
AlphaFold2 Update and Perspectives
 and
and 
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Foreword
2. Introduction
2.1. Proteins and 3D Structures
2.2. Protein Structure Prediction
2.3. Recent Protein Structure Prediction Methods
3. Deep Learning in Structural Bioinformatics
4. Current Identified AlphaFold2 Limits
4.1. Introduction
4.2. Law and Order
4.3. Few Pertinent Quantifications
- (i)
- The first work, coming directly from the EBI with the help of DeepMind, made available structural models of 220 million sequences (including proteomes of interest, UniProt, and other sequences) [46,47]. With the analysis of the pLDDT, they clearly show that a third of the human proteome is of atomistic quality; 58% corresponds to a correct fold, and there is no clear information on remaining 42%. This tendency is also found on several of the other available proteomes. Of course, this result may be partly due to disordered areas, but they are considered in smaller numbers. Clearly, a significant number of transmembrane protein structures are not correctly predicted [56]. It was recently confirmed with the analysis of nearly 700,000 domains provided by AF2 that only 52% of models were appropriate for analysis with the new CATH-annotation tool [57].
- (ii)
- The second work came from a large academic consortium that has independently evaluated the advancements provided by this methodology compared to a recognized comparative modelling tool, namely SwissModel [19], with its own repository [58]. They selected 21 model species, corresponding to more than 365,000 proteins, i.e., twice the number of experimental structures and six times the number of unique proteins in PDB. They analysed the SwissModel repository for 11 model species and compared it with the AF2 database. On average, the predicted models of AF2 provide longer predictions (+44% of residues). Looking at high-quality regions (pLDDT > 90), an average of around 25% of the residues of the proteomes of the 11 model species are covered by AF2 with novel (not present in SwissModel repository) and confident predictions [59]. This very elegant and rigorous study also shows, similar to the previous analysis, that a large number of proteins are still not reachable. The surprise for non-specialists is that they are not only transmembrane proteins but also globular ones.
- (iii)
- The third study comprises the analysis of the local conformation of proteins and shows that, globally, the results are very good. However, in a surprising way, some local conformations observed in a recurrent way within all the proteins are, in a way, systematically associated with particularly low confidence scores [60]. These conformations are PolyProline II helices (important for protein–proline interaction) [61], γ-turns (present in many loops), and ω angles in cis conformation (often associated with Proline) [62]. This analysis also shows that there would be an under-representation of sheets and beta compared to what should be observed. In addition, those β-like forms are present in large numbers and would only ask to be able to form sheets.
- (iv)
- The fourth study focuses on the position of the side chains, a complex subject due to their large panel of motions. The quality of the predictions at this level is still largely perfectible [63].
- (a)
- An undeniable strength of AlphaFold 2 compared to its previous version is that it is made available in the form of a usable and stable GitHub, which does not require overly expensive and powerful computers. In addition, several academic groups provide their own AlphaFold system, called CollabFold, which can be used free of charge by the scientific community, but with a less rich database of protein structures compared to the real AlphaFold.
- (b)
- AlphaFold2 can quickly model more protein than previous approaches and, on average, with better quality. However, the attainable/usable protein number is lower than we would have expected from the assertion that “a 50-year-old problem was solved”.
- (c)
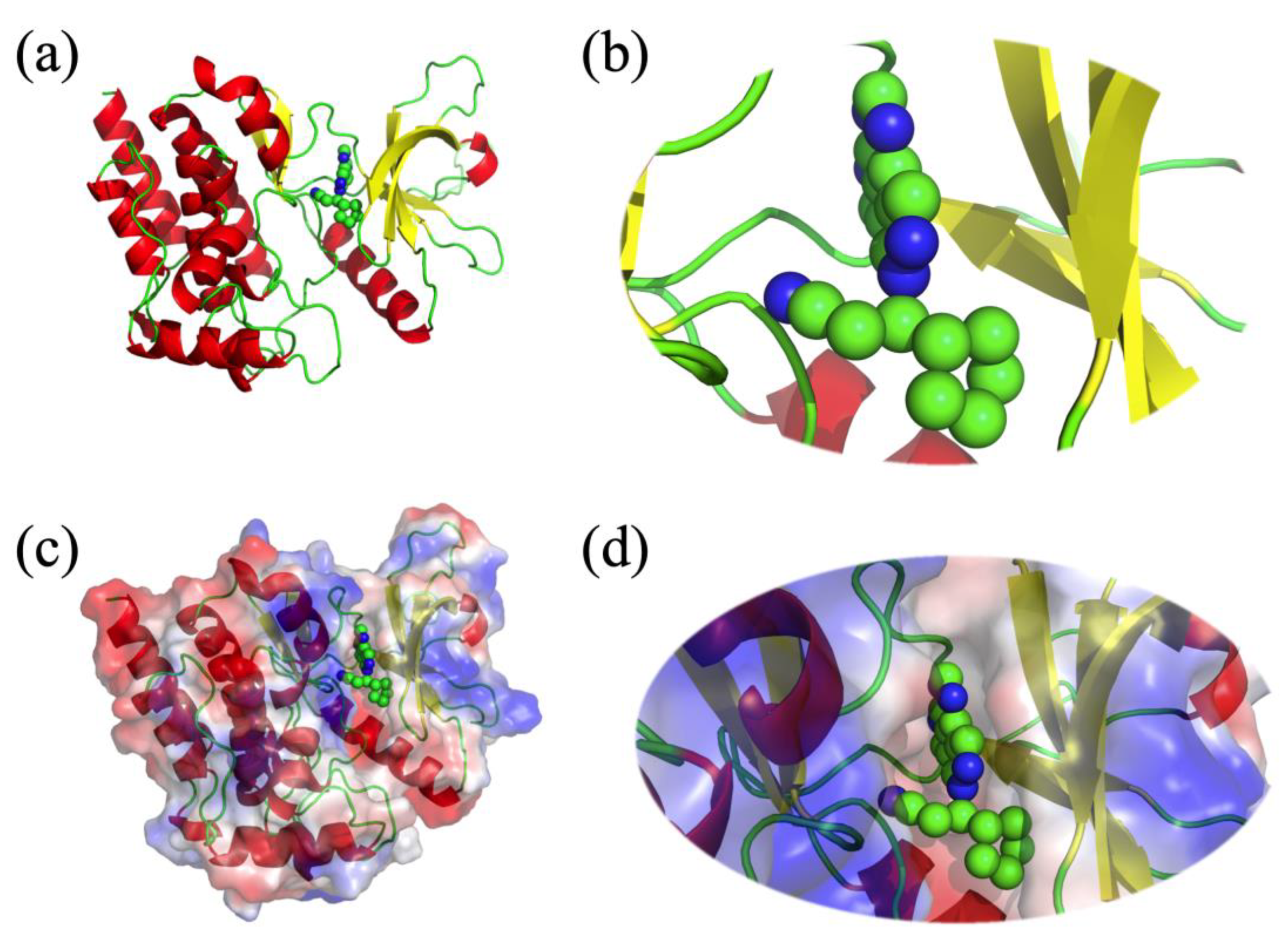
- One AF2 limitation that strongly affects the biomedical field is the poor quality of transmembrane protein models, whereas the confidence indices of transmembrane segments can be of good quality. The overall predicted topology is not compatible with its insertion within a membrane bilayer. Figure 2 shows the AF2 model proposed for Atypical Chemokine Receptor 1 (ACKR1), previously named Duffy Antigen for Chemokine. ACKR1 is a seven TM protein associated with malarial Plasmodium vivax infection [64,65]. This model is incomplete, but segments are present. However, it is not possible to insert it in a membrane bilayer, e.g., with CHARMM-GUI [66]. Indeed, its topology does not allow any recognition of this portion as transmembrane by the webserver.
- (d)
- Another limitation is that most proteins have ions and co-factors (such as FAD and NADPH); however, AlphaFold 2 has not been trained to take them into account. In addition, in a certain number of cases, it is impossible to add them or to dock them. Thus, an external tool, called AlphaFill, has been dedicated to place them and add post-translational modifications such as glycosylation (often essential for the protein functions) on the models [68].
- (e)
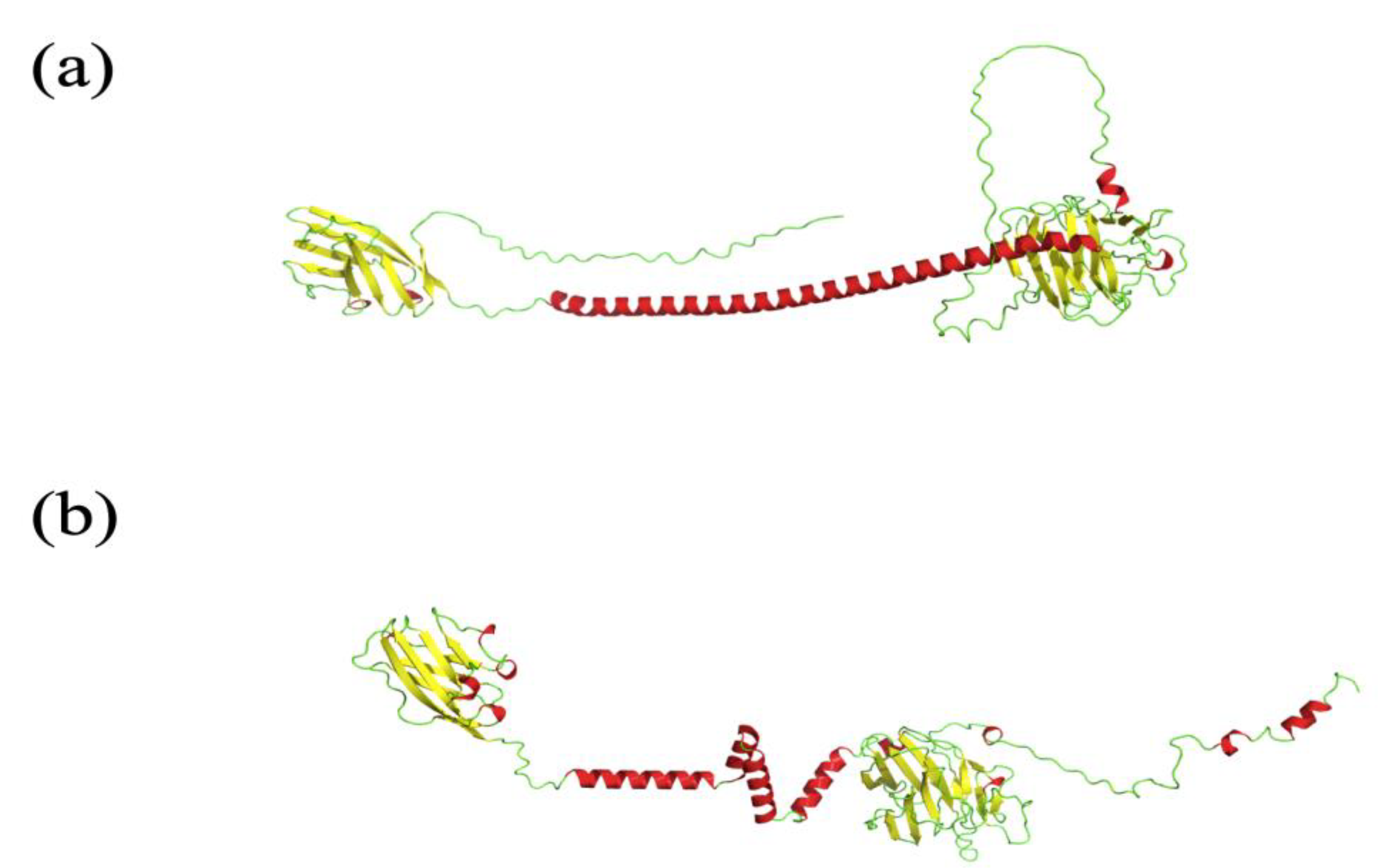
- As noticed, AlphaFold2 is also pertinent for underlying intrinsic disordered regions (IDRs). Nonetheless, sometimes AF2 provides protein models with regions that look like IDRs but are in reality not disordered. Figure 3 presents the E3 ubiquitin-protein ligase PPP1R11 (UniProt ID O60927) AF2 model underlined by Thornton and collaborators in [69]. This model is considered as a poor-quality model because this protein is a globular one, and the model presents what looks like a disordered protein.
5. Perspectives
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Service, R. Breakthrough of the Year—Protein Structures for All. Science, 16 December 2021. Available online: https://www.science.org/content/article/breakthrough-2021 (accessed on 15 March 2023).
- Knapp, A. 2023 Breakthrough Prizes Announced: Deepmind’s Protein Folders Awarded $3 Million. Forbes, 22 September 2022. Available online: https://www.forbes.com/sites/alexknapp/2022/09/22/2023-breakthrough-prizes-announced-deepminds-protein-folders-awarded-3-million/ (accessed on 15 March 2023).
- Perrigo, B. Mapping Life—DeepMind AlphaFold. Time, 10 November 2022. Available online: https://time.com/collection/best-inventions-2022/6229912/deepmind-alphafold/ (accessed on 15 March 2023).
- Callaway, E. ‘It will change everything’: DeepMind’s AI makes gigantic leap in solving protein structures. Nature 2020, 588, 203–204. [Google Scholar] [CrossRef]
- Sample, I. DeepMind AI Cracks 50-Year-Old Problem of Protein Folding. Guardian 2020. Available online: https://www.theguardian.com/technology/2020/nov/2030/deepmind-ai-cracks-2050-year-old-problem-of-biology-research (accessed on 15 March 2023).
- Rabbani, G.; Baig, M.H.; Ahmad, K.; Choi, I. Protein-protein Interactions and their Role in Various Diseases and their Prediction Techniques. Curr. Protein Pept. Sci. 2018, 19, 948–957. [Google Scholar] [CrossRef]
- Davis, R.R.; Li, B.; Yun, S.Y.; Chan, A.; Nareddy, P.; Gunawan, S.; Ayaz, M.; Lawrence, H.R.; Reuther, G.W.; Lawrence, N.J.; et al. Structural Insights into jak2 Inhibition by Ruxolitinib, Fedratinib, and Derivatives Thereof. J. Med. Chem. 2021, 64, 2228–2241. [Google Scholar] [CrossRef]
- McLornan, D.P.; Pope, J.E.; Gotlib, J.; Harrison, C.N. Current and future status of jak inhibitors. Lancet 2021, 398, 803–816. [Google Scholar] [CrossRef]
- Delano, W.L. The Pymol Molecular Graphics System on World Wide Web. 2013. Available online: http://www.pymol.org (accessed on 15 March 2023).
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The Protein Data Bank. Acta Cryst. D Biol. Cryst. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer, E.F., Jr.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The Protein Data Bank: A computer-based archival file for macromolecular structures. J. Mol. Biol. 1977, 112, 535–542. [Google Scholar] [CrossRef]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Yeh, L.S. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2023, 51, D523–D531. [Google Scholar]
- Thakur, M.; Bateman, A.; Brooksbank, C.; Freeberg, M.; Harrison, M.; Hartley, M.; Keane, T.; Kleywegt, G.; Leach, A.; Levchenko, M.; et al. Embl’s European Bioinformatics Institute (embl-ebi) in 2022. Nucleic Acids Res. 2023, 51, D9–D17. [Google Scholar] [CrossRef]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef]
- Pieper, U.; Eswar, N.; Stuart, A.C.; Ilyin, V.A.; Sali, A. Modbase, a database of annotated comparative protein structure models. Nucleic Acids Res. 2002, 30, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Melo, F.; Sali, A. Fold assessment for comparative protein structure modeling. Protein Sci. 2007, 16, 2412–2426. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using modeller. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. Swiss-model: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [PubMed]
- Schwede, T.; Kopp, J.; Guex, N.; Peitsch, M.C. Swiss-model: An automated protein homology-modeling server. Nucleic Acids Res. 2003, 31, 3381–3385. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef]
- Bystroff, C.; Shao, Y. Fully automated ab initio protein structure prediction using i-sites, hmmstr and rosetta. Bioinformatics 2002, 18 (Suppl. S1), S54–S61. [Google Scholar] [CrossRef]
- Kosinski, J.; Cymerman, I.A.; Feder, M.; Kurowski, M.A.; Sasin, J.M.; Bujnicki, J.M. A “FRankenstein’s monster” approach to comparative modeling: Merging the finest fragments of Fold-Recognition models and iterative model refinement aided by 3D structure evaluation. Proteins 2003, 53 (Suppl. S6), 369–379. [Google Scholar] [CrossRef]
- Baker, D.; Sali, A. Protein structure prediction and structural genomics. Science 2001, 294, 93–96. [Google Scholar] [CrossRef]
- Roy, A.; Kucukural, A.; Zhang, Y. I-tasser: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef]
- Zhang, Y. Interplay of I-tasser and quark for template-based and ab initio protein structure prediction in casp10. Proteins 2014, 82 (Suppl. S2), 175–187. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The i-tasser Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [PubMed]
- Bradley, P.; Malmström, L.; Qian, B.; Schonbrun, J.; Chivian, D.; Kim, D.E.; Meiler, J.; Misura, K.M.; Baker, D. Free modeling with Rosetta in casp6. Proteins 2005, 61 (Suppl. S7), 128–134. [Google Scholar] [CrossRef] [PubMed]
- Kinch, L.N.; Li, W.; Monastyrskyy, B.; Kryshtafovych, A.; Grishin, N.V. Evaluation of free modeling targets in casp11 and roll. Proteins 2016, 84 (Suppl. S1), 51–66. [Google Scholar] [CrossRef]
- Leman, J.K.; Weitzner, B.D.; Lewis, S.M.; Adolf-Bryfogle, J.; Alam, N.; Alford, R.F.; Aprahamian, M.; Baker, D.; Barlow, K.A.; Barth, P.; et al. Macromolecular modeling and design in Rosetta: Recent methods and frameworks. Nat. Methods 2020, 17, 665–680. [Google Scholar] [CrossRef]
- AlQuraishi, M. AlphaFold at casp13. Bioinformatics 2019, 35, 4862–4865. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Protein structure prediction using multiple deep neural networks in the 13th Critical Assessment of Protein Structure Prediction (casp13). Proteins 2019, 87, 1141–1148. [Google Scholar] [CrossRef]
- Senior, A.W.; Evans, R.; Jumper, J.; Kirkpatrick, J.; Sifre, L.; Green, T.; Qin, C.; Žídek, A.; Nelson, A.W.R.; Bridgland, A.; et al. Improved protein structure prediction using potentials from deep learning. Nature 2020, 577, 706–710. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Applying and improving AlphaFold at casp14. Proteins 2021, 89, 1711–1721. [Google Scholar] [CrossRef]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic level protein structure with a language model. bioRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Mosimann, S.; Meleshko, R.; James, M.N. A critical assessment of comparative molecular modeling of tertiary structures of proteins. Proteins 1995, 23, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Jumper, J.; Hassabis, D. Protein structure predictions to atomic accuracy with AlphaFold. Nat. Methods 2022, 19, 11–12. [Google Scholar] [CrossRef] [PubMed]
- Marcu, Ş.B.; Tăbîrcă, S.; Tangney, M. An Overview of Alphafold’s Breakthrough. Front. Artif. Intell. 2022, 5, 875587. [Google Scholar] [CrossRef] [PubMed]
- Fersht, A.R. AlphaFold—A Personal Perspective on the Impact of Machine Learning. J. Mol. Biol. 2021, 433, 167088. [Google Scholar] [CrossRef]
- Skolnick, J.; Gao, M.; Zhou, H.; Singh, S. AlphaFold 2: Why It Works and Its Implications for Understanding the Relationships of Protein Sequence, Structure, and Function. J. Chem. Inf. Model. 2021, 61, 4827–4831. [Google Scholar] [CrossRef]
- Method of the Year 2021: Protein structure prediction. Nat. Methods 2022, 19, 1. [CrossRef]
- Evans, R.; O’Neill, M.; Pritzel, A.; Antropova, N.; Senior, A.; Green, T.; Žídek, A.; Bates, R.; Blackwell, S.; Yim, J.; et al. Protein complex prediction with AlphaFold-Multimer. bioRxiv 2021. [Google Scholar] [CrossRef]
- Mirdita, M.; Schütze, K.; Moriwaki, Y.; Heo, L.; Ovchinnikov, S.; Steinegger, M. ColabFold—Making protein folding accessible to all. Nat. Methods 2022, 19, 679–682. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.; Adler, J.; Wu, Z.; Green, T.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Du, Z.; Su, H.; Wang, W.; Ye, L.; Wei, H.; Peng, Z.; Anishchenko, I.; Baker, D.; Yang, J. The trRosetta server for fast and accurate protein structure prediction. Nat. Protoc. 2021, 16, 5634–5651. [Google Scholar] [CrossRef]
- Al-Janabi, A. Has DeepMind’s AlphaFold solved the protein folding problem? BioTechniques 2022, 72, 73–76. [Google Scholar] [CrossRef] [PubMed]
- DeForte, S.; Uversky, V.N. Order, Disorder, and Everything in Between. Molecules 2016, 21, 1090. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Turzo, S.B.A.; Seffernick, J.T.; Kim, S.S.; Lindert, S. Prediction of Intrinsic Disorder Using Rosetta ResidueDisorder and AlphaFold2. J. Phys. Chem. B 2022, 126, 8439–8446. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.; Li, D.W.; Brüschweiler, R. Predicting protein flexibility with AlphaFold. Proteins 2023, 91, 847–855. [Google Scholar] [CrossRef]
- Piovesan, D.; Monzon, A.M.; Tosatto, S.C.E. Intrinsic protein disorder and conditional folding in AlphaFoldDB. Protein Sci. 2022, 31, e4466. [Google Scholar] [CrossRef]
- Bruley, A.; Bitard-Feildel, T.; Callebaut, I.; Duprat, E. A sequence-based foldability score combined with AlphaFold2 predictions to disentangle the protein order/disorder continuum. Proteins 2023, 91, 466–484. [Google Scholar] [CrossRef]
- Bruley, A.; Mornon, J.P.; Duprat, E.; Callebaut, I. Digging into the 3D Structure Predictions of AlphaFold2 with Low Confidence: Disorder and Beyond. Biomolecules 2022, 12, 1467. [Google Scholar] [CrossRef]
- Azzaz, F.; Yahi, N.; Chahinian, H.; Fantini, J. The Epigenetic Dimension of Protein Structure Is an Intrinsic Weakness of the AlphaFold Program. Biomolecules 2022, 12, 1527. [Google Scholar] [CrossRef] [PubMed]
- Bordin, N.; Sillitoe, I.; Nallapareddy, V.; Rauer, C.; Lam, S.D.; Waman, V.P.; Sen, N.; Heinzinger, M.; Littmann, M.; Kim, S.; et al. AlphaFold2 reveals commonalities and novelties in protein structure space for 21 model organisms. Commun. Biol. 2023, 6, 160. [Google Scholar] [CrossRef] [PubMed]
- Bienert, S.; Waterhouse, A.; de Beer, T.A.; Tauriello, G.; Studer, G.; Bordoli, L.; Schwede, T. The swiss-model Repository-new features and functionality. Nucleic Acids Res. 2017, 45, D313–D319. [Google Scholar] [CrossRef] [PubMed]
- Akdel, M.; Pires, D.E.V.; Pardo, E.P.; Jänes, J.; Zalevsky, A.O.; Mészáros, B.; Bryant, P.; Good, L.L.; Laskowski, R.A.; Pozzati, G.; et al. A structural biology community assessment of AlphaFold2 applications. Nat. Struct. Mol. Biol. 2022, 29, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- de Brevern, A.G. An agnostic analysis of the human AlphaFold2 proteome using local protein conformations. Biochimie 2022, 207, 11–19. [Google Scholar] [CrossRef]
- Narwani, T.J.; Santuz, H.; Shinada, N.; Melarkode Vattekatte, A.; Ghouzam, Y.; Srinivasan, N.; Gelly, J.C.; de Brevern, A.G. Recent advances on polyproline II. Amino Acids 2017, 49, 705–713. [Google Scholar] [CrossRef]
- Craveur, P.; Joseph, A.P.; Poulain, P.; de Brevern, A.G.; Rebehmed, J. Cis-trans isomerization of omega dihedrals in proteins. Amino Acids 2013, 45, 279–289. [Google Scholar] [CrossRef]
- Shiono, D.; Yoshidome, T. AlphaFold-predicted Protein Structure vs Experimentally Obtained Protein Structure: An Emphasis on the Side Chains. J. Phys. Soc. Jpn. 2022, 91, 064804. [Google Scholar] [CrossRef]
- Horuk, R. The Duffy Antigen Receptor for Chemokines darc/ackr1. Front. Immunol. 2015, 6, 279. [Google Scholar] [CrossRef]
- de Brevern, A.G.; Wong, H.; Tournamille, C.; Colin, Y.; Le Van Kim, C.; Etchebest, C. A structural model of a seven-transmembrane helix receptor: The Duffy antigen/receptor for chemokine (darc). Biochim. Biophys. Acta 2005, 1724, 288–306. [Google Scholar] [CrossRef]
- Jo, S.; Cheng, X.; Lee, J.; Kim, S.; Park, S.J.; Patel, D.S.; Beaven, A.H.; Lee, K.I.; Rui, H.; Park, S.; et al. Charmm-gui 10 years for biomolecular modeling and simulation. J. Comput. Chem. 2017, 38, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Yeh, L.S. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar]
- Hekkelman, M.L.; de Vries, I.; Joosten, R.P.; Perrakis, A. AlphaFill: Enriching AlphaFold models with ligands and cofactors. Nat. Methods 2023, 20, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Thornton, J.M.; Laskowski, R.A.; Borkakoti, N. AlphaFold heralds a data-driven revolution in biology and medicine. Nat. Med. 2021, 27, 1666–1669. [Google Scholar] [CrossRef] [PubMed]
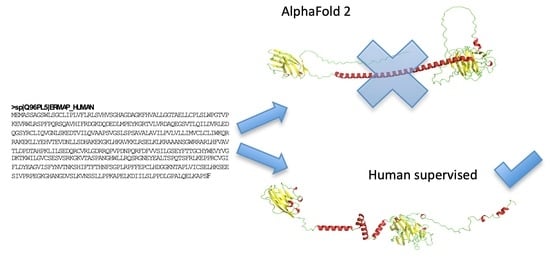
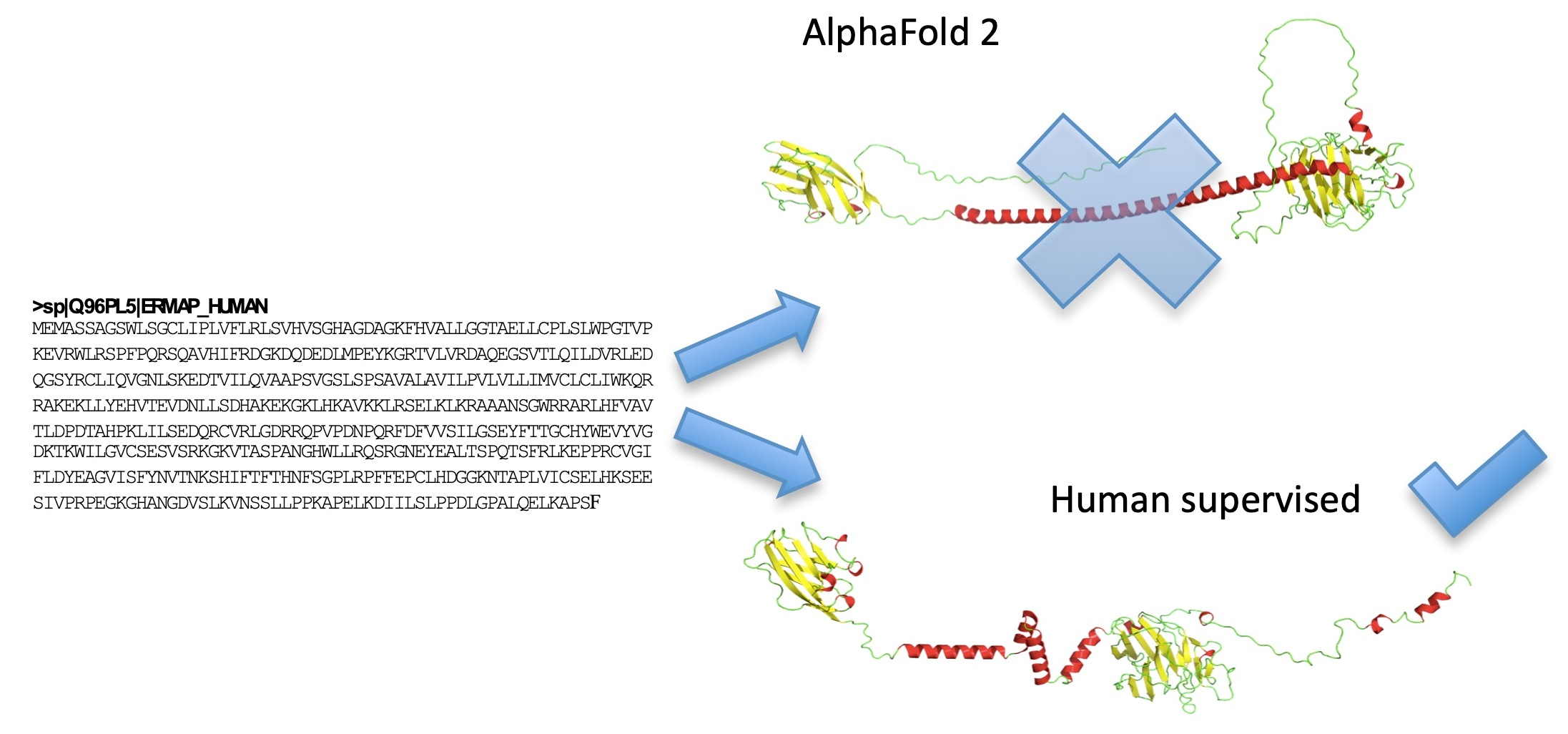
- Floch, A.; Lomas-Francis, C.; Vege, S.; Burgos, A.; Hoffman, R.; Cusick, R.; de Brevern, A.G.; Westhoff, C.M. Two new Scianna variants causing loss of high prevalence antigens: Ermap model and 3D analysis of the antigens. Transfusion 2023, 63, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Diwan, G.D.; Gonzalez-Sanchez, J.C.; Apic, G.; Russell, R.B. Next Generation Protein Structure Predictions and Genetic Variant Interpretation. J. Mol. Biol. 2021, 433, 167180. [Google Scholar] [CrossRef] [PubMed]
- McBride, J.M.; Polev, K.; Reinharz, V.; Grzybowski, B.A.; Tlusty, T. AlphaFold2 can predict single-mutation effects on structure and phenotype. arXiv 2022, arXiv:2022.04.14.488301. [Google Scholar]
- Terwilliger, T.C.; Liebschner, D.; Croll, T.I.; Williams, C.J.; McCoy, A.J.; Poon, B.K.; Afonine, P.V.; Oeffner, R.D.; Richardson, J.S.; Read, R.J.; et al. AlphaFold predictions: Great hypotheses but no match for experiment. bioRxiv 2022. [Google Scholar] [CrossRef]
- Pak, M.A.; Markhieva, K.A.; Novikova, M.S.; Petrov, D.S.; Vorobyev, I.S.; Maksimova, E.S.; Kondrashov, F.A.; Ivankov, D.N. Using AlphaFold to predict the impact of single mutations on protein stability and function. bioRxiv 2021. [Google Scholar] [CrossRef]
- Song, B.; Luo, X.; Luo, X.; Liu, Y.; Niu, Z.; Zeng, X. Learning spatial structures of proteins improves protein-protein interaction prediction. Brief. Bioinform. 2022, 23, bbab558. [Google Scholar] [CrossRef]
- Iqbal, S.; Ge, F.; Li, F.; Akutsu, T.; Zheng, Y.; Gasser, R.B.; Yu, D.J.; Webb, G.I.; Song, J. Prost: AlphaFold2-aware Sequence-Based Predictor to Estimate Protein Stability Changes upon Missense Mutations. J. Chem. Inf. Model. 2022, 62, 4270–4282. [Google Scholar] [CrossRef] [PubMed]
- Swapna, L.S.; Mahajan, S.; de Brevern, A.G.; Srinivasan, N. Comparison of tertiary structures of proteins in protein-protein complexes with unbound forms suggests prevalence of allostery in signalling proteins. BMC Struct. Biol. 2012, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Craveur, P.; Joseph, A.P.; Esque, J.; Narwani, T.J.; Noël, F.; Shinada, N.; Goguet, M.; Leonard, S.; Poulain, P.; Bertrand, O.; et al. Protein flexibility in the light of structural alphabets. Front. Mol. Biosci. 2015, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Narwani, T.J.; Craveur, P.; Shinada, N.K.; Floch, A.; Santuz, H.; Vattekatte, A.M.; Srinivasan, N.; Rebehmed, J.; Gelly, J.C.; Etchebest, C.; et al. Discrete analyses of protein dynamics. J. Biomol. Struct. Dyn. 2020, 38, 2988–3002. [Google Scholar] [CrossRef]
- Degiacomi, M.T. Coupling Molecular Dynamics and Deep Learning to Mine Protein Conformational Space. Structure 2019, 27, 1034–1040.e3. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Jiao, Y.; Shi, C.; Zhang, Y. Deep learning-based molecular dynamics simulation for structure-based drug design against SARS-CoV-2. Comput. Struct. Biotechnol. J. 2022, 20, 5014–5027. [Google Scholar] [CrossRef]
- Nussinov, R.; Zhang, M.; Liu, Y.; Jang, H. AlphaFold, Artificial Intelligence (AI), and Allostery. J. Phys. Chem. B 2022, 126, 6372–6383. [Google Scholar] [CrossRef]
- Chakravarty, D.; Porter, L.L. AlphaFold2 fails to predict protein fold switching. Protein Sci. 2022, 31, e4353. [Google Scholar] [CrossRef]
- Barbarin-Bocahu, I.; Graille, M. The X-ray crystallography phase problem solved thanks to AlphaFold and RoseTTAFold models: A case-study report. Acta Crystallogr. Sect. D Struct. Biol. 2022, 78, 517–531. [Google Scholar] [CrossRef]
- Bond, P.S.; Cowtan, K.D. ModelCraft: An advanced automated model-building pipeline using Buccaneer. Acta Crystallogr. Sect. D Struct. Biol. 2022, 78, 1090–1098. [Google Scholar] [CrossRef]
- Simpkin, A.J.; Thomas, J.M.H.; Keegan, R.M.; Rigden, D.J. MrParse: Finding homologues in the PDB and the EBI AlphaFold database for molecular replacement and more. Acta Crystallogr. Sect. D Struct. Biol. 2022, 78, 553–559. [Google Scholar] [CrossRef] [PubMed]
- de Brevern, A.G.; Floch, A.; Barrault, A.; Martret, J.; Bodivit, G.; Djoudi, R.; Pirenne, F.; Tournamille, C. Alloimmunization risk associated with amino acid 223 substitution in the RhD protein: Analysis in the light of molecular modeling. Transfusion 2018, 58, 2683–2692. [Google Scholar] [CrossRef] [PubMed]
- Floch, A.; Pirenne, F.; Barrault, A.; Chami, B.; Toly-Ndour, C.; Tournamille, C.; de Brevern, A.G. Insights into anti-D formation in carriers of Rhd variants through studies of 3D intraprotein interactions. Transfusion 2021, 61, 1286–1301. [Google Scholar] [CrossRef] [PubMed]
- Floch, A.; Lomas-Francis, C.; Vege, S.; Brennan, S.; Shakarian, G.; de Brevern, A.G.; Westhoff, C.M. A novel high-prevalence antigen in the Lutheran system, luga (lu24), and an updated, full-length 3D bcam model. Transfusion 2023, 63, 798–807. [Google Scholar] [CrossRef]
- Shao, C.; Bittrich, S.; Wang, S.; Burley, S.K. Assessing pdb macromolecular crystal structure confidence at the individual amino acid residue level. Structure 2022, 30, 1385–1394.e3. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tourlet, S.; Radjasandirane, R.; Diharce, J.; de Brevern, A.G. AlphaFold2 Update and Perspectives. BioMedInformatics 2023, 3, 378-390. https://doi.org/10.3390/biomedinformatics3020025
Tourlet S, Radjasandirane R, Diharce J, de Brevern AG. AlphaFold2 Update and Perspectives. BioMedInformatics. 2023; 3(2):378-390. https://doi.org/10.3390/biomedinformatics3020025
Chicago/Turabian StyleTourlet, Sébastien, Ragousandirane Radjasandirane, Julien Diharce, and Alexandre G. de Brevern. 2023. "AlphaFold2 Update and Perspectives" BioMedInformatics 3, no. 2: 378-390. https://doi.org/10.3390/biomedinformatics3020025