

Transcriptome Analysis Identifies Tumor Immune Microenvironment Signaling Networks Supporting Metastatic Castration-Resistant Prostate Cancer
 ,
,  , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Samples and Quality Control Measures
2.2. Normalization and Gene Expression Quantification
2.3. Co-Expression Network Analysis and Module Identification
2.4. Enrichment Analysis, Differential Expression Analysis, and Hub Gene Identification
2.5. Statistical Analysis
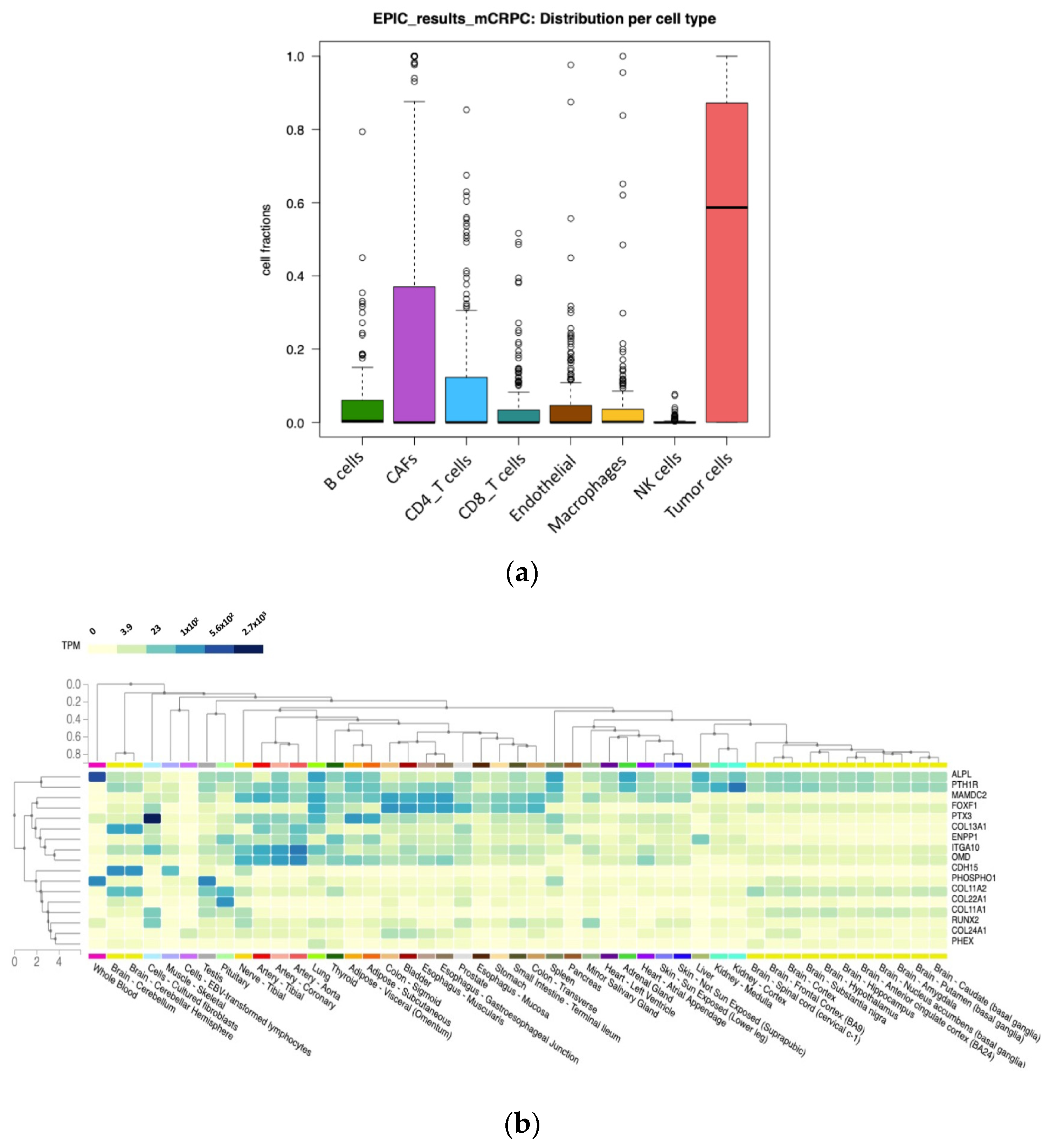
2.6. Transcriptome Deconvolution and Tissue Expression Correlation Analysis
3. Results
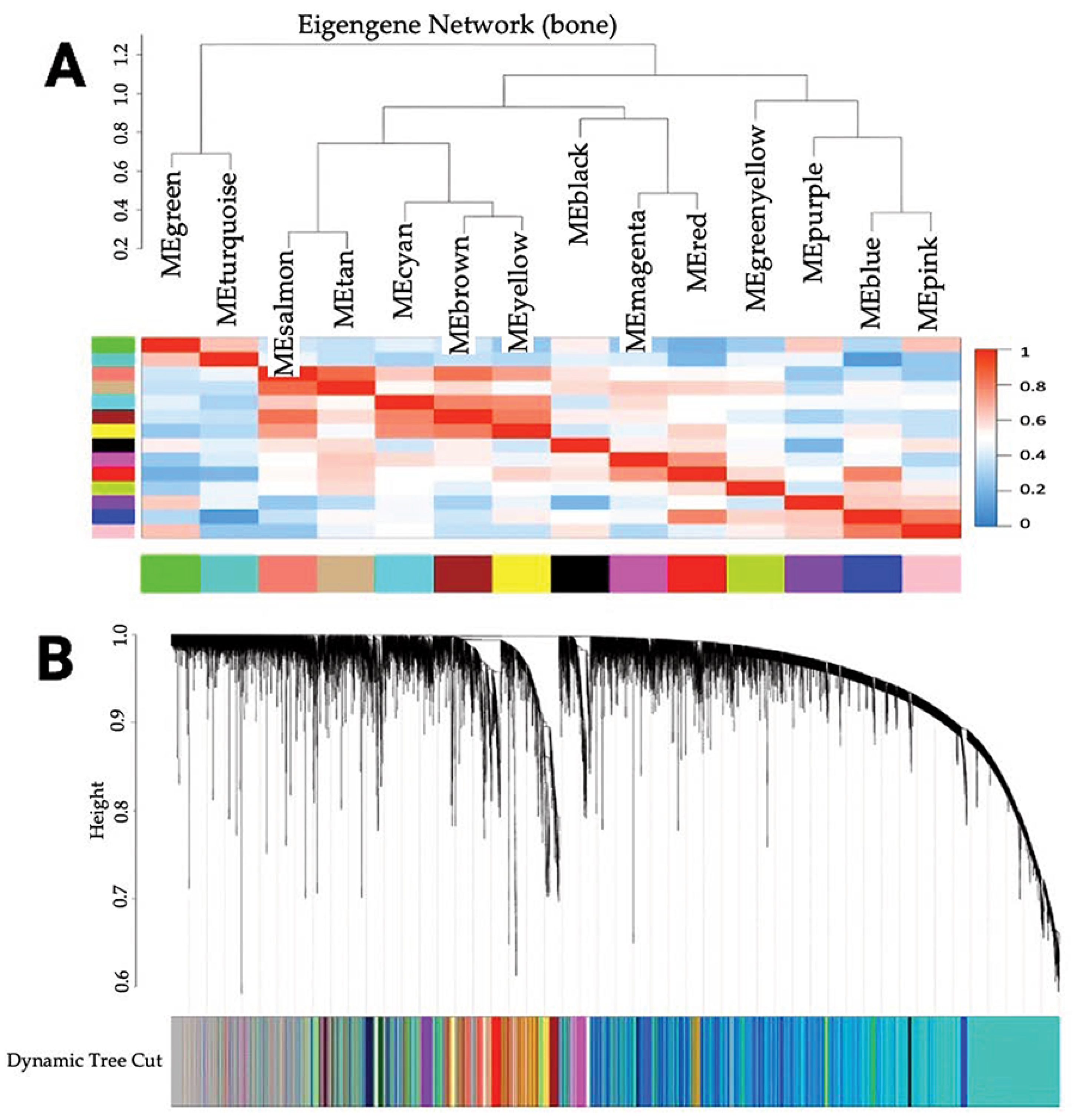
3.1. Identification of Co-Expressed Genes
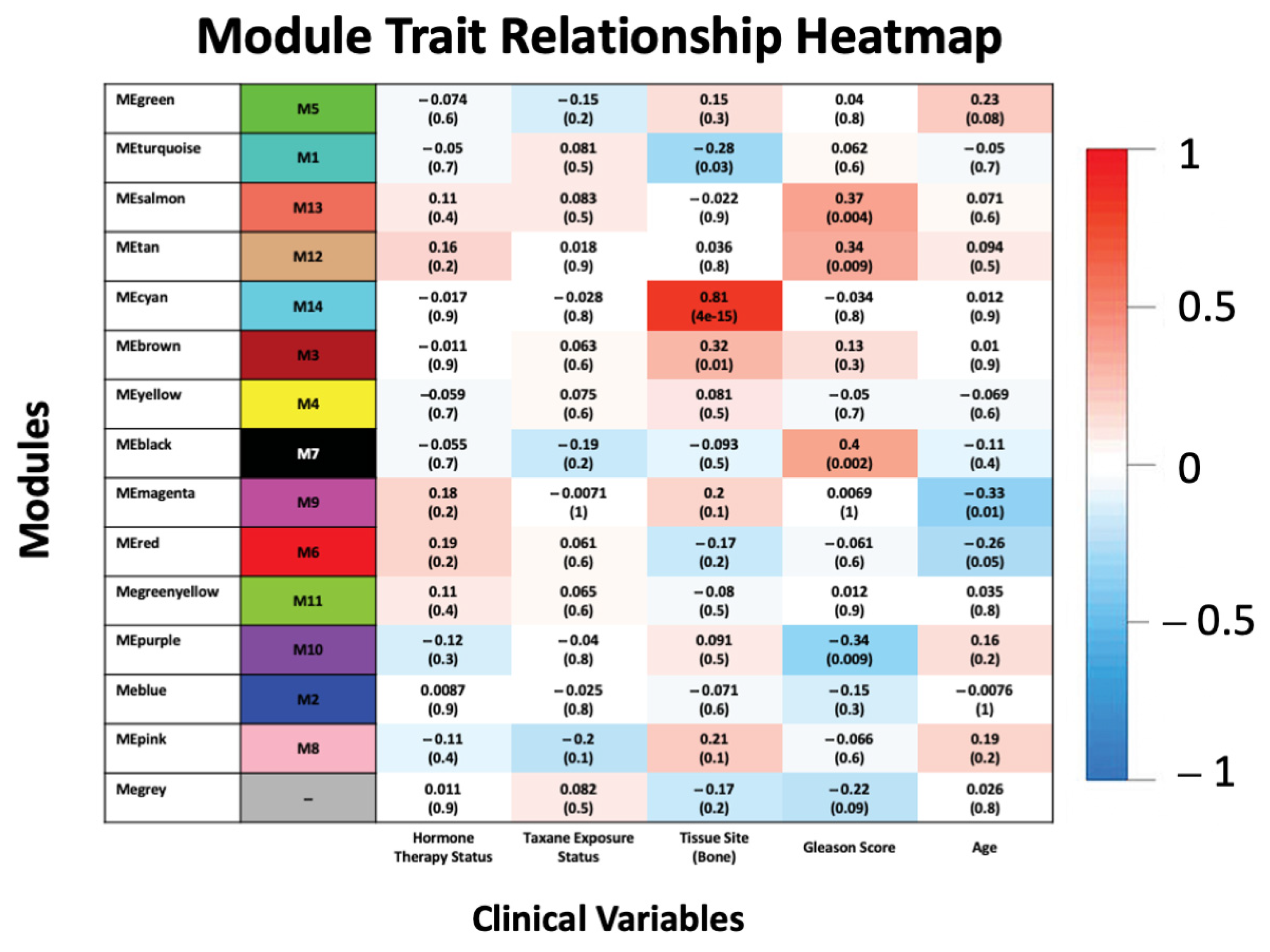
3.2. Association of Modules with Clinical Traits
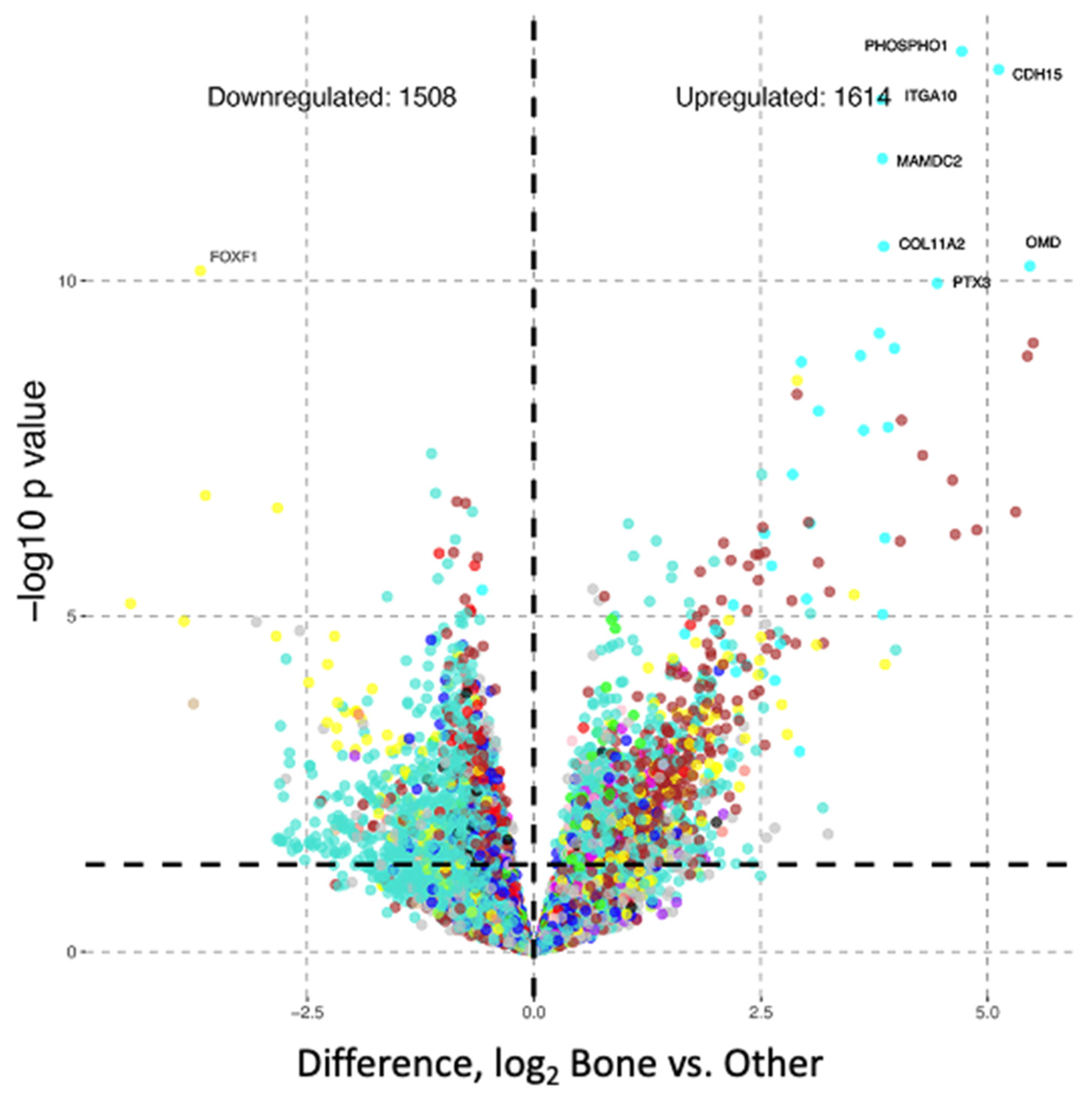
3.3. Differential Gene Expression Analysis
3.4. Enrichment Analysis of Biological Features
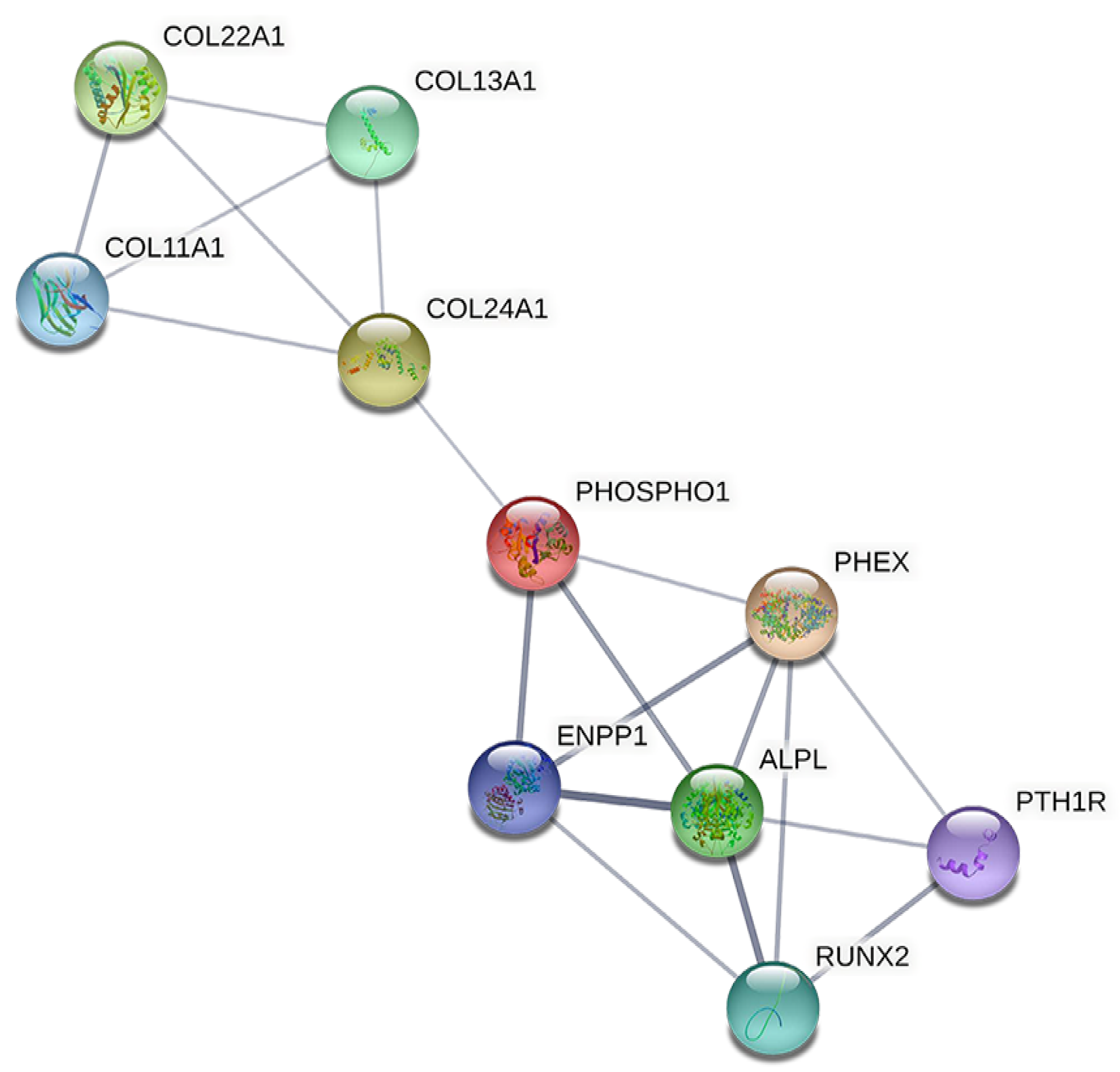
3.5. Identification of Hub Genes
3.6. Transcriptome Deconvolution and Tissue Gene Expression Analysis
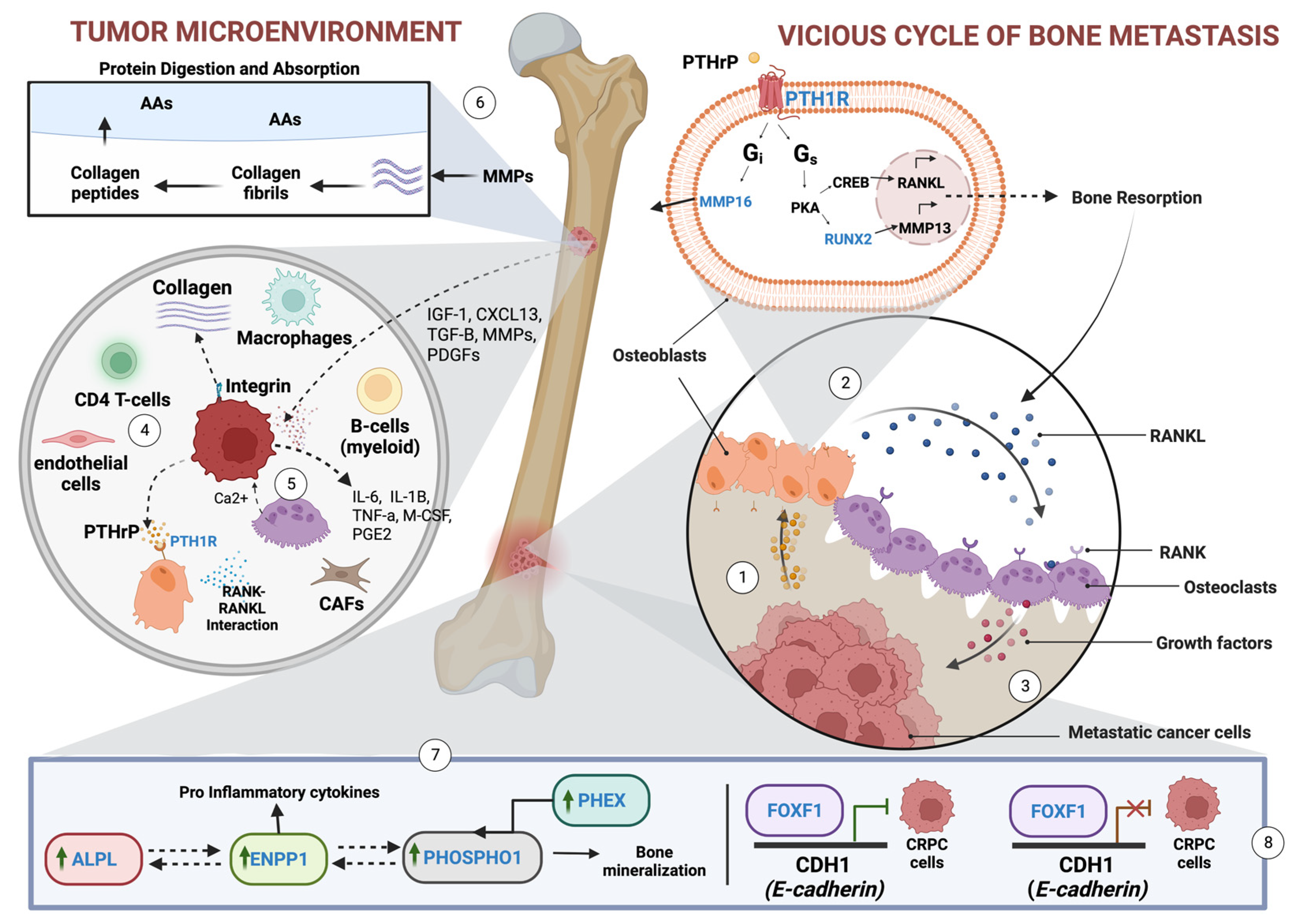
4. Discussion
4.1. Key Findings in the Study
4.2. Hub Genes Not Previously Associated with mCRPC
4.3. Hub Genes Previously Associated with mCRPC
4.3.1. ALPL and RUNX2
4.3.2. ENPP1
4.3.3. PTH1R
4.4. FOXF1
4.5. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Singh, S.K.; Lillard, J.W., Jr.; Singh, R. Molecular basis for prostate cancer racial disparities. Front. Biosci. 2017, 22, 428–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwan, E.M.; Wyatt, A.W. Androgen receptor genomic alterations and treatment resistance in metastatic prostate cancer. Prostate 2022, 82 (Suppl. S1), S25–S36. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.-M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.-M.; Montgomery, B.; Taplin, M.-E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abida, W.; Cyrta, J.; Heller, G.; Prandi, D.; Armenia, J.; Coleman, I.; Cieslik, M.; Benelli, M.; Robinson, D.; Van Allen, E.M.; et al. Genomic correlates of clinical outcome in advanced prostate cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 11428–11436. [Google Scholar] [CrossRef] [Green Version]
- Ohandjo, A.Q.; Liu, Z.; Dammer, E.B.; Dill, C.D.; Griffen, T.L.; Carey, K.M.; Hinton, D.E.; Meller, R.; Lillard, J.W., Jr. Transcriptome Network Analysis Identifies CXCL13-CXCR5 Signaling Modules in the Prostate Tumor Immune Microenvironment. Sci. Rep. 2019, 9, 14963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- prad_su2c_2019. Available online: https://github.com/cBioPortal/datahub/tree/master/public/prad_su2c_2019 (accessed on 7 October 2022).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.; Smyth, G.K.; Shi, W. feature Counts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Horvath, S. A General Framework for Weighted Gene Co-Expression Network Analysis. Stat. Appl. Genet. Mol. Biol. 2005, 4, Article17. [Google Scholar] [CrossRef] [PubMed]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Wang, J.; Peng, X.; Liu, K.; Zhang, C.; Zeng, X.; Lai, Y. Comprehensive analysis of biomarkers for prostate cancer based on weighted gene co-expression network analysis. Medicine 2020, 99, e19628. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Tan, Q.; Kir, J.; Liu, D.; Bryant, D.; Guo, Y.; Stephens, R.; Baseler, M.W.; Lane, H.C.; et al. DAVID Bioinformatics Resources: Expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic Acids Res. 2007, 35, W169–W175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef]
- Otasek, D.; Morris, J.H.; Bouças, J.; Pico, A.R.; Demchak, B. Cytoscape Automation: Empowering workflow-based network analysis. Genome Biol. 2019, 20, 185. [Google Scholar] [CrossRef] [Green Version]
- Chin, C.-H.; Chen, S.-H.; Wu, H.-H.; Ho, C.-W.; Ko, M.-T.; Lin, C.-Y. cytoHubba: Identifying hub objects and sub-networks from complex interactome. BMC Syst. Biol. 2014, 8 (Suppl. S4), S11. [Google Scholar] [CrossRef] [Green Version]
- Li, C.Y.; Cai, J.-H.; Tsai, J.J.P.; Wang, C.C.-N. Identification of Hub Genes Associated with Development of Head and Neck Squamous Cell Carcinoma by Integrated Bioinformatics Analysis. Front. Oncol. 2020, 10, 681. [Google Scholar] [CrossRef]
- Ge, C.; Zhao, G.; Li, Y.; Li, H.; Zhao, X.; Pannone, G.; Bufo, P.; Santoro, A.; Sanguedolce, F.; Tortorella, S.; et al. Role of Runx2 phosphorylation in prostate cancer and association with metastatic disease. Oncogene 2016, 35, 366–376. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Xu, H.; Wang, W.; Li, S.; Li, H.; Li, T.; Zhang, W.; Yu, X.; Liu, L. The role of collagen in cancer: From bench to bedside. J. Transl. Med. 2019, 17, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Yang, H.; Chai, J.; Xing, L. RUNX2 as a promising therapeutic target for malignant tumors. Cancer Manag. Res. 2021, 13, 2539–2548. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.R.; Snaith, A.E.; Marino, D.; Cheng, X.; Lwin, S.T.; Orriss, I.R.; Hamdy, F.C.; Edwards, C.M. Tumour-derived alkaline phosphatase regulates tumour growth, epithelial plasticity and disease-free survival in metastatic prostate cancer. Br. J. Cancer 2017, 116, 227–236. [Google Scholar] [CrossRef] [PubMed]
- Onyedibe, K.I.; Wang, M.; Sintim, H.O. ENPP1, an Old Enzyme with New Functions, and Small Molecule Inhibitors—A STING in the Tale of ENPP1. Molecules 2019, 24, 4192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberts, F.; Zhu, D.; Farquharson, C.; Macrae, V.E. ENPP1 in the Regulation of Mineralization and Beyond. Trends Biochem. Sci. 2019, 44, 616–628. [Google Scholar] [CrossRef]
- Li, J.; Karaplis, A.C.; Huang, D.C.; Siegel, P.M.; Camirand, A.; Yang, X.F.; Muller, W.J.; Kremer, R. PTHrP drives breast tumor initiation, progression, and metastasis in mice and is a potential therapy target. J. Clin. Investig. 2011, 121, 4655–4669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Wang, B. PTH1R-CaSR Cross Talk: New Treatment Options for Breast Cancer Osteolytic Bone Metastases. Int. J. Endocrinol. 2018, 2018, 7120979. [Google Scholar] [CrossRef]
- Bergwitz, C.; Jüppner, H. Regulation of Phosphate Homeostasis by PTH, Vitamin D, and FGF23. Annu. Rev. Med. 2010, 61, 91–104. [Google Scholar] [CrossRef] [Green Version]
- Morcos, M.W.; Al-Jallad, H.; Li, J.; Farquharson, C.; Millan, J.L.; Hamdy, R.C.; Murshed, M. PHOSPHO1 is essential for normal bone fracture healing: An Animal Study. Bone Joint Res. 2018, 7, 397–405. [Google Scholar] [CrossRef]
- Dillon, S.; Staines, K.A.; Millán, J.L.; Farquharson, C. How to Build a Bone: PHOSPHO1, Biomineralization, and Beyond. JBMR Plus 2019, 3, e10202. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Fu, Y. Comprehensive Analysis and Identification of an Immune-Related Gene Signature with Prognostic Value for Prostate Cancer. Int. J. Gen. Med. 2021, 14, 2931–2942. [Google Scholar] [CrossRef]
- Renema, N.; Navet, B.; Heymann, M.F.; Lezot, F.; Heymann, D. RANK-RANKL signalling in cancer. Biosci. Rep. 2016, 36, e00366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Thulin, M.H.; Damber, J.-E.; Welén, K. The roles of RUNX2 and osteoclasts in regulating expression of steroidogenic enzymes in castration-resistant prostate cancer cells. Mol. Cell. Endocrinol. 2021, 535, 111380. [Google Scholar] [CrossRef]
- Kim, B.; Kim, H.; Jung, S.; Moon, A.; Noh, D.Y.; Lee, Z.H.; Kim, H.J.; Kim, H.H. A CTGF-RUNX2-RANKL Axis in Breast and Prostate Cancer Cells Promotes Tumor Progression in Bone. J. Bone Miner. Res. 2020, 35, 155–166. [Google Scholar] [CrossRef]
- Ashe, H.; Krakowiak, P.; Hasterok, S.; Sleppy, R.; Roller, D.G.; Gioeli, D. Role of the runt-related transcription factor (RUNX) family in prostate cancer. FEBS J. 2021, 288, 6112–6126. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X. Interactions between cancer cells and bone microenvironment promote bone metastasis in prostate cancer. Cancer Commun. 2019, 39, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Duran, M.A.; Dhanota, N.; Chatila, W.K.; Bettigole, S.E.; Kwon, J.; Sriram, R.K.; Humphries, M.P.; Salto-Tellez, M.; James, J.A.; et al. Metastasis and Immune Evasion from Extracellular cGAMP Hydrolysis. Cancer Discov. 2021, 11, 1212–1220. [Google Scholar] [CrossRef]
- Gao, Z.-G.; Jacobson, K.A. A2B Adenosine Receptor and Cancer. Int. J. Mol. Sci. 2019, 20, 5139. [Google Scholar] [CrossRef] [Green Version]
- Mousavi, S.; Panjehpour, M.; Izadpanahi, M.H.; Aghaei, M. Expression of adenosine receptor subclasses in malignant and adjacent normal human prostate tissues. Prostate 2015, 75, 735–747. [Google Scholar] [CrossRef]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef]
- Tamura, M.; Sasaki, Y.; Koyama, R.; Takeda, K.; Idogawa, M.; Tokino, T. Forkhead transcription factor FOXF1 is a novel target gene of the p53 family and regulates cancer cell migration and invasiveness. Oncogene 2014, 33, 4837–4846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stand Up to Cancer East Coast Prostate Cancer Research Group. Available online: https://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000915.v2.p2 (accessed on 1 May 2020).
- Tryka, K.A.; Hao, L.; Sturcke, A.; Jin, Y.; Wang, Z.Y.; Ziyabari, L.; Lee, M.; Popova, N.; Sharopova, N.; Kimura, M.; et al. NCBI’s Database of Genotypes and Phenotypes: dbGaP. Nucleic Acids Res. 2014, 42, D975–D979. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Variable | Total Number | |
|---|---|---|
| Total | 60 | |
| Age | Median (range) | 65 (50–85) |
| UNK | 8 | |
| Tissue Site | ||
| Bone | 15 | |
| Lymph Node | 34 | |
| Liver | 11 | |
| Abiraterone and Enzalutamide Exposure (Hormone Therapy) Status | ||
| Naïve | 31 | |
| Exposed | 25 | |
| UNK | 4 | |
| Taxane Exposure Status | ||
| Naïve | 37 | |
| Exposed | 21 | |
| UNK | 2 | |
| Gleason Score | ||
| 6 | 3 | |
| 7 | 8 | |
| 8 | 8 | |
| 9 | 16 | |
| 10 | 5 | |
| UNK | 20 |
| Pathway | Genes | % | p-Value | Benjamini–Hochberg Value |
|---|---|---|---|---|
| Protein digestion and absorption | COL11A1 COL13A1 COL22A1 COL24A1 | 10.8 | 2.2 × 10−3 | 1.3 × 10−1 |
| Parathyroid hormone synthesis, secretion, and action | RUNX2 MMP16 PTH1R | 8.1 | 8.6 × 10−1 | 8.6 × 10−1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McKinney, L.P.; Singh, R.; Jordan, I.K.; Varambally, S.; Dammer, E.B.; Lillard, J.W., Jr. Transcriptome Analysis Identifies Tumor Immune Microenvironment Signaling Networks Supporting Metastatic Castration-Resistant Prostate Cancer. Onco 2023, 3, 81-95. https://doi.org/10.3390/onco3020007
McKinney LP, Singh R, Jordan IK, Varambally S, Dammer EB, Lillard JW Jr. Transcriptome Analysis Identifies Tumor Immune Microenvironment Signaling Networks Supporting Metastatic Castration-Resistant Prostate Cancer. Onco. 2023; 3(2):81-95. https://doi.org/10.3390/onco3020007
Chicago/Turabian StyleMcKinney, Lawrence P., Rajesh Singh, I. King Jordan, Sooryanarayana Varambally, Eric B. Dammer, and James W. Lillard, Jr. 2023. "Transcriptome Analysis Identifies Tumor Immune Microenvironment Signaling Networks Supporting Metastatic Castration-Resistant Prostate Cancer" Onco 3, no. 2: 81-95. https://doi.org/10.3390/onco3020007