
Conformational Dependence of the First Hyperpolarizability of the Li@B10H14 in Solution


Abstract
:
1. Introduction
2. Computational Details
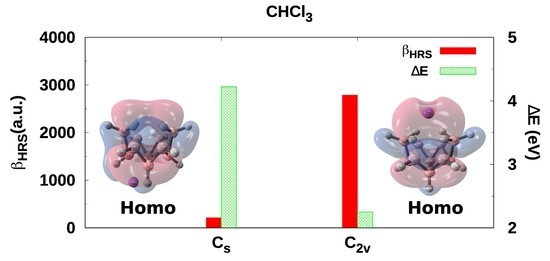
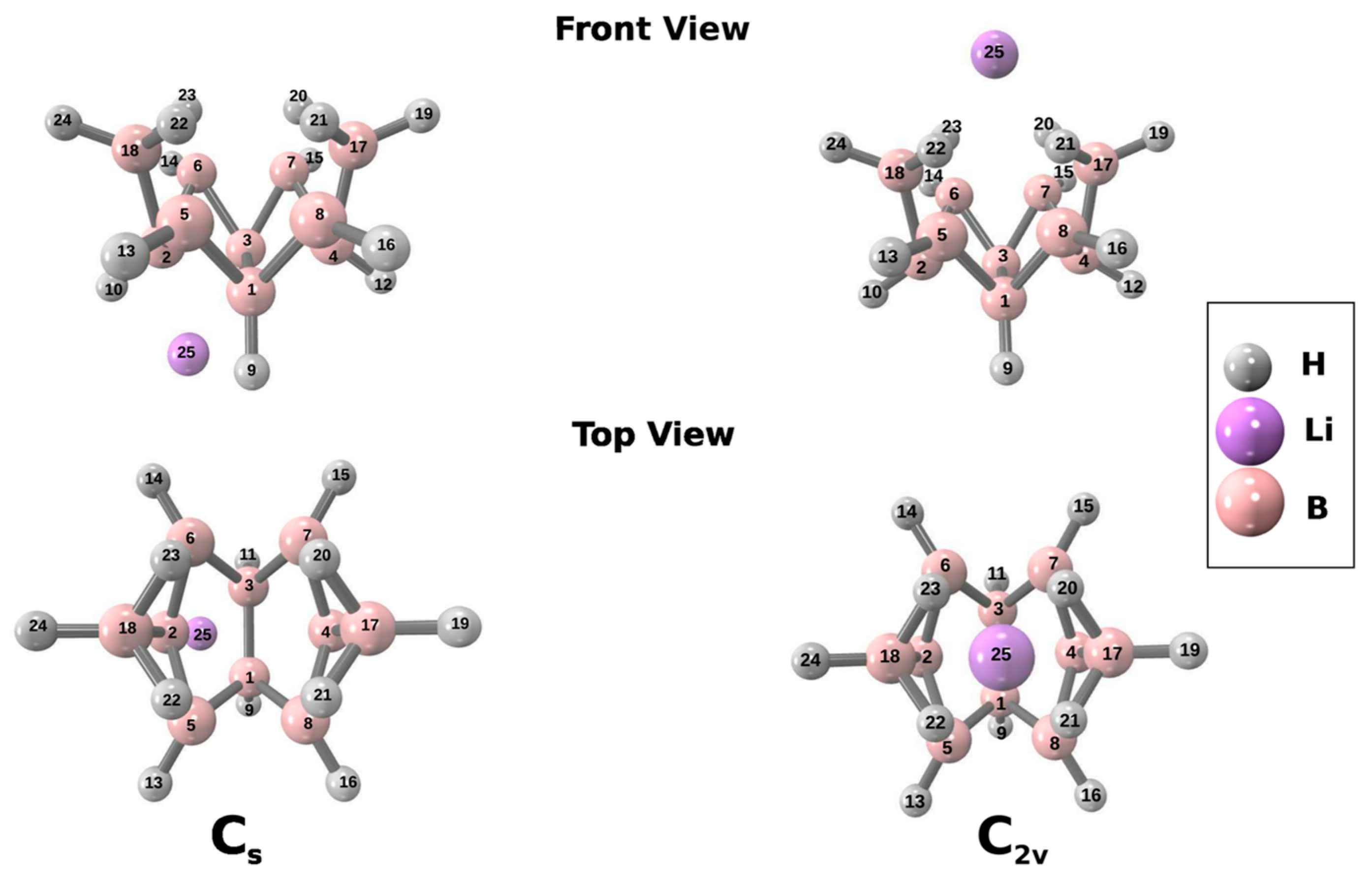
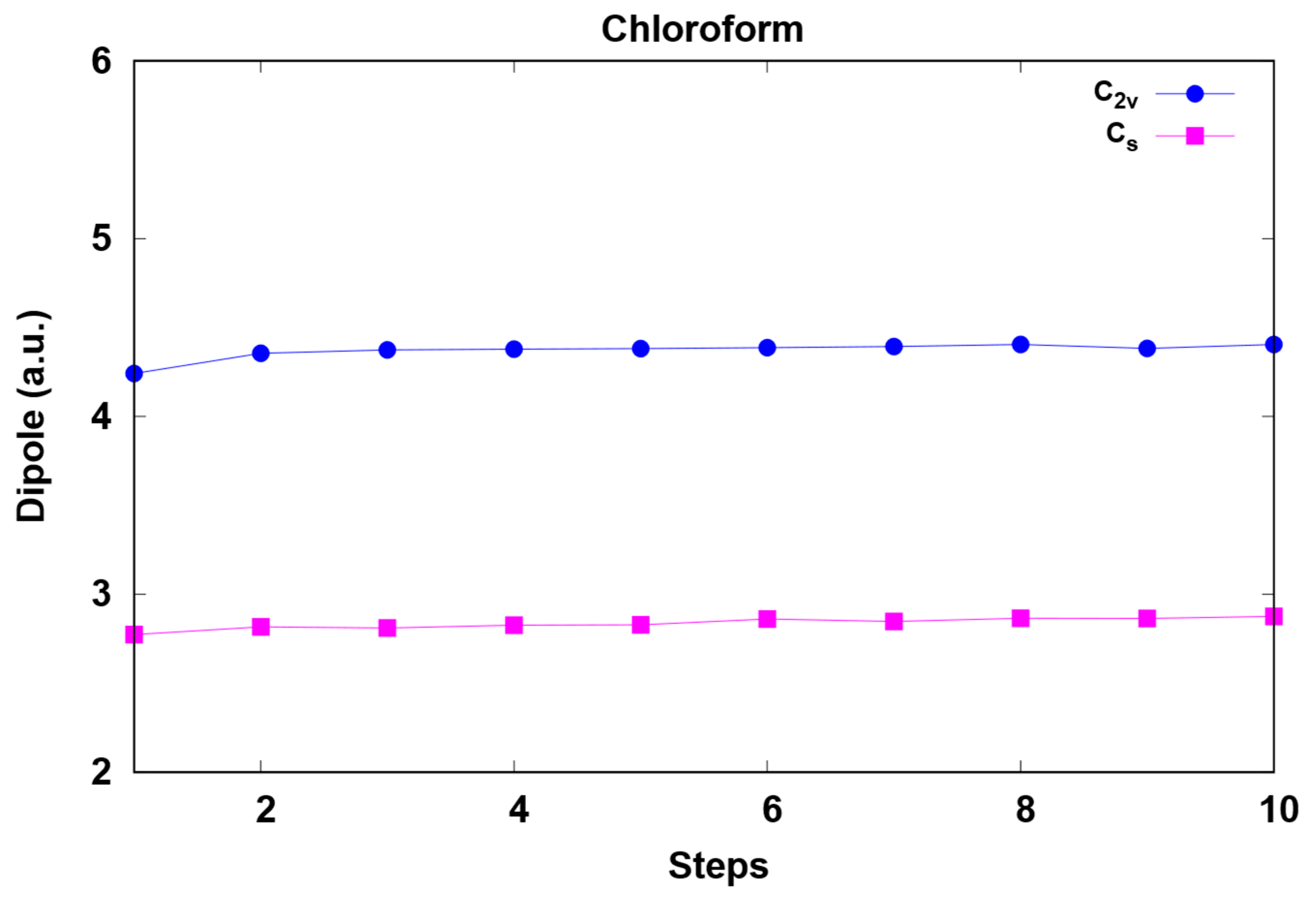
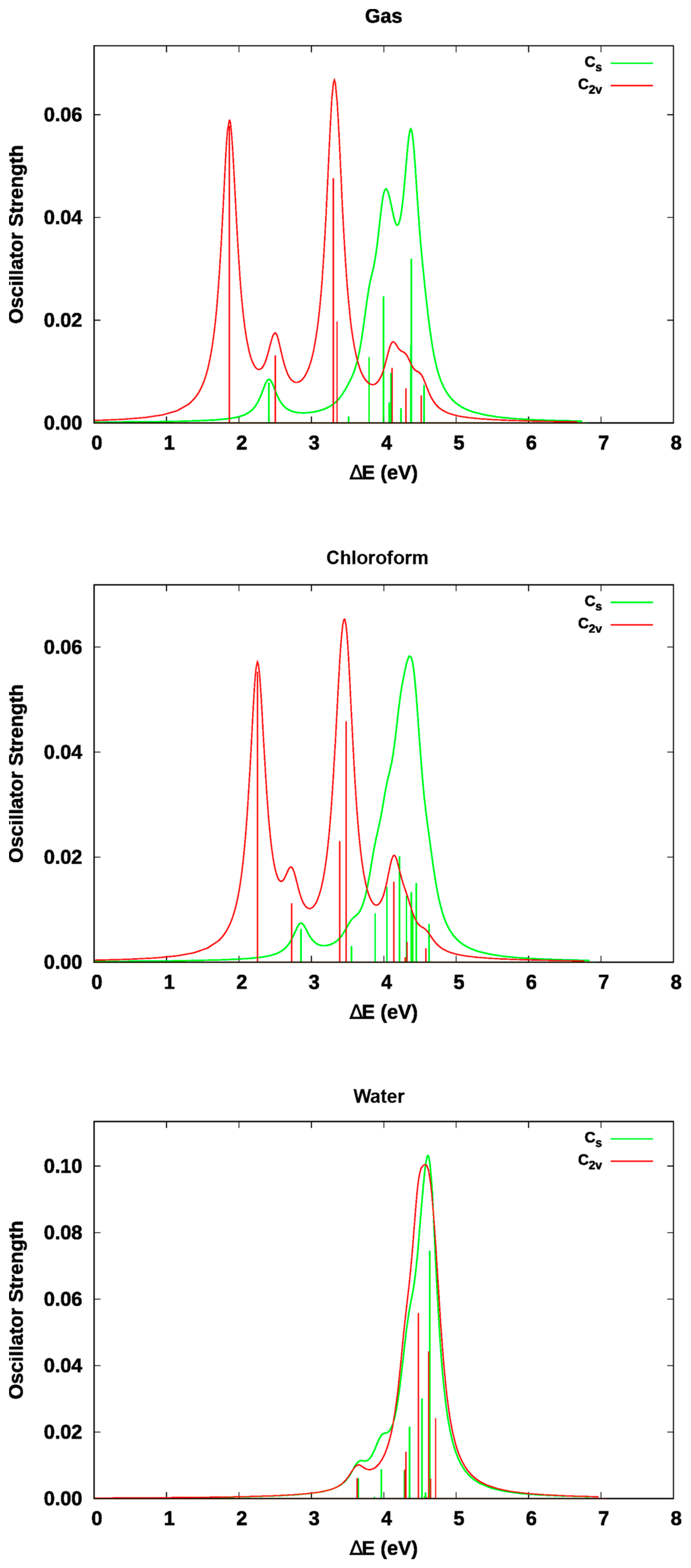
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dye, J.L. Electrons as Anions. Science 2003, 301, 607–608. [Google Scholar] [CrossRef]
- Dye, J.L. Electrides: Early Examples of Quantum Confinement. Acc. Chem. Res. 2009, 42, 1564–1572. [Google Scholar] [CrossRef]
- Chen, W.; Li, Z.R.; Wu, D.; Li, Y.; Sun, C.C.; Gu, F.L. The Structure and the Large Nonlinear Optical Properties of Li@calix[4]Pyrrole. J. Am. Chem. Soc. 2005, 127, 10977–10981. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, Z.R.; Wu, D.; Li, R.Y.; Sun, C.C. Theoretical Investigation of the Large Nonlinear Optical Properties of (HCN)n Clusters with Li Atom. J. Phys. Chem. B 2005, 109, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.L.; Li, Z.R.; Wu, D.; Wang, B.Q.; Li, Y.; Gu, F.L.; Aoki, Y. Structures and Large NLO Responses of New Electrides: Li-Doped Fluorocarbon Chain. J. Am. Chem. Soc. 2007, 129, 2967–2970. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.J.; Li, Z.R.; Wang, F.F.; Luo, C.; Ma, F.; Wu, D.; Wang, Q.; Huang, X.R. A Dependence on the Petal Number of the Static and Dynamic First Hyperpolarizability for Electride Molecules: Many-Petal-Shaped Li-Doped Cyclic Polyamines. J. Phys. Chem. A 2009, 113, 2961–2966. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.-L.; Li, Z.R.; Wu, D.; Ma, F.; Li, Z.J.; Gu, F.L. Lithiation and Li-Doped Effects of [5]Cyclacene on the Static First Hyperpolarizability. J. Phys. Chem. C 2009, 113, 4984–4986. [Google Scholar] [CrossRef]
- Liu, Z.B.; Zhou, Z.J.; Li, Y.; Li, Z.R.; Wang, R.; Li, Q.Z.; Li, Y.; Jia, F.Y.; Wang, Y.F.; Li, Z.J.; et al. Push-Pull Electron Effects of the Complexant in a Li Atom Doped Molecule with Electride Character: A New Strategy to Enhance the First Hyperpolarizability. Phys. Chem. Chem. Phys. 2010, 12, 10562–10568. [Google Scholar] [CrossRef]
- Xu, H.L.; Sun, S.L.; Muhammad, S.; Su, Z.M. Three-Propeller-Blade-Shaped Electride: Remarkable Alkali-Metal-Doped Effect on the First Hyperpolarizability. Theor. Chem. Acc. 2011, 128, 241–248. [Google Scholar] [CrossRef]
- Zhong, R.L.; Xu, H.L.; Li, Z.R.; Su, Z.M. Role of Excess Electrons in Nonlinear Optical Response. J. Phys. Chem. Lett. 2015, 6, 612–619. [Google Scholar] [CrossRef]
- Oliveira, I.M.; Castro, M.A.; Leão, S.A.; Fonseca, T.L.; Pontes, R.B. Li4C4H2N2: A Molecule with Large Hyperpolarizabilities and Electride Characteristic. Int. J. Quantum Chem. 2018, 118, e25661. [Google Scholar] [CrossRef]
- Matsuishi, S.; Toda, Y.; Miyakawa, M.; Hayashi, K.; Kamiya, T.; Hirano, M.; Tanaka, I.; Hosono, H. High-Density Electron Anions in a Nanoporous Single Crystal: [Ca24Al28O64]4+ (4e−). Science 2003, 301, 626–629. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, S.; Xu, H.; Liao, Y.; Kan, Y.; Su, Z. Quantum Mechanical Design and Structure of the Li@B10H14 Basket with a Remarkably Enhanced Electro-Optical Response. J. Am. Chem. Soc. 2009, 131, 11833–11840. [Google Scholar] [CrossRef]
- Muhammad, S.; Xu, H.; Su, Z. Capturing a Synergistic Effect of a Conical Push and an Inward Pull in Fluoro Derivatives of Li@B10H14 Basket: Toward a Higher Vertical Ionization Potential and Nonlinear Optical Response. J. Phys. Chem. A 2011, 115, 923–931. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Gong, J.; Li, S.; Zhang, J.; Qiu, Y.; Zhang, G. Second-Order NLO Responses of Two-Cavity Inorganic Electrides Li: N@B20H26 (n = 1, 2): Evolutions with Increasing Excess Electron Number and Various B-B Connection Sites of B20H26. Phys. Chem. Chem. Phys. 2017, 19, 2557–2566. [Google Scholar] [CrossRef] [PubMed]
- Brandão, I.; Fonseca, T.L.; Georg, H.C.; Castro, M.A.; Pontes, R.B. Assessing the Structure and First Hyperpolarizability of Li@B10H14 in Solution: A Sequential QM/MM Study Using the ASEC-FEG Method. Phys. Chem. Chem. Phys. 2020, 22, 17314–17324. [Google Scholar] [CrossRef]
- Medved, M.; Demissie, T.B.; McKee, M.L.; Hnyk, D. The Behavior of a Paramagnetic System in Electric and Magnetic Fields as Exemplified by Revisiting Li@B10H14. Phys. Chem. Chem. Phys. 2017, 19, 12229–12236. [Google Scholar] [CrossRef]
- Okuyama-Yoshida, N.; Nagaoka, M.; Yamabe, T. Transition-State Optimization on Free Energy Surface: Toward Solution Chemical Reaction Ergodography. Int. J. Quantum Chem. 1998, 70, 95–103. [Google Scholar] [CrossRef]
- Okuyama-Yoshida, N.; Kataoka, K.; Nagaoka, M.; Yamabe, T. Structure Optimization via Free Energy Gradient Method: Application to Glycine Zwitterion in Aqueous Solution. J. Chem. Phys. 2000, 113, 3519–3524. [Google Scholar] [CrossRef]
- Hirao, H.; Nagae, Y.; Nagaoka, M. Transition-State Optimization by the Free Energy Gradient Method: Application to Aqueous-Phase Menshutkin Reaction between Ammonia and Methyl Chloride. Chem. Phys. Lett. 2001, 348, 350–356. [Google Scholar] [CrossRef]
- Coutinho, K.; Georg, H.C.; Fonseca, T.L.; Ludwig, V.; Canuto, S. An Efficient Statistically Converged Average Configuration for Solvent Effects. Chem. Phys. Lett. 2007, 437, 148–152. [Google Scholar] [CrossRef]
- Georg, H.C.; Canuto, S. Electronic Properties of Water in Liquid Environment: A Sequential QM/MM Study Using the Free Energy Gradient Method. J. Phys. Chem. B 2012, 116, 11247–11254. [Google Scholar] [CrossRef] [PubMed]
- Franco, L.R.; Brandão, I.; Fonseca, T.L.; Georg, H.C. Elucidating the Structure of Merocyanine Dyes with the ASEC-FEG Method. Phenol Blue in Solution. J. Chem. Phys. 2016, 145, 194301. [Google Scholar] [CrossRef] [PubMed]
- Brandão, I.; Franco, L.R.; Fonseca, T.L.; Castro, M.A.; Georg, H.C. Confirming the Relationship between First Hyperpolarizability and the Bond Length Alternation Coordinate for Merocyanine Dyes. J. Chem. Phys. 2017, 146, 224505. [Google Scholar] [CrossRef] [PubMed]
- Valverde, D.; Georg, H.C.; Canuto, S. Free-Energy Landscape of the SN2 Reaction CH3Br + Cl− –> CH3Cl + Br- in Different Liquid Environments. J. Phys. Chem. B 2022, 126, 3685–3692. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, K.; Canuto, S. Solvent Effects from a Sequential Monte Carlo-Quantum Mechanical Approach. Adv. Quantum Chem. 1997, 28, 89–105. [Google Scholar] [CrossRef]
- Coutinho, K.; Canuto, S. Solvent Effects in Emission Spectroscopy: A Monte Carlo Quantum Mechanics Study of the n←π* Shift of Formaldehyde in Water. J. Chem. Phys. 2000, 113, 9132–9139. [Google Scholar] [CrossRef]
- Fonseca, T.L.; Coutinho, K.; Canuto, S. Probing Supercritical Water with the n-π* Transition of Acetone: A Monte Carlo/Quantum Mechanics Study. J. Chem. Phys. 2007, 126, 034508. [Google Scholar] [CrossRef]
- Fonseca, T.L.; Georg, H.C.; Coutinho, K.; Canuto, S. Polarization and Spectral Shift of Benzophenone in Supercritical Water. J. Phys. Chem. A 2009, 113, 5112–5118. [Google Scholar] [CrossRef]
- Fonseca, T.L.; Coutinho, K.; Canuto, S. Hydrogen Bond Interactions between Acetone and Supercritical Water. Phys. Chem. Chem. Phys. 2010, 12, 6660–6665. [Google Scholar] [CrossRef]
- Junior, L.A.; Colherinhas, G.; Fonseca, T.L.; Castro, M.A. Solvent Effects on the First Hyperpolarizability of Retinal Derivatives. Chem. Phys. Lett. 2014, 598, 43–47. [Google Scholar] [CrossRef]
- Oliveira, L.B.A.; Colherinhas, G.; Fonseca, T.L.; Castro, M.A. Spectroscopic Properties of Vitamin E Models in Solution. Chem. Phys. Lett. 2015, 628, 49–53. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Grigera, J.R.; Straatsma, T.P. The missing term in effective pair potentials. J. Phys. Chem. 1987, 91, 6269–6271. [Google Scholar] [CrossRef]
- Guissani, Y.; Guillot, B. A computer simulation study of the liquid– vapor coexistence curve of water. J. Chem. Phys. 1993, 98, 8221–8235. [Google Scholar] [CrossRef]
- McDonald, N.A.; Carlson, H.A.; Jorgensen, W.L. Free energies of solvation in chloroform and water from a linear response approach. J. Phys. Org. Chem. 1997, 10, 563–576. [Google Scholar] [CrossRef]
- Besler, B.H.; Merz, K.M.; Kollman, P.A. Atomic charges derived from semiempirical methods. J. Comput. Chem. 1990, 11, 431–439. [Google Scholar] [CrossRef]
- Clays, K.; Persoons, A. Hyper-Rayleigh Scattering in Solution. Phys. Rev. Lett. 1991, 66, 2980–2983. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B. GAUSSIAN 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Naves, E.S.; Castro, M.A.; Fonseca, T.L. Dynamic (Hyper)Polarizabilities of the Ozone Molecule: Coupled Cluster Calculations Including Vibrational Corrections. J. Chem. Phys. 2011, 134, 054315. [Google Scholar] [CrossRef]
- Silveira, O.; Castro, M.A.; Fonseca, T.L. Vibrational Corrections to the First Hyperpolarizability of the Lithium Salt of Pyridazine Li-H3C4N2. J. Chem. Phys. 2013, 138, 074312. [Google Scholar] [CrossRef]
- Silveira, O.; Castro, M.A.; Leão, S.A.; Fonseca, T.L. Second Hyperpolarizabilities of the Lithium Salt of Pyridazine Li-H3C4N2 and Lithium Salt Electride Li-H3C4N2. Chem. Phys. Lett. 2015, 633, 241–246. [Google Scholar] [CrossRef]
- Castet, F.; Bogdan, E.; Plaquet, A.; Ducasse, L.; Champagne, B.; Rodriguez, V. Reference Molecules for Nonlinear Optics: A Joint Experimental and Theoretical Investigation. J. Chem. Phys. 2012, 136, 024506. [Google Scholar] [CrossRef]
- Iikura, H.; Tsuneda, T.; Yanai, T.; Hirao, K. A Long-Range Correction Scheme for Generalized-Gradient-Approximation Exchange Functionals. J. Chem. Phys. 2001, 115, 3540–3544. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A New Hybrid Exchange-Correlation Functional Using the Coulomb-Attenuating Method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Schultz, N.E.; Truhlar, D.G. Design of Density Functionals by Combining the Method of Constraint Satisfaction with Parametrization for Thermochemistry, Thermochemical Kinetics, and Noncovalent Interactions. J. Chem. Theory Comput. 2006, 2, 364–382. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Function. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef] [Green Version]
- Cezar, H.M.; Canuto, S.; Coutinho, K. DICE: A Monte Carlo Code for Molecular Simulation Including the Configurational Bias Monte Carlo Method. J. Chem. Inf. Model. 2020, 60, 3472–3488. [Google Scholar] [CrossRef]
- Wang, F.F.; Li, Z.R.; Wu, D.; Wang, B.Q.; Li, Y.; Li, Z.J.; Chen, W.; Yu, G.T.; Gu, F.L.; Aoki, Y. Structures and Considerable Static First Hyperpolarizabilities: New Organic Alkalides (M+@n6adz)M′- (M, M′ = Li, Na, K; n = 2, 3) with Cation inside and Anion Outside of the Cage Complexants. J. Phys. Chem. B 2008, 112, 1090–1094. [Google Scholar] [CrossRef]
- Oudar, J.L.; Chemla, D.S.; Batifol, E. Optical Nonlinearities of Various Substituted Benzene Molecules in the Liquid State, and Comparison with Solid State Nonlinear Susceptibilities. J. Chem. Phys. 1977, 67, 1626–1635. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Length | Gas-Phase | Chloroform (Converged Value) | Water (Last Step Value) |
|---|---|---|---|
| B1-Li | 2.256 | 2.295 | 3.150 |
| B2-Li | 2.244 | 2.281 | 3.230 |
| B3-Li | 2.256 | 2.295 | 3.228 |
| H9-Li | 2.021 | 2.051 | 2.679 |
| H10-Li | 1.957 | 1.980 | 2.764 |
| H11-Li | 2.021 | 2.051 | 2.981 |
| H23–Li | 2.043 | 2.169 | 2.819 |
| Conformer | Gas-Phase | Chloroform (Converged Value) | Water (Last Step Value) |
|---|---|---|---|
| Cs | 2.53 (2.55 2) | 2.91 | 6.56 |
| C2v | 3.59 1 (3.72 2) | 4.59 1 | 9.42 1 |
| Conformer | βHRS (a.u.) | DR | ||||
|---|---|---|---|---|---|---|
| Gas-Phase | Chloroform (Converged Value) | Water (Last Step Value) | Gas-Phase | Chloroform (Converged Value) | Water (Last Step Value) | |
| Cs | 504 | 212 | 154 | 4.94 | 4.02 | 7.23 |
| C2v 1 | 6214 | 2786 | 64 | 6.20 | 6.52 | 3.57 |
| Cs Conformer | C2v Conformer 1 | |||||
|---|---|---|---|---|---|---|
| Method | Gas-Phase | Chloroform (Converged Value) | Water (Last Step Value) | Gas-Phase | Chloroform (Converged Value) | Water (Last Step Value) |
| LC-BLYP | 4.58 (0.0302) | 4.48 (0.0439) | 4.90 (0.0723) | 2.23 (0.0504) | 2.59 (0.0505) | 3.81 (0.0069) |
| M05-2X | 4.10 (0.0300) | 4.21 (0.0331) | 4.58 (0.0773) | 2.28 (0.0503) | 2.64 (0.0494) | 3.55 (0.0065) |
| M06-2X | 3.65 (0.0211) | 3.95 (0.0225) | 4.30 (0.0351) | 1.92 (0.0573) | 2.43 (0.0522) | 3.44 (0.0011) |
| CAM-B3LYP | 4.38 (0.0319) | 4.22 (0.0201) | 4.64 (0.0744) | 1.87 (0.0578) | 2.25 (0.0553) | 3.63 (0.0061) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brandão, I.; Fonseca, T.L.; Georg, H.C.; Castro, M.A.; Pontes, R.B. Conformational Dependence of the First Hyperpolarizability of the Li@B10H14 in Solution. Liquids 2023, 3, 159-167. https://doi.org/10.3390/liquids3010012
Brandão I, Fonseca TL, Georg HC, Castro MA, Pontes RB. Conformational Dependence of the First Hyperpolarizability of the Li@B10H14 in Solution. Liquids. 2023; 3(1):159-167. https://doi.org/10.3390/liquids3010012
Chicago/Turabian StyleBrandão, Idney, Tertius L. Fonseca, Herbert C. Georg, Marcos A. Castro, and Renato B. Pontes. 2023. "Conformational Dependence of the First Hyperpolarizability of the Li@B10H14 in Solution" Liquids 3, no. 1: 159-167. https://doi.org/10.3390/liquids3010012