
Whole Genome Sequencing Evidences High Rates of Relapse in Clostridioides difficile Infection Caused by the Epidemic Ribotype 106

Abstract
:1. Introduction
2. Methods
2.1. Study Design and Clostridioides Difficile Isolation
2.2. Molecular Characterization of the Isolates
2.3. Whole-Genome Sequencing and Analysis
2.4. Antimicrobial Susceptibility and Mechanisms of Resistance Analysis
3. Results
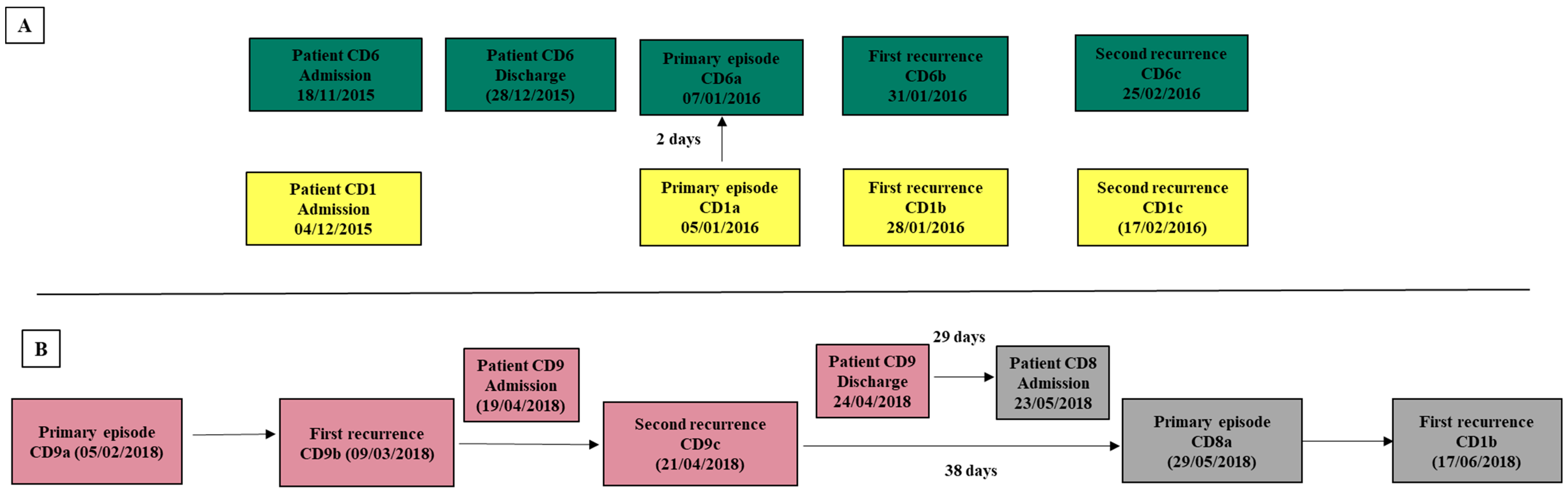
3.1. Patients and Isolates Data Collection
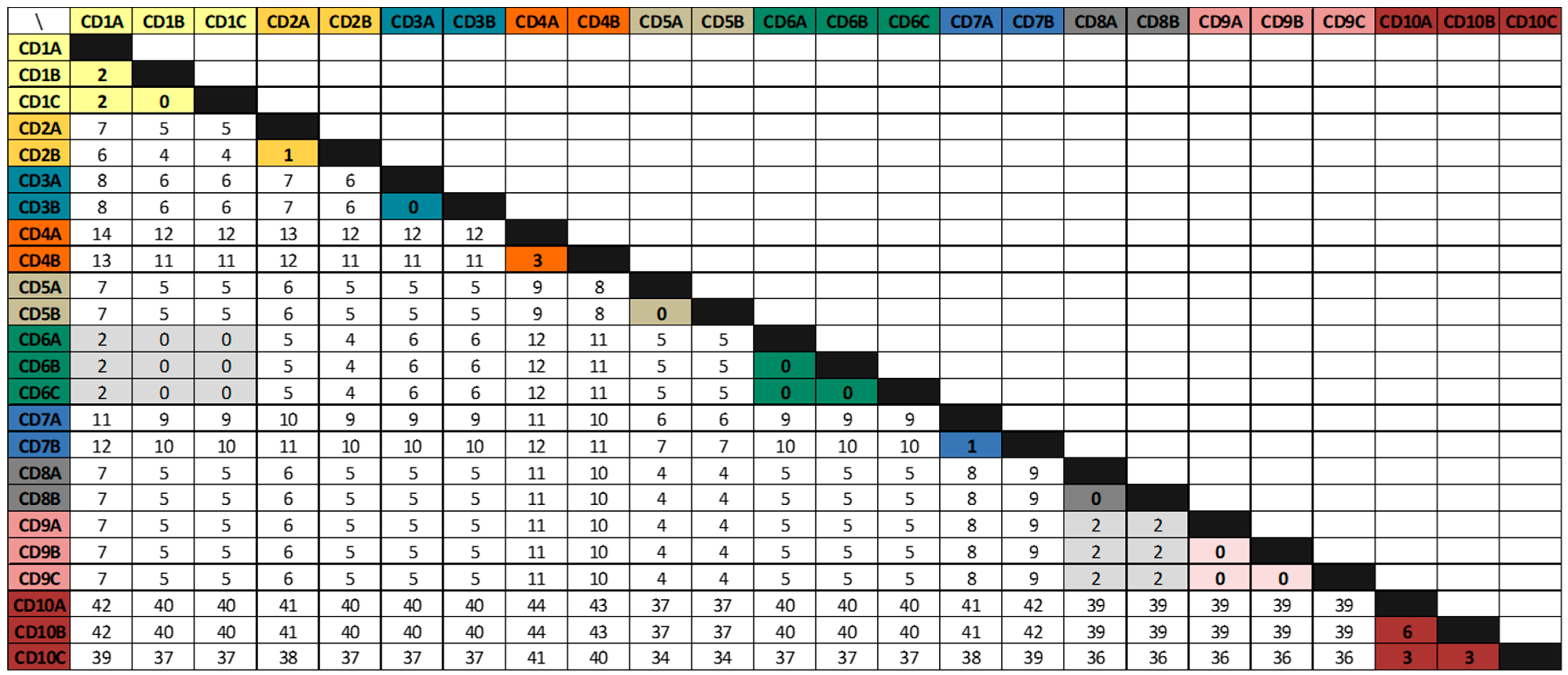
3.2. Whole-Genome Sequencing Analysis
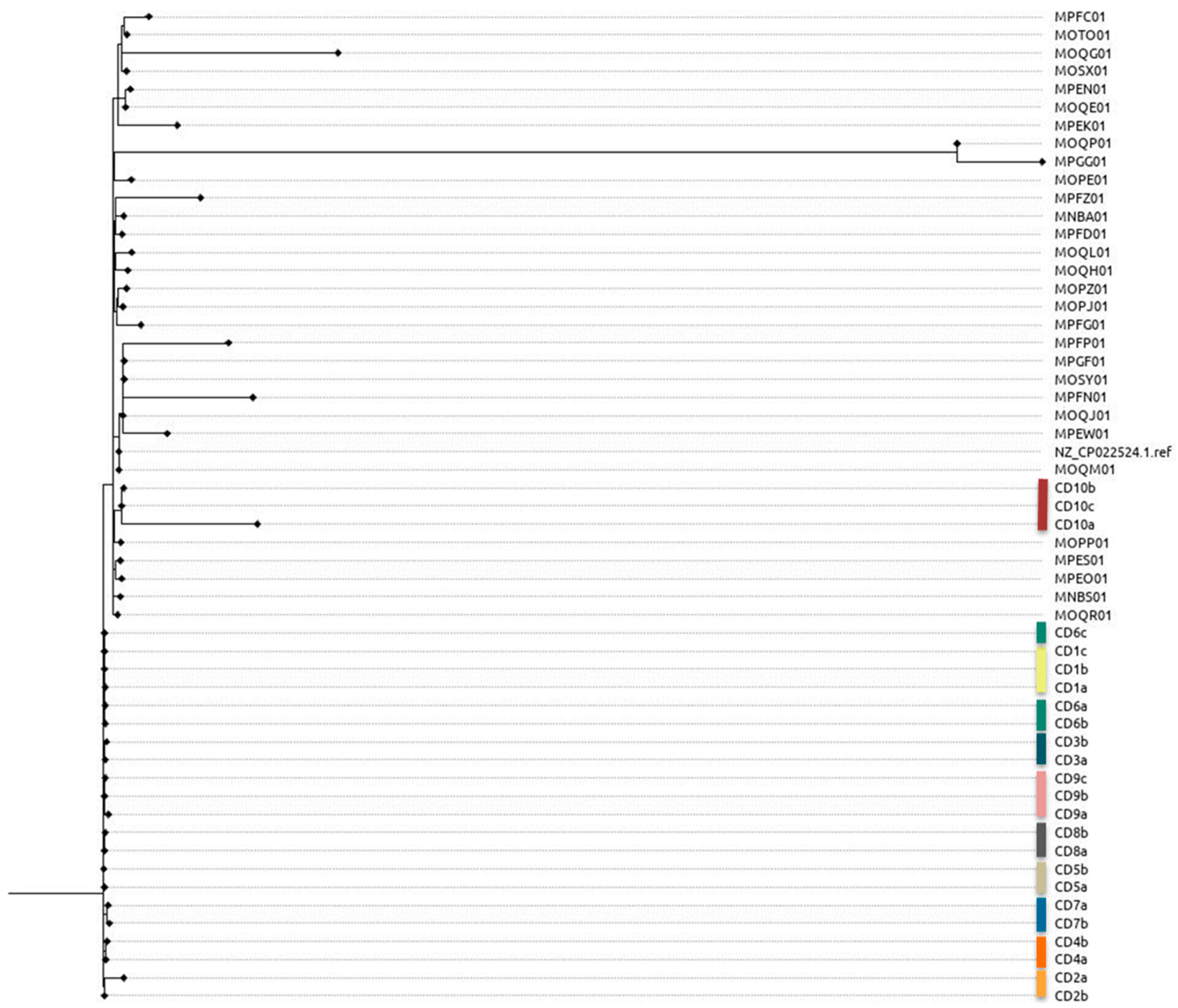
3.3. Phylogenetic Analysis
3.4. Antimicrobial Susceptibility and Mechanisms of Resistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Carlson, T.J.; Blasingame, D.; Gonzales-Luna, A.J.; Alnezary, F.; Garey, K.W. Clostridioides difficile ribotype 106: A systematic review of the antimicrobial susceptibility, genetics, and clinical outcomes of this common worldwide strain. Anaerobe 2020, 62, 102142. [Google Scholar] [CrossRef] [PubMed]
- Wilcox, M.H.; Shetty, N.; Fawley, W.N.; Shemko, M.; Coen, P.; Birtles, A.; Cairns, M.; Curran, M.D.; Dodgson, K.J.; Green, S.M.; et al. Changing epidemiology of Clostridium difficile infection following the introduction of a national ribotyping-based surveillance scheme in England. Clin. Infect. Dis. 2012, 55, 1056–1063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kociolek, L.K.; Patel, S.J.; Shulman, S.T.; Gerding, D.N. Molecular epidemiology of Clostridium difficile infections in children: A retrospective cohort study. Infect. Control. Hosp. Epidemiol. 2015, 36, 445–451. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Healthcare-Associated Infections—Community Interface (HAIC). Clostridioides difficile infection (CDI) Tracking. Available online: https://www.cdc.gov/hai/eip/cdiff-tracking.html (accessed on 11 May 2020).
- Roxas, B.A.P.; Roxas, J.L.; Claus-Walker, R.; Harishankar, A.; Mansoor, A.; Anwar, F.; Jillella, S.; Williams, A.; Lindsey, J.; Elliott, S.P.; et al. Phylogenomic analysis of Clostridioides difficile ribotype 106 strains reveals novel genetic islands and emergent phenotypes. Sci. Rep. 2020, 10, 22135. [Google Scholar] [CrossRef]
- Alcalá, L.; Reigadas, E.; Marín, M.; Martín, A.; Catalán, P.; Bouza, E. Spanish Clostridium difficile Study Group. Impact of clinical awareness and diagnostic tests on the underdiagnosis of Clostridium difficile infection. Eur. J. Clin. Microbiol. Infect. Dis. 2015, 34, 1515–1525. [Google Scholar] [CrossRef]
- Weber, I.; Riera, E.; Déniz, C.; Pérez, J.L.; Oliver, A.; Mena, A. Molecular epidemiology and resistance profiles of Clostridium difficile in a tertiary care hospital in Spain. Int. J. Med. Microbiol. 2013, 303, 128–133. [Google Scholar] [CrossRef]
- Suárez-Bode, L.; Barrón, R.; Pérez, J.L.; Mena, A. Increasing prevalence of the epidemic ribotype 106 in healthcare facility-associated and community-associated Clostridioides difficile infection. Anaerobe 2019, 55, 124–129. [Google Scholar] [CrossRef]
- García-Fernández, S.; Frentrup, M.; Steglich, M.; Gonzaga, A.; Cobo, M.; López-Fresneña, N.; Cobo, J.; Morosini, M.I.; Cantón, R.; Del Campo, R.; et al. Whole-genome sequencing reveals nosocomial Clostridioides difficile transmission and a previously unsuspected epidemic scenario. Sci. Rep. 2019, 9, 6959. [Google Scholar] [CrossRef] [Green Version]
- Vohra, P.; Poxton, I.R. Comparison of toxin and spore production in clinically relevant strains of Clostridium difficile. Microbiology (Reading) 2011, 157, 1343–1353. [Google Scholar] [CrossRef] [Green Version]
- Vohra, P.; Poxton, I.R. Efficacy of decontaminants and disinfectants against Clostridium difficile. J. Med. Microbiol. 2011, 60, 1218–1224. [Google Scholar] [CrossRef]
- Kelly, C.P.; LaMont, J.T. Clostridium difficile—More difficult than ever. N. Engl. J. Med. 2010, 359, 1932–1940, Erratum in N. Engl. J. Med. 2010, 363, 1585. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Miyajima, F.; Roberts, P.; Ellison, L.; Pickard, D.J.; Martin, M.J.; Connor, T.R.; Harris, S.R.; Fairley, D.; Bamford, K.B.; et al. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat. Genet. 2013, 45, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Dingle, K.E.; Didelot, X.; Quan, T.P.; Eyre, D.W.; Stoesser, N.; Golubchik, T.; Harding, R.M.; Wilson, D.J.; Griffiths, D.; Vaughan, A.; et al. Modernising Medical Microbiology Informatics Group. Effects of control interventions on Clostridium difficile infection in England: An observational study. Lancet Infect. Dis. 2017, 17, 411–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mutlu, E.; Wroe, A.J.; Sanchez-Hurtado, K.; Brazier, J.S.; Poxton, I.R. Molecular characterization and antimicrobial susceptibility patterns of Clostridium difficile strains isolated from hospitals in south-east Scotland. J. Med. Microbiol. 2007, 56, 921–929. [Google Scholar] [CrossRef] [Green Version]
- Solomon, K.; Fanning, S.; McDermott, S.; Murray, S.; Scott, L.; Martin, A.; Skally, M.; Burns, K.; Kuijper, E.; Fitzpatrick, F.; et al. PCR ribotype prevalence and molecular basis of macrolide-lincosamide-streptogramin B (MLSB) and fluoroquinolone resistance in Irish clinical Clostridium difficile isolates. J. Antimicrob. Chemother. 2011, 66, 1976–1982. [Google Scholar] [CrossRef]
- Kociolek, L.K.; Gerding, D.N.; Hecht, D.W.; Ozer, E.A. Comparative genomics analysis of Clostridium difficile epidemic strain DH/NAP11/106. Microbes Infect. 2018, 20, 245–253. [Google Scholar] [CrossRef]
- Baines, S.D.; O’Connor, R.; Saxton, K.; Freeman, J.; Wilcox, M.H. Activity of vancomycin against epidemic Clostridium difficile strains in a human gut model. J. Antimicrob. Chemother. 2009, 63, 520–525. [Google Scholar] [CrossRef] [Green Version]
- Eyre, D.W.; Cule, M.L.; Wilson, D.J.; Griffiths, D.; Vaughan, A.; O’Connor, L.; Ip, C.L.; Golubchik, T.; Batty, E.M.; Finney, J.M.; et al. Diverse sources of C. difficile infection identified on whole-genome sequencing. N. Engl. J. Med. 2013, 369, 1195–1205. [Google Scholar] [CrossRef] [Green Version]
- Janezic, S.; Rupnik, M. Development and Implementation of Whole Genome Sequencing-Based Typing Schemes for Clostridioides difficile. Front. Public Health 2019, 7, 309. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control. European Surveillance of Clostridioides (Clostridium) Difficile Infections. Surveillance Protocol Version 2.4; ECDC: Stockholm, Sweden, 2019.
- Fawley, W.N.; Knetsch, C.W.; MacCannell, D.R.; Harmanus, C.; Du, T.; Mulvey, M.R.; Paulick, A.; Anderson, L.; Kuijper, E.J.; Wilcox, M.H. Development and validation of an internationally-standardized, high-resolution capillary gel-based electrophoresis PCR-ribotyping protocol for Clostridium difficile. PLoS ONE 2015, 10, e0118150. [Google Scholar] [CrossRef]
- Indra, A.; Huhulescu, S.; Schneeweis, M.; Hasenberger, P.; Kernbichler, S.; Fiedler, A.; Wewalka, G.; Allerberger, F.; Kuijper, E.J. Characterization of Clostridium difficile isolates using capillary gel electrophoresis-based PCR ribotyping. J. Med. Microbiol. 2008, 57, 1377–1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus sequence typing of total-genome-sequenced bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef]
- Zankari, E.; Allesøe, R.; Joensen, K.G.; Cavaco, L.M.; Lund, O.; Aarestrup, F.M. PointFinder: A novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J. Antimicrob. Chemother. 2017, 72, 2764–2768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Córdova, P.; Mora-Uribe, P.; Reyes-Ramírez, R.; Cofré-Araneda, G.; Orozco-Aguilar, J.; Brito-Silva, C.; Mendoza-León, M.J.; Kuehne, S.A.; Minton, N.P.; Pizarro-Guajardo, M.; et al. Entry of spores into intestinal epithelial cells contributes to recurrence of Clostridioides difficile infection. Nat. Commun. 2021, 12, 1140. [Google Scholar] [CrossRef] [PubMed]
- Knight, D.R.; Elliott, B.; Chang, B.J.; Perkins, T.T.; Riley, T.V. Diversity and Evolution in the Genome of Clostridium difficile. Clin. Microbiol. Rev. 2015, 28, 721–741. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.; Cunningham, S.; Pu, M.; Lennon, R.J.; Dens Higano, J.; Jeraldo, P.; Sampathkumar, P.; Shannon, S.; Kashyap, P.C.; Patel, R. Clostridioides difficile Whole-genome Sequencing Differentiates Relapse With the Same Strain From Reinfection With a New Strain. Clin. Infect. Dis. 2021, 72, 806–813. [Google Scholar] [CrossRef]
- Balaji, A.; Ozer, E.A.; Kociolek, L.K. Clostridioides difficile Whole-Genome Sequencing Reveals Limited Within-Host Genetic Diversity in a Pediatric Cohort. J. Clin. Microbiol. 2019, 57, e00559-19. [Google Scholar] [CrossRef] [Green Version]
- Behroozian, A.A.; Chludzinski, J.P.; Lo, E.S.; Ewing, S.A.; Waslawski, S.; Newton, D.W.; Young, V.B.; Aronoff, D.M.; Walk, S.T. Detection of mixed populations of Clostridium difficile from symptomatic patients using capillary-based polymerase chain reaction ribotyping. Infect. Control. Hosp. Epidemiol. 2013, 34, 961–966. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Patients IDs | Age | Gender | Primary Episodes | First Recurrence | Second Recurrence | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Acquisition | Isolate | Isolate | Day a | SNVs b | Isolate | Day a | SNVs b | |||
| CD1 | 83 | F | HCFA-ICD | CD1a | CD1b | 23 | 2 | CD1c | 20 | 0 |
| CD2 | 58 | F | HCFA-ICD | CD2a | CD2b | 127 * | 1 | |||
| CD3 | 34 | M | Indeterminate | CD3a | CD3b | 78 * | 0 | |||
| CD4 | 61 | M | CA-ICD | CD4a | CD4b | 24 | 3 | |||
| CD5 | 92 | F | CA-ICD | CD5a | CD5b | 24 | 0 | |||
| CD6 | 87 | F | HCFA-ICD | CD6a | CD6b | 24 | 0 | CD6c | 25 | 0 |
| CD7 | 2 | M | CA-ICD | CD7a | CD7b | 24 | 1 | |||
| CD8 | 38 | F | CA-ICD | CD8a | CD8b | 19 | 0 | |||
| CD9 | 69 | M | CA-ICD | CD9a | CD9b | 32 | 0 | CD9c | 43 | 0 |
| CD10 | 64 | M | CA-ICD | CD10a | CD10b | 43 | 6 | CD10c | 34 | 3 |
| Isolate | Position | Reference Allele | Isolate Allele | Gene | Protein | Amino Acid Change |
|---|---|---|---|---|---|---|
| CD7b | 3,118,341 | G | A | slpA | S-layer protein SlpA | Ala166Val |
| CD10b | 1,645,698 | G | T | CGC51_ RS07795 | response regulator transcription factor | His162Asn |
| CD10b | 1,986,896 | G | T | CGC51_ RS09380 | 3-dehydroquinate synthase | Ala117Ser |
| CD10b | 2,458,725 | C | A | CGC51_ RS11610 | cell division protein FtsK | Gly420Stop |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Suárez-Bode, L.; López-Causapé, C.; Arcay, R.M.; Oliver, A.; Mena, A. Whole Genome Sequencing Evidences High Rates of Relapse in Clostridioides difficile Infection Caused by the Epidemic Ribotype 106. Appl. Microbiol. 2023, 3, 64-75. https://doi.org/10.3390/applmicrobiol3010005
Suárez-Bode L, López-Causapé C, Arcay RM, Oliver A, Mena A. Whole Genome Sequencing Evidences High Rates of Relapse in Clostridioides difficile Infection Caused by the Epidemic Ribotype 106. Applied Microbiology. 2023; 3(1):64-75. https://doi.org/10.3390/applmicrobiol3010005
Chicago/Turabian StyleSuárez-Bode, Loreto, Carla López-Causapé, Ricardo M. Arcay, Antonio Oliver, and Ana Mena. 2023. "Whole Genome Sequencing Evidences High Rates of Relapse in Clostridioides difficile Infection Caused by the Epidemic Ribotype 106" Applied Microbiology 3, no. 1: 64-75. https://doi.org/10.3390/applmicrobiol3010005