all liquids [31,46] and does not affect the ΔGC magnitude.
3. Formation of Host–Guest Complexes
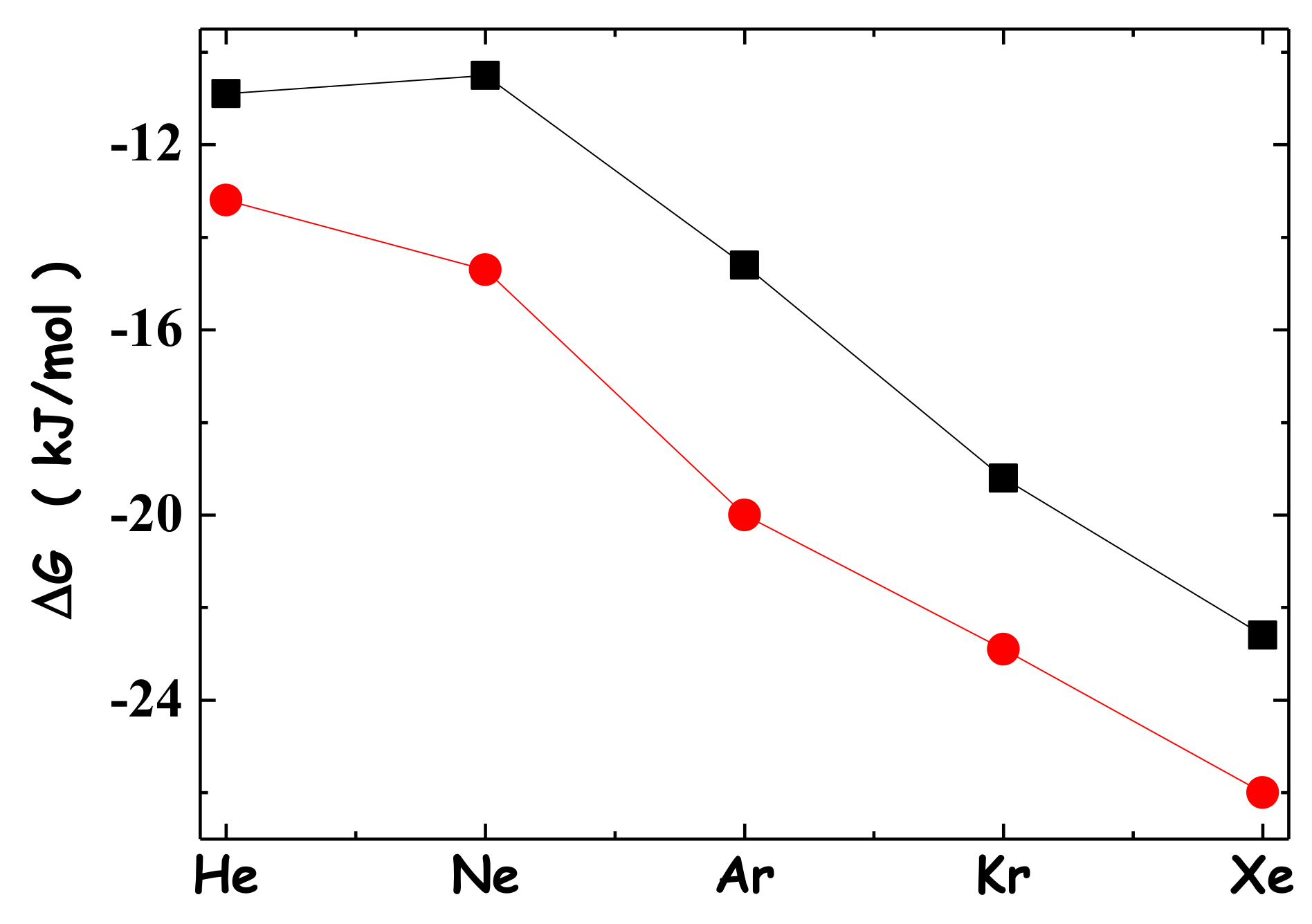
It is interesting that noble gases are able to bind macrocyclic hosts in aqueous solutions. In particular, thanks to specialized NMR experiments, it has been possible to measure the binding constants of noble gases to cucurbit[5]uril, a rigid, synthetic, and water-soluble macrocyclic host [47]. Specifically, at 22 °C, Kb(in M−1) = 87 for He, 72 for Ne, 360 for Ar, 2390 for Kr and 8700 for Xe. These numbers imply that the binding process is spontaneous, and thus, there is the need to identify the driving force of this host–guest recognition [47,48,49]. It is important to underscore that the inner part of cucurbit[5]uril proved not to be filled by water molecules, on the basis of both specialized NMR measurements and MD simulations [47] (note that the inner part of cucurbit[5]uril has a volume of 68 Å3 and can host very few water molecules, considering that the van der Waals volume of a water molecule is 11.5 Å3). Researchers calculated with great accuracy, at DFT level, the dispersion energetic attractions of noble gases in bulk water and in the inner part of cucurbit[5]uril. The unexpected result was that the magnitude of attractive dispersion interactions was larger in bulk water than in the inner part of the rigid macrocyclic host [47]. As a consequence, researchers turned their attention to the reversible work of cavity creation. The transfer of a noble gas atom from water to the inner part of cucurbit[5]uril implies the following steps: the switching-off of the energetic dispersion attractions with water, the closure of the cavity in water, the creation of the cavity in the macrocyclic host interior, and the switching-on of the energetic dispersion attractions with the host.
However, the reversible work to create a cavity in the inner part of cucurbit[5]uril is zero because this region does not contain water molecules (i.e., it is empty); in addition, as a first approximation, the magnitude of the energetic dispersion attractions of a noble gas atom in bulk water and in the host interior can be assumed to be equal. This implies ΔG(binding) ≈ −ΔGC(H2O), and the driving force is given by the decrease in solvent-excluded volume for cavity closure in water (i.e., leading to a gain in configurational–translational entropy of water molecules).
The experimental ΔG(binding) values of noble gases to cucurbit[5]uril are reported in Figure 2, together with the values of minus ΔGC(H2O), calculated via classic SPT formulas and listed in Table 1. One could say that the agreement between the two sets of numbers is better than expected, considering their totally different origin. In the original article, the authors calculated ΔGC(H2O) by means of computer simulations, they also considered the contribution of the difference in energetic dispersion attractions between the bulk water and the host interior, and obtained a good agreement with the experimental data [47]. This example demonstrates the pivotal role played by the reversible work of cavity creation in driving host–guest recognition phenomena in water [50,51,52].
4. Conformational Stability of Globular Proteins
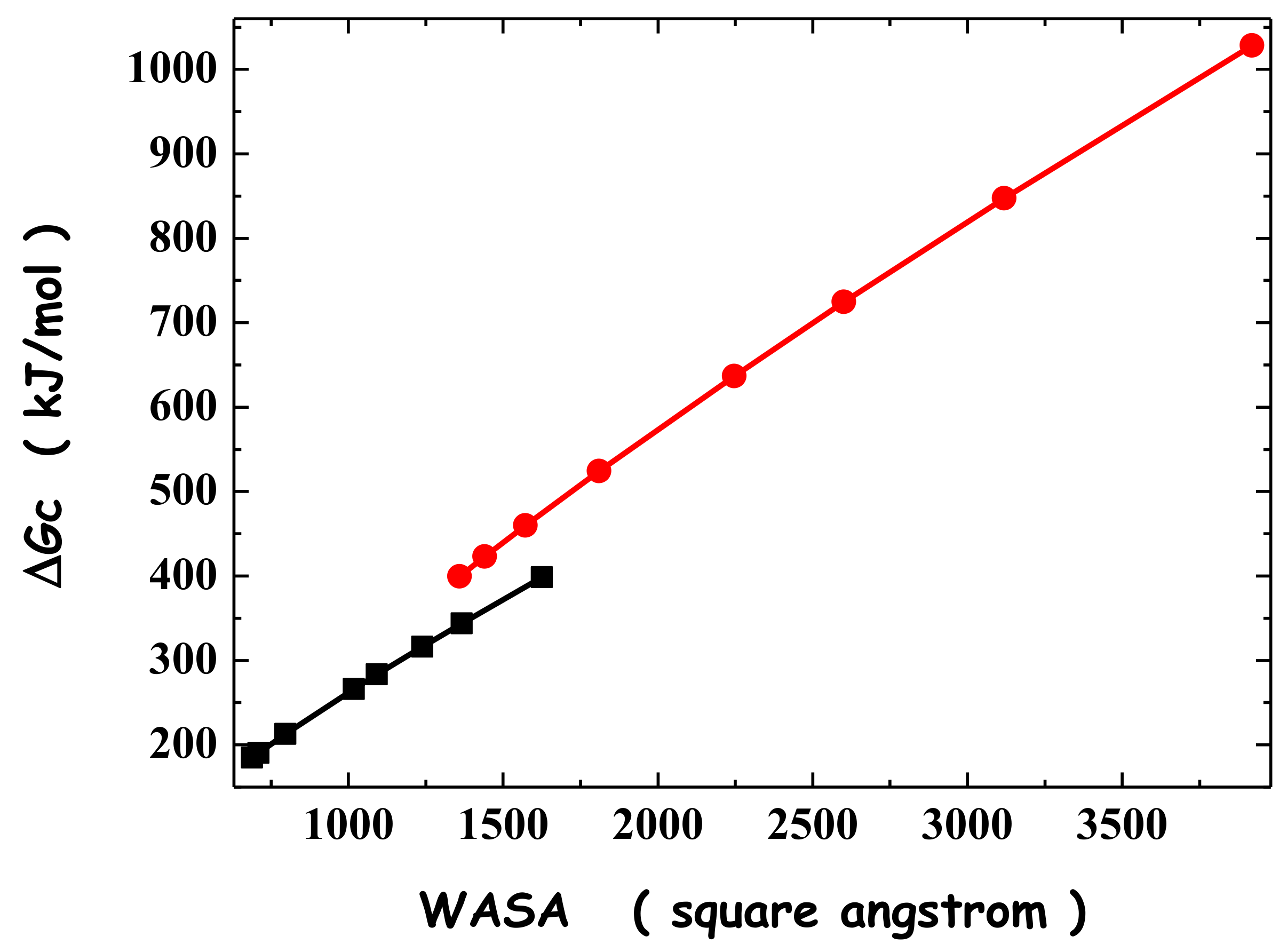
The geometric explanation for the solvent-excluded volume effect implies that the ΔGC magnitude has to increase if the cavity shape is changed, by keeping its van der Waals volume fixed, VvdW, and increasing its WASA. This is a fundamental point. Passing from a spherical cavity to several prolate spherocylinders with the same VvdW of the sphere, it is possible to test the rightness of the geometric arguments. Classic SPT analytical formulas allow for the calculation of ΔGC for both spherical and prolate spherocylindrical cavities. Therefore, the test can readily be completed and the results have confirmed that ΔGC increases with cavity WASA, even though VvdW is fixed [24,53]. This holds true also with ΔGC calculated by means of computer simulations [54,55]. The results of classic SPT calculations in water, at 25 °C and 1 atm, for two sets of cavities, the first starting with a sphere of 6 Å radius and the second starting with a sphere of 9 Å radius, are listed in Table 3. It is evident that on lengthening the prolate spherocylinder, WASA increases and ΔGC also increases. The plot of ΔGC versus WASA, constructed with the numbers of Table 3, is shown in Figure 3. The ΔGC values scale linearly with cavity WASA, but the line slope is not unique; the slope magnitude depends upon the VvdW of the cavity. In fact, the largest spherocylinder of the first set has a WASA larger than that of the smallest spherocylinder of the second set, but the order is reversed in the case of ΔGC values (see Table 3 and Figure 3). This means also that the cavity VvdW plays a role [53,56,57,58].
Anyway, the trend of ΔGC versus cavity WASA is important to shed light on the driving force of protein folding and on the main factor responsible for the conformational stability of globular proteins. Experimental measurements have proved that the difference in molecular volume between the folded state and the unfolded state ensemble is negligibly small [59,60]. Thus, the folding process can be viewed as a collapse from a set of elongated conformations toward a compact, almost spherical one, keeping the volume occupied by the polypeptide chain fixed [24,25]. Such a collapse is characterized by a large WASA decrease; that means a large ΔGC decrease, which corresponds to a significant gain in the configurational–translational entropy of water molecules. The numbers listed in the last column of Table 3 indicate that a large negative Gibbs free energy change is associated, at 25 °C and 1 atm, with the collapse from the longest spherocylinder to the sphere. Polypeptide chains are flexible and can populate different conformations, producing markedly different solvent-excluded volume effects. Water molecules push these chains to assume compact conformations in order to gain configuration–translational entropy. This is the geometric-molecular basis of what is called the hydrophobic effect, considered to be the main determinant of the conformational stability of globular proteins [24,25].
5. Conclusions
In the present article, I have tried to show that the solvent-excluded volume effect associated with cavity creation in all liquids (that are a condensed state of the matter) allows one to devise a common and general theoretical approach to rationalize several disparate phenomena occurring in liquids. In particular, the ΔGC(SPT) values are able to rationalize the low solubility of noble gases in water and its entropic origin, the driving force of the recognition between noble gases and cucurbit[5]uril in water, and, last but not least, a reliable driving force for protein folding and stability.
Funding
This research was funded by Università degli Studi del Sannio, FRA 2022.
Conflicts of Interest
The author declares no conflict of interest.
References
- Ben-Naim, A. Water and Aqueous Solutions; Springer US: Boston, MA, USA, 1974. [Google Scholar]
- Pratt, L.R.; Chandler, D. Theory of the hydrophobic effect. J. Chem. Phys. 1977, 67, 3683–3704. [Google Scholar] [CrossRef]
- Lee, B. The physical origin of the low solubility of nonpolar solutes in water. Biopolymers 1985, 24, 813–823. [Google Scholar] [CrossRef]
- Ben-Naim, A. Solvation Thermodynamics; Plenum Press: New York, NY, USA, 1987. [Google Scholar]
- Yu, H.; Karplus, M. A thermodynamic analysis of solvation. J. Chem. Phys. 1988, 89, 2366–2379. [Google Scholar] [CrossRef]
- Pohorille, A.; Pratt, L.R. Cavities in molecular liquids and the theory of hydrophobic solubilities. J. Am. Chem. Soc. 1990, 112, 5066–5074. [Google Scholar] [CrossRef]
- Blokzijl, W.; Engberts, J.B.F.N. Hydrophobic Effects. Opinions and Facts. Angew. Chem. Int. Ed. 1993, 32, 1545–1579. [Google Scholar] [CrossRef]
- Guillot, B.; Guissani, Y. A computer simulation study of the temperature dependence of the hydrophobic hydration. J. Chem. Phys. 1993, 99, 8075–8094. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular Interactions in Solution: An Overview of Methods Based on Continuous Distributions of the Solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Beutler, T.C.; Béguelin, D.R.; van Gunsteren, W.F. Free energy of cavity formation in solvent: Computational, methodological, and physical aspects. J. Chem. Phys. 1995, 102, 3787–3793. [Google Scholar] [CrossRef]
- Hummer, G.; Garde, S.; García, A.E.; Paulaitis, M.E.; Pratt, L.R. Hydrophobic Effects on a Molecular Scale. J. Phys. Chem. B 1998, 102, 10469–10482. [Google Scholar] [CrossRef] [Green Version]
- Lum, K.; Chandler, D.; Weeks, J.D. Hydrophobicity at Small and Large Length Scales. J. Phys. Chem. B 1999, 103, 4570–4577. [Google Scholar] [CrossRef]
- Lazaridis, T. Solvent Size vs Cohesive Energy as the Origin of Hydrophobicity. Acc. Chem. Res. 2001, 34, 931–937. [Google Scholar] [CrossRef]
- Southall, N.T.; Dill, K.A.; Haymet, A.D.J. A View of the Hydrophobic Effect. J. Phys. Chem. B 2002, 106, 521–533. [Google Scholar] [CrossRef]
- Pratt, L.R.; Pohorille, A. Hydrophobic Effects and Modeling of Biophysical Aqueous Solution Interfaces. Chem. Rev. 2002, 102, 2671–2692. [Google Scholar] [CrossRef]
- Benzi, C.; Cossi, M.; Improta, R.; Barone, V. Building cavities in a fluid of spherical or rod-like particles: A contribution to the solvation free energy in isotropic and anisotropic polarizable continuum model. J. Comput. Chem. 2005, 26, 1096–1105. [Google Scholar] [CrossRef]
- Chandler, D. Interfaces and the driving force of hydrophobic assembly. Nature 2005, 437, 640–647. [Google Scholar] [CrossRef]
- Ashbaugh, H.S.; Pratt, L.R. Contrasting Nonaqueous against Aqueous Solvation on the Basis of Scaled-Particle Theory. J. Phys. Chem. B 2007, 111, 9330–9336. [Google Scholar] [CrossRef]
- Ben-Amotz, D.; Underwood, R. Unraveling Water’s Entropic Mysteries: A Unified View of Nonpolar, Polar, and Ionic Hydration. Acc. Chem. Res. 2008, 41, 957–967. [Google Scholar] [CrossRef]
- Otto, S. The role of solvent cohesion in nonpolar solvation. Chem. Sci. 2013, 4, 2953–2959. [Google Scholar] [CrossRef] [Green Version]
- Ben-Amotz, D. Water-Mediated Hydrophobic Interactions. Annu. Rev. Phys. Chem. 2016, 67, 617–638. [Google Scholar] [CrossRef]
- Soda, K. Solvent Exclusion Effect Predicted by the Scaled Particle Theory as an Important Factor of the Hydrophobic Effect. J. Phys. Soc. Jpn. 1993, 62, 1782–1793. [Google Scholar] [CrossRef]
- Tang, K.E.; Bloomfield, V.A. Excluded Volume in Solvation: Sensitivity of Scaled-Particle Theory to Solvent Size and Density. Biophys. J. 2000, 79, 2222–2234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merlino, A.; Pontillo, N.; Graziano, G. A driving force for polypeptide and protein collapse. Phys. Chem. Chem. Phys. 2017, 19, 751–756. [Google Scholar] [CrossRef]
- Graziano, G. On the mechanism of cold denaturation. Phys. Chem. Chem. Phys. 2014, 16, 21755–21767. [Google Scholar] [CrossRef]
- Lee, B.; Richards, F. The interpretation of protein structures: Estimation of static accessibility. J. Mol. Biol. 1971, 55, 379-IN4. [Google Scholar] [CrossRef]
- Reiss, H. Scaled Particle Methods in the Statistical Thermodynamics of Fluids. In Advances in Chemical Physics; Prigogine, I., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2007; pp. 1–84. [Google Scholar]
- Lebowitz, J.L.; Helfand, E.; Praestgaard, E. Scaled Particle Theory of Fluid Mixtures. J. Chem. Phys. 1965, 43, 774–779. [Google Scholar] [CrossRef]
- Pierotti, R.A. A scaled particle theory of aqueous and nonaqueous solutions. Chem. Rev. 1976, 76, 717–726. [Google Scholar] [CrossRef]
- Gallicchio, E.; Kubo, M.M.; Levy, R.M. Enthalpy−Entropy and Cavity Decomposition of Alkane Hydration Free Energies: Numerical Results and Implications for Theories of Hydrophobic Solvation. J. Phys. Chem. B 2000, 104, 6271–6285. [Google Scholar] [CrossRef]
- Graziano, G. Contrasting the hydration thermodynamics of methane and methanol. Phys. Chem. Chem. Phys. 2019, 21, 21418–21430. [Google Scholar] [CrossRef]
- Wilhelm, E.; Battino, R. Thermodynamic functions of the solubilities of gases in liquids at 25. deg. Chem. Rev. 1973, 73, 1–9. [Google Scholar] [CrossRef]
- Krause, D.; Benson, B.B. The solubility and isotopic fractionation of gases in dilute aqueous solution. IIa. solubilities of the noble gases. J. Solut. Chem. 1989, 18, 823–873. [Google Scholar] [CrossRef]
- Graziano, G. On the temperature dependence of hydration thermodynamics for noble gases. Phys. Chem. Chem. Phys. 1999, 1, 1877–1886. [Google Scholar] [CrossRef]
- Wilhelm, E.; Battino, R. Estimation of Lennard-Jones (6,12) Pair Potential Parameters from Gas Solubility Data. J. Chem. Phys. 1971, 55, 4012–4017. [Google Scholar] [CrossRef]
- Graziano, G. Salting out of methane by sodium chloride: A scaled particle theory study. J. Chem. Phys. 2008, 129, 084506. [Google Scholar] [CrossRef] [PubMed]
- Graziano, G. Scaled Particle Theory Study of the Length Scale Dependence of Cavity Thermodynamics in Different Liquids. J. Phys. Chem. B 2006, 110, 11421–11426. [Google Scholar] [CrossRef] [PubMed]
- Head-Gordon, T.; Hura, G. Water Structure from Scattering Experiments and Simulation. Chem. Rev. 2002, 102, 2651–2670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sedov, I.; Magsumov, T. The Gibbs free energy of cavity formation in a diverse set of solvents. J. Chem. Phys. 2020, 153, 134501. [Google Scholar] [CrossRef]
- Buchanan, P.; Aldiwan, N.; Soper, A.; Creek, J.; Koh, C. Decreased structure on dissolving methane in water. Chem. Phys. Lett. 2005, 415, 89–93. [Google Scholar] [CrossRef]
- Graziano, G.; Lee, B. On the Intactness of Hydrogen Bonds around Nonpolar Solutes Dissolved in Water. J. Phys. Chem. B 2005, 109, 8103–8107. [Google Scholar] [CrossRef]
- Bowron, D.T.; Filipponi, A.; Lobban, C.; Finney, J.L. Temperature-induced disordering of the hydrophobic hydration shell of Kr and Xe. Chem. Phys. Lett. 1998, 293, 33–37. [Google Scholar] [CrossRef]
- Kim, J.; Tian, Y.; Wu, J. Thermodynamic and Structural Evidence for Reduced Hydrogen Bonding among Water Molecules near Small Hydrophobic Solutes. J. Phys. Chem. B 2015, 119, 12108–12116. [Google Scholar] [CrossRef]
- Irudayam, S.J.; Henchman, R. Solvation theory to provide a molecular interpretation of the hydrophobic entropy loss of noble-gas hydration. J. Phys. Condens. Matter 2010, 22, 284108. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Besford, Q.A.; Mulvaney, T.; Gray-Weale, A. Order and correlation contributions to the entropy of hydrophobic solvation. J. Chem. Phys. 2015, 142, 114117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B. A procedure for calculating thermodynamic functions of cavity formation from the pure solvent simulation data. J. Chem. Phys. 1985, 83, 2421–2425. [Google Scholar] [CrossRef]
- He, S.; Biedermann, F.; Vankova, N.; Zhechkov, L.; Heine, T.; Hoffman, R.E.; De Simone, A.; Duignan, T.T.; Nau, W.M. Cavitation energies can outperform dispersion interactions. Nat. Chem. 2018, 10, 1252–1257. [Google Scholar] [CrossRef]
- Biedermann, F.; Uzunova, V.D.; Scherman, O.A.; Nau, W.M.; De Simone, A. Release of High-Energy Water as an Essential Driving Force for the High-Affinity Binding of Cucurbit[n]urils. J. Am. Chem. Soc. 2012, 134, 15318–15323. [Google Scholar] [CrossRef] [PubMed]
- Biedermann, F.; Nau, W.; Schneider, H.-J. The Hydrophobic Effect Revisited-Studies with Supramolecular Complexes Imply High-Energy Water as a Noncovalent Driving Force. Angew. Chem. Int. Ed. 2014, 53, 11158–11171. [Google Scholar] [CrossRef]
- Setny, P.; Baron, R.; McCammon, J.A. How Can Hydrophobic Association Be Enthalpy Driven? J. Chem. Theory Comput. 2010, 6, 2866–2871. [Google Scholar] [CrossRef]
- Dzubiella, J. How Interface Geometry Dictates Water’s Thermodynamic Signature in Hydrophobic Association. J. Stat. Phys. 2011, 145, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Graziano, G. Molecular driving forces of the pocket–ligand hydrophobic association. Chem. Phys. Lett. 2012, 533, 95–99. [Google Scholar] [CrossRef]
- Graziano, G. The Gibbs energy cost of cavity creation depends on geometry. J. Mol. Liq. 2015, 211, 1047–1051. [Google Scholar] [CrossRef]
- Wallqvist, A.; Berne, B.J. Molecular Dynamics Study of the Dependence of Water Solvation Free Energy on Solute Curvature and Surface Area. J. Phys. Chem. 1995, 99, 2885–2892. [Google Scholar] [CrossRef] [Green Version]
- Patel, A.; Varilly, P.; Chandler, D. Fluctuations of Water near Extended Hydrophobic and Hydrophilic Surfaces. J. Phys. Chem. B 2010, 114, 1632–1637. [Google Scholar] [CrossRef] [Green Version]
- Sosso, G.C.; Caravati, S.; Rotskoff, G.; Vaikuntanathan, S.; Hassanali, A. On the Role of Nonspherical Cavities in Short Length-Scale Density Fluctuations in Water. J. Phys. Chem. A 2016, 121, 370–380. [Google Scholar] [CrossRef] [Green Version]
- Ansari, N.; Laio, A.; Hassanali, A.A. Spontaneously Forming Dendritic Voids in Liquid Water Can Host Small Polymers. J. Phys. Chem. Lett. 2019, 10, 5585–5591. [Google Scholar] [CrossRef]
- Azizi, K.; Laio, A.; Hassanali, A. Model Folded Hydrophobic Polymers Reside in Highly Branched Voids. J. Phys. Chem. Lett. 2022, 13, 183–189. [Google Scholar] [CrossRef]
- Royer, C.A. Revisiting volume changes in pressure-induced protein unfolding. Biochim. et Biophys. Acta (BBA)—Protein Struct. Mol. Enzym. 2002, 1595, 201–209. [Google Scholar] [CrossRef]
- Chalikian, T.V. Volumetric Properties of Proteins. Annu. Rev. Biophys. Biomol. Struct. 2003, 32, 207–235. [Google Scholar] [CrossRef]
Figure 1.
Trend of ΔGC versus the cavity diameter for water, CCl4, and C6H6 calculated by means of classic SPT, at 25 °C and 1 atm. The data necessary to perform the calculations are reported in the notes of Table 1.
Figure 1.
Trend of ΔGC versus the cavity diameter for water, CCl4, and C6H6 calculated by means of classic SPT, at 25 °C and 1 atm. The data necessary to perform the calculations are reported in the notes of Table 1.

Figure 2.
Experimental ΔG(binding) values of noble gases to cucurbit[5]uril, measured via NMR at 22 °C (black filled squares) [47], contrasted with minus the ΔGC values for noble gases in water, calculated via classic SPT analytical formulas, and listed in column eight of Table 1 (red filled circles).
Figure 2.
Experimental ΔG(binding) values of noble gases to cucurbit[5]uril, measured via NMR at 22 °C (black filled squares) [47], contrasted with minus the ΔGC values for noble gases in water, calculated via classic SPT analytical formulas, and listed in column eight of Table 1 (red filled circles).

Figure 3.
Plot of ΔGC versus WASA for the two sets of cavities listed in Table 3 (in each set, all of the cavities have the same van der Waals volume). The two lines simply connect the points; they are not the result of a linear regression.
Figure 3.
Plot of ΔGC versus WASA for the two sets of cavities listed in Table 3 (in each set, all of the cavities have the same van der Waals volume). The two lines simply connect the points; they are not the result of a linear regression.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Experimental thermodynamic data for the solvation [32,33,34], according to the Ben–Naim standard (i.e., the transfer from a fixed position in the gas phase to a fixed position in the liquid phase), at 25 °C and 1 atm, of noble gases in water (a), CCl4 (b), and C6H6 (c); the values of the hard sphere diameter and the Lennard–Jones energy parameter come from [35,36] with small modifications; the values of ΔGC are calculated by means of classic SPT analytical formulas [27,28]; those of Ea are calculated by means of Pierotti’s analytical formula [29].
Table 1.
Experimental thermodynamic data for the solvation [32,33,34], according to the Ben–Naim standard (i.e., the transfer from a fixed position in the gas phase to a fixed position in the liquid phase), at 25 °C and 1 atm, of noble gases in water (a), CCl4 (b), and C6H6 (c); the values of the hard sphere diameter and the Lennard–Jones energy parameter come from [35,36] with small modifications; the values of ΔGC are calculated by means of classic SPT analytical formulas [27,28]; those of Ea are calculated by means of Pierotti’s analytical formula [29].
| σ Å | ε/k K | ΔH∙ kJ mol−1 | ΔS∙ J K−1mol−1 | ΔG∙ kJ mol−1 | ΔGC kJ mol−1 | Ea kJ mol−1 | ΔGC + Ea kJ mol−1 | ||
|---|---|---|---|---|---|---|---|---|---|
| a | He | 2.6 | 6 | 1.8 | −32.5 | 11.5 | 13.2 | −1.6 | 11.6 |
| Ne | 2.8 | 28 | −1.3 | −41.9 | 11.2 | 14.7 | −3.9 | 10.8 | |
| Ar | 3.4 | 125 | −9.6 | −60.4 | 8.4 | 20.0 | −11.3 | 8.7 | |
| Kr | 3.7 | 175 | −13.0 | −66.7 | 6.9 | 22.9 | −15.4 | 7.5 | |
| Xe | 4.0 | 230 | −16.8 | −74.8 | 5.5 | 26.0 | −20.2 | 5.8 | |
| b | Ar | 3.4 | 110 | 2.1 | −2.0 | 2.7 | 14.1 | −11.7 | 2.4 |
| c | Ar | 3.4 | 110 | 2.8 | −2.3 | 3.5 | 15.3 | −12.3 | 3.0 |
| Kr | 3.7 | 165 | −0.2 | −3.0 | 0.7 | 17.3 | −16.6 | 0.7 | |
| Xe | 3.4 | 240 | −5.5 | −8.7 | −2.9 | 19.4 | −22.1 | −2.7 |
Additional data used to perform the calculations [37]. Water: σ1 = 2.8 Å; v1 = 18.07 cm3·mol−1; ξ = 0.383; αP = 0.257·10−3·K−1; ε/k = 120 K. Carbon tetrachloride: σ1 = 5.37 Å; v1 = 97.09 cm3·mol−1; ξ = 0.503; αP = 1.226·10−3·K−1; ε/k = 530 K. Benzene: σ1 = 5.26 Å; v1 = 89.41 cm3·mol−1; ξ = 0.513; αP = 1.22·10−3·K−1; ε/k = 530 K.
Table 2.
Enthalpy and entropy changes associated with cavity creation in water (a), CCl4 (b), and C6H6 (c), calculated by means of the classic SPT relationships at 25 °C and 1 atm, to be compared with the reorganization enthalpy change and the total solvation entropy change, respectively.
Table 2.
Enthalpy and entropy changes associated with cavity creation in water (a), CCl4 (b), and C6H6 (c), calculated by means of the classic SPT relationships at 25 °C and 1 atm, to be compared with the reorganization enthalpy change and the total solvation entropy change, respectively.
| ΔHC kJ·mol−1 | ΔH∙ − Ea kJ·mol−1 | ΔSC J·K−1·mol−1 | ΔS∙ J·K−1·mol−1 | ||
|---|---|---|---|---|---|
| a | He | 2.1 | 3.4 | −37.2 | −32.5 |
| Ne | 2.3 | 2.6 | −41.6 | −41.9 | |
| Ar | 3.2 | 2.1 | −56.0 | −60.4 | |
| Kr | 3.7 | 2.4 | −64.1 | −66.7 | |
| Xe | 4.3 | 3.4 | −72.8 | −74.8 | |
| b | Ar | 13.4 | 13.8 | −2.3 | −2.0 |
| c | Ar | 14.9 | 15.1 | −1.3 | −2.3 |
| Kr | 17.1 | 16.4 | −0.7 | −3.0 | |
| Xe | 19.5 | 16.6 | 0.3 | −8.7 |
Table 3.
ΔGC estimates associated with the creation of prolate spherocylindrical cavities, at 25 °C and 1 atm, in a hard sphere fluid, with the experimental density of water and particle diameter σ = 2.8 Å. By keeping the cavity VvdW fixed at the volume of 6 Å and 9 Å radius spheres, respectively (i.e., 904.8 Å3 and 3053.6 Å3, respectively), the ΔGC numbers have been calculated on varying the cylindrical length by means of the classic SPT analytic formulas. The first row of the A and B sections contains the numbers for the two spherical cavities.
Table 3.
ΔGC estimates associated with the creation of prolate spherocylindrical cavities, at 25 °C and 1 atm, in a hard sphere fluid, with the experimental density of water and particle diameter σ = 2.8 Å. By keeping the cavity VvdW fixed at the volume of 6 Å and 9 Å radius spheres, respectively (i.e., 904.8 Å3 and 3053.6 Å3, respectively), the ΔGC numbers have been calculated on varying the cylindrical length by means of the classic SPT analytic formulas. The first row of the A and B sections contains the numbers for the two spherical cavities.
| a Å | l Å | WASAC Å2 | ΔGC kJ mol−1 | |
|---|---|---|---|---|
| A | 6.0 | - - | 688.1 | 184.7 |
| 5.0 | 4.85 | 709.7 | 190.5 | |
| 4.0 | 12.67 | 796.3 | 212.8 | |
| 3.0 | 28.00 | 1017.4 | 266.1 | |
| 2.8 | 33.00 | 1092.5 | 283.4 | |
| 2.5 | 42.75 | 1238.7 | 316.1 | |
| 2.3 | 51.37 | 1366.3 | 343.9 | |
| 2.0 | 69.31 | 1625.9 | 398.3 | |
| B | 9.0 | - - | 1359.2 | 399.3 |
| 7.0 | 10.50 | 1440.9 | 422.9 | |
| 6.0 | 19.00 | 1571.5 | 459.6 | |
| 5.0 | 32.21 | 1810.0 | 524.2 | |
| 4.0 | 55.41 | 2246.5 | 636.9 | |
| 3.5 | 74.69 | 2601.2 | 724.5 | |
| 3.0 | 104.01 | 3118.7 | 847.3 | |
| 2.5 | 152.15 | 3919.5 | 1028.3 |
The geometric formulas for a prolate spherocylinder of radius a and cylindrical length l are: VvdW = (4/3)π·a3 + π·l·a2 and WASA = 4π(a + rw)2 + 2π·l·(a + rw), where rw is the radius of water molecules, fixed at 1.4 Å; by setting l = 0, such formulas become right for a sphere of radius a.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Graziano, G. The Relevance of Cavity Creation for Several Phenomena Occurring in Water. Liquids 2023, 3, 57-65. https://doi.org/10.3390/liquids3010006
AMA Style
Graziano G. The Relevance of Cavity Creation for Several Phenomena Occurring in Water. Liquids. 2023; 3(1):57-65. https://doi.org/10.3390/liquids3010006
Chicago/Turabian StyleGraziano, Giuseppe. 2023. "The Relevance of Cavity Creation for Several Phenomena Occurring in Water" Liquids 3, no. 1: 57-65. https://doi.org/10.3390/liquids3010006