
Evaluation of Transmembrane Protein Structural Models Using HPMScore

 , ,
, , 
Abstract
:
1. Introduction
2. Materials and Methods
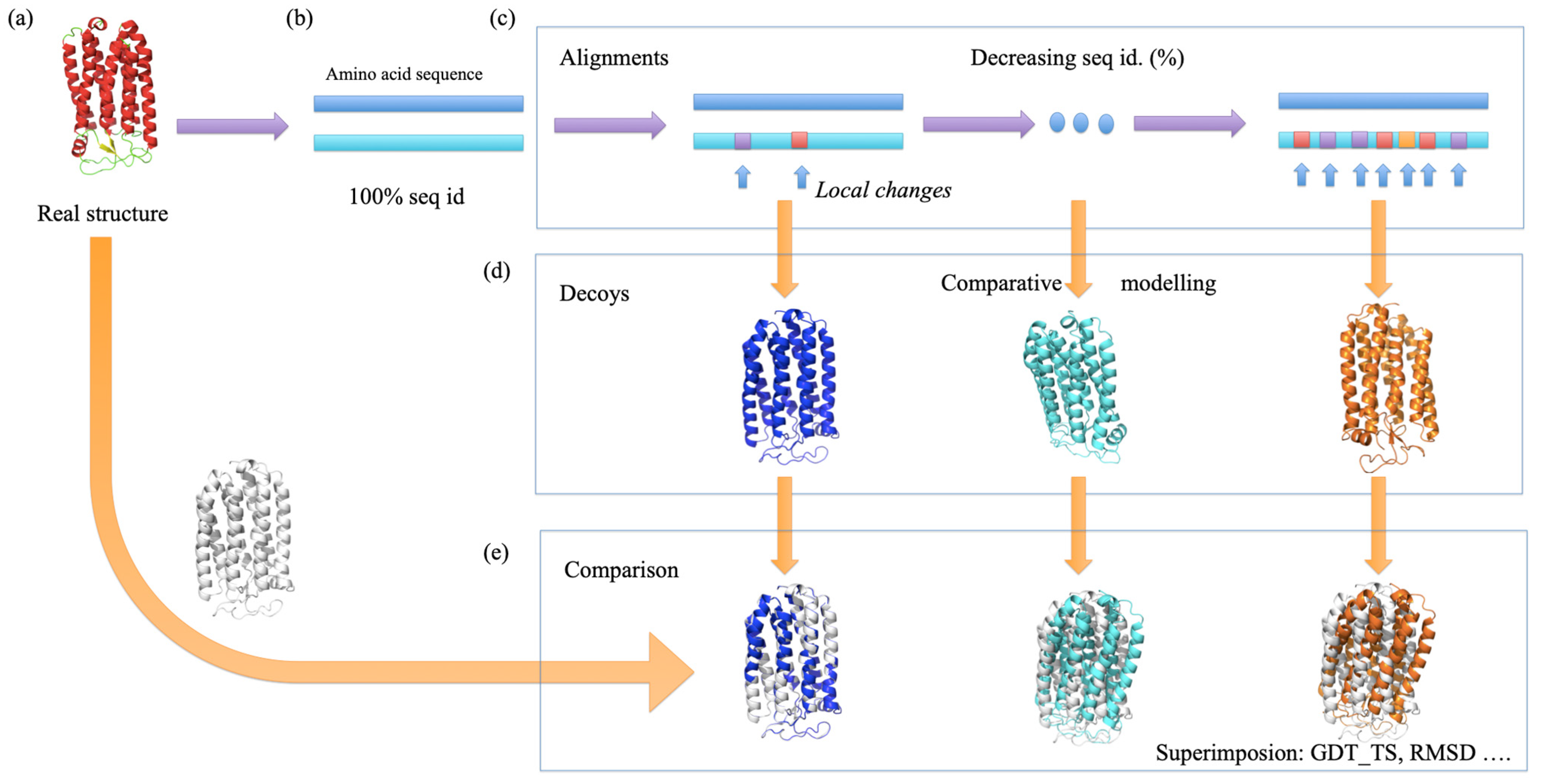
2.1. Protein Structure Dataset
2.2. Generation of Alternative Structural Models
2.3. Assessment Scores
2.4. Data Analyses
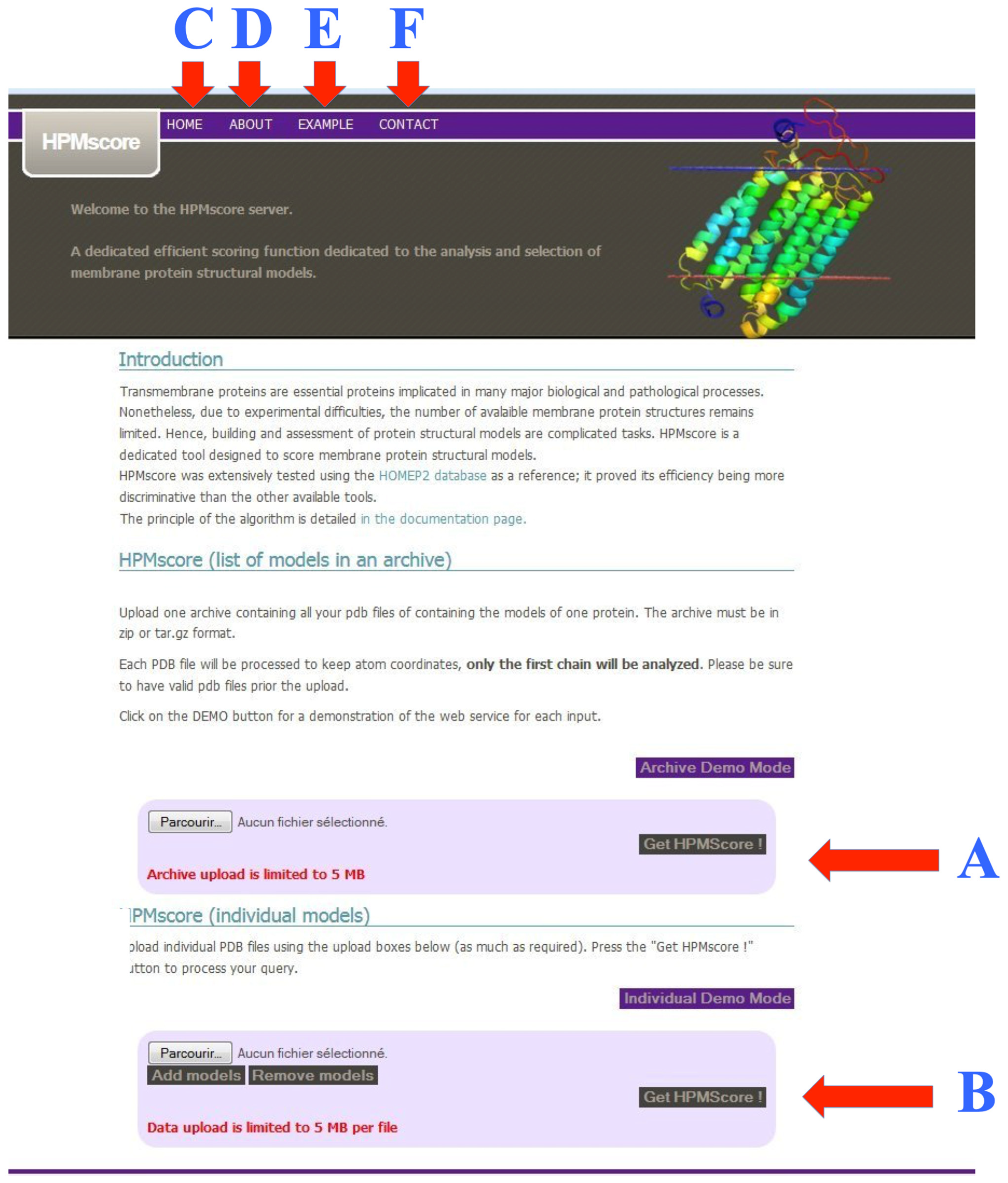
2.5. Scripting and Web Server of HPMScore
3. Results
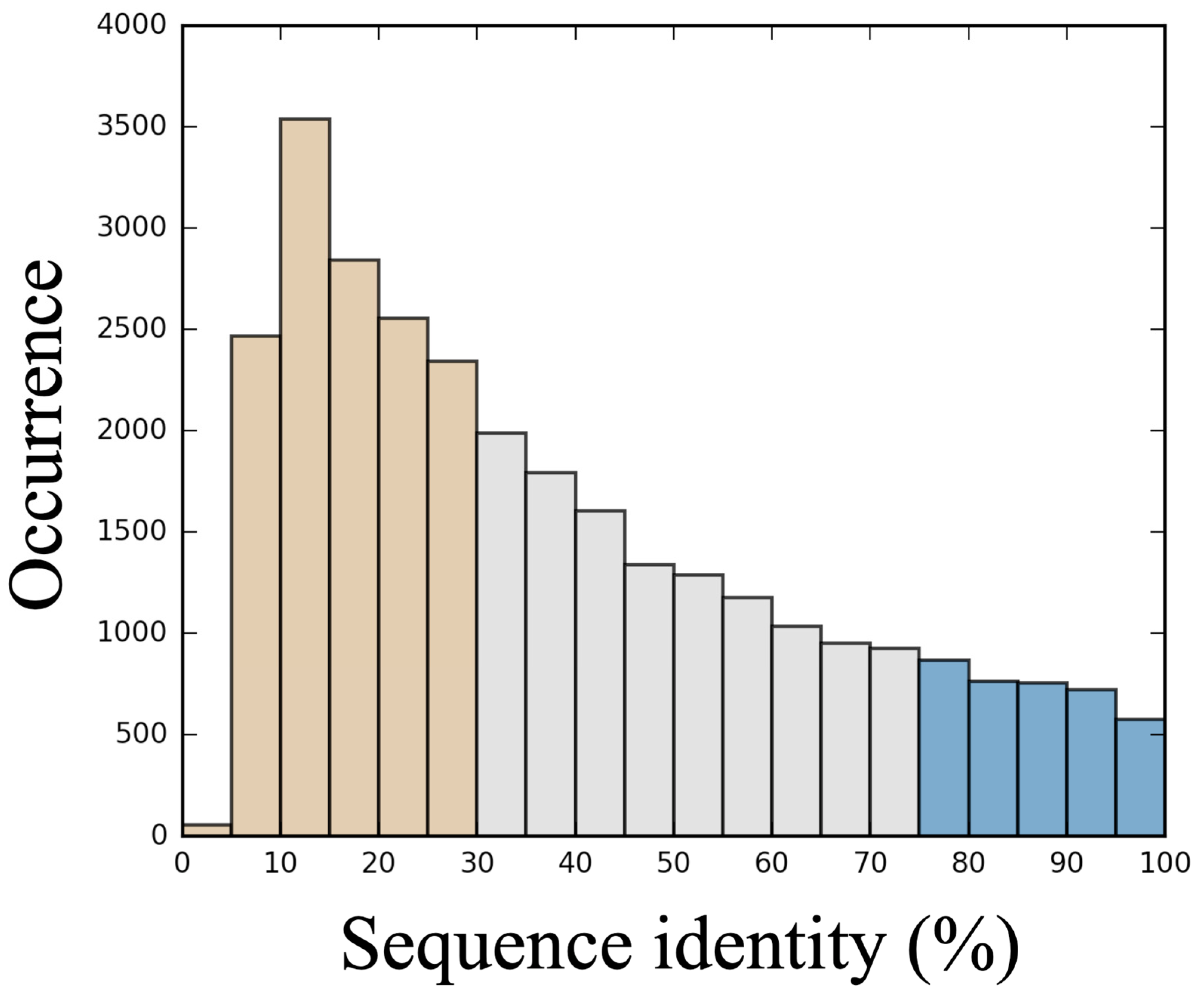
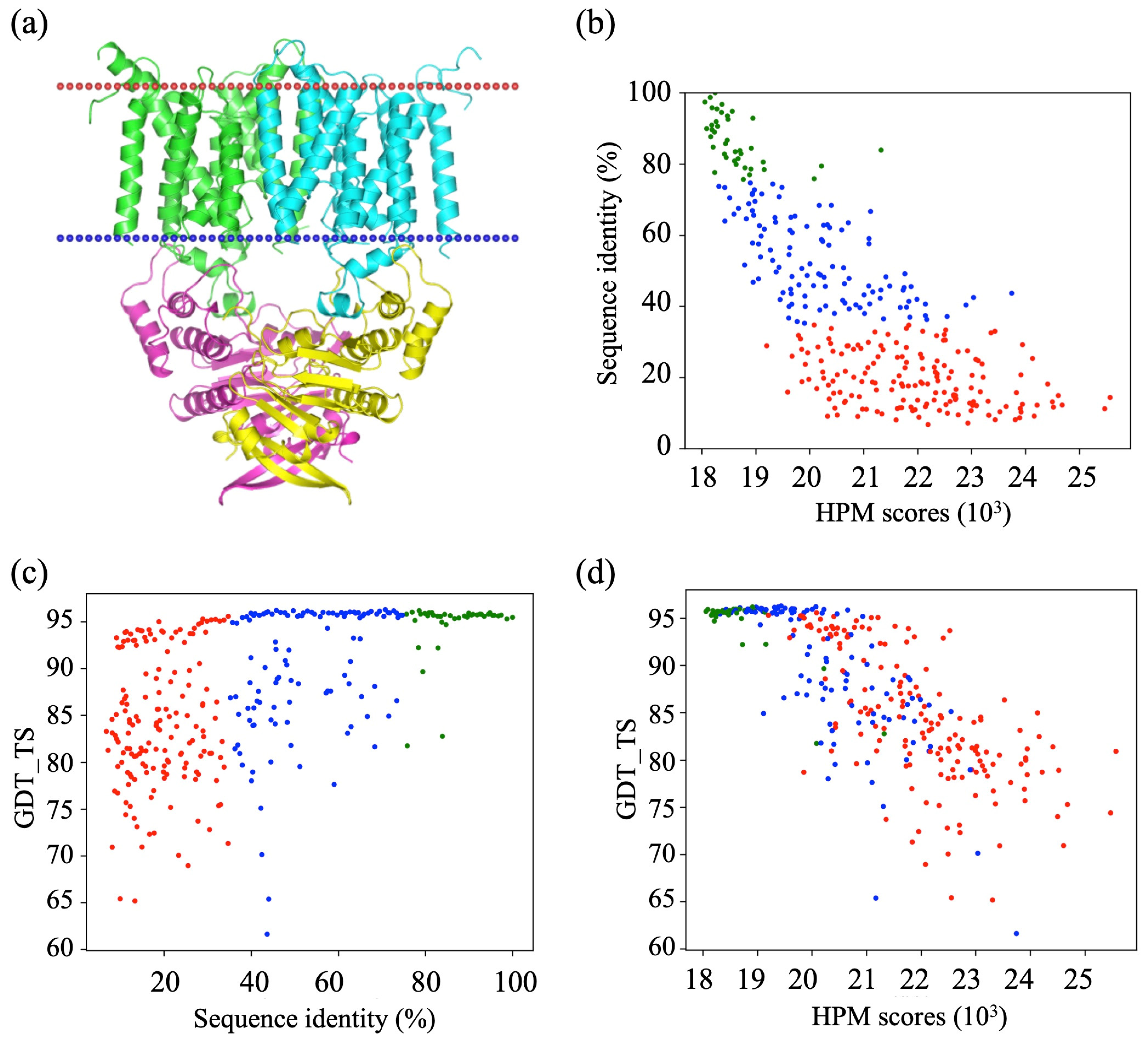
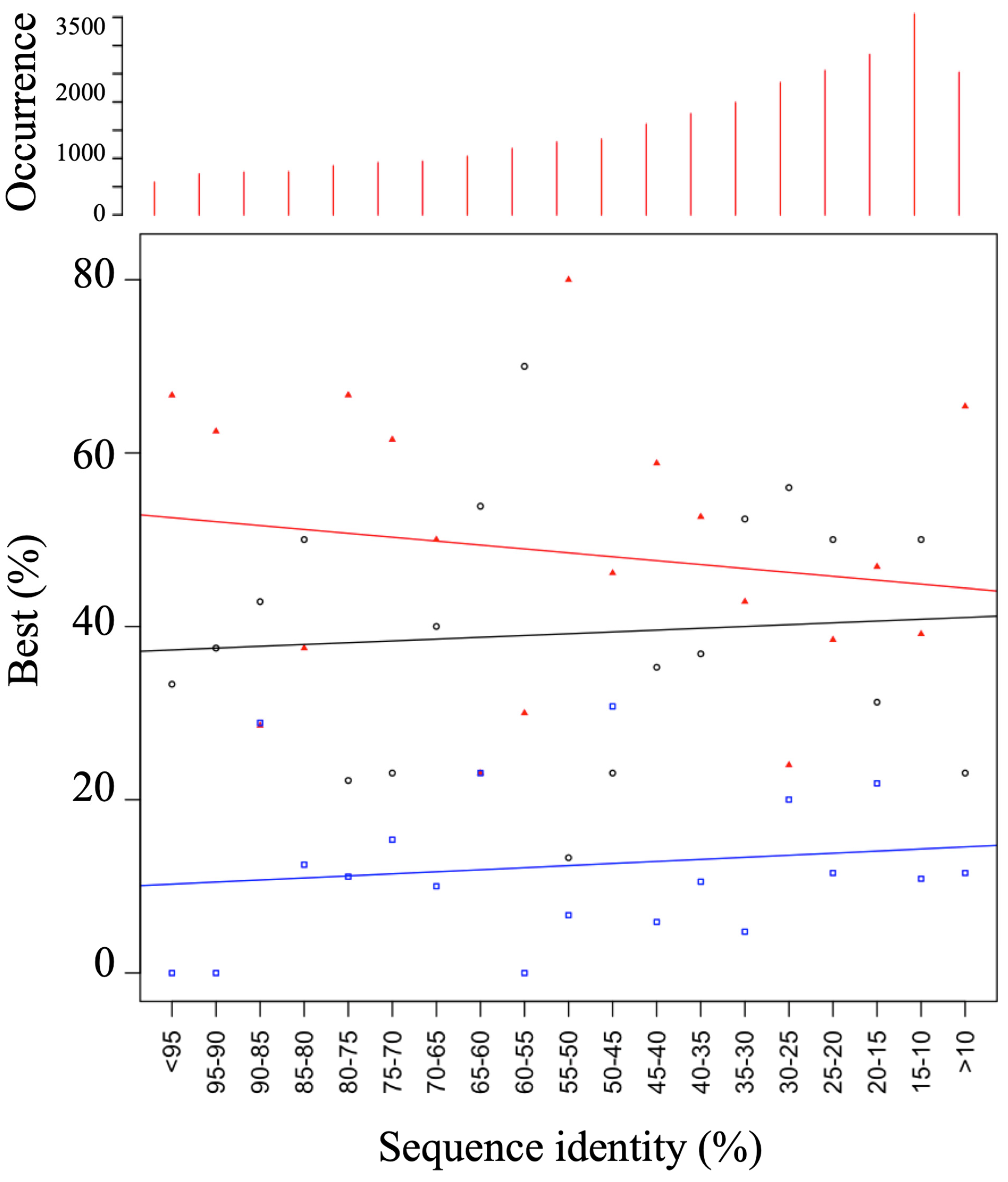
3.1. Generation of a Set of Structural Models for Sequences with Various Sequence Identities with Templates
3.2. HPM Selects Better Models Than DOPE
3.3. Assessment of Protein Model Quality
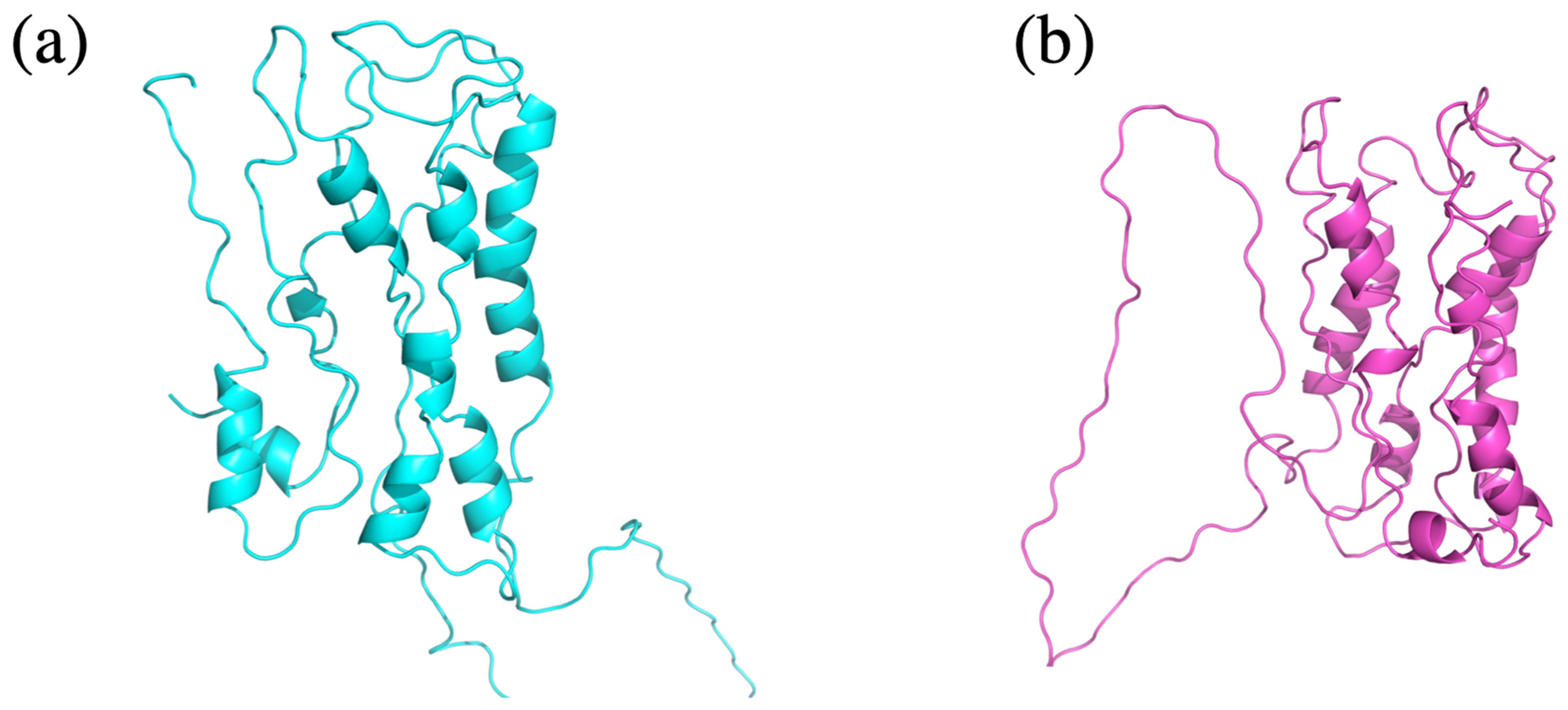
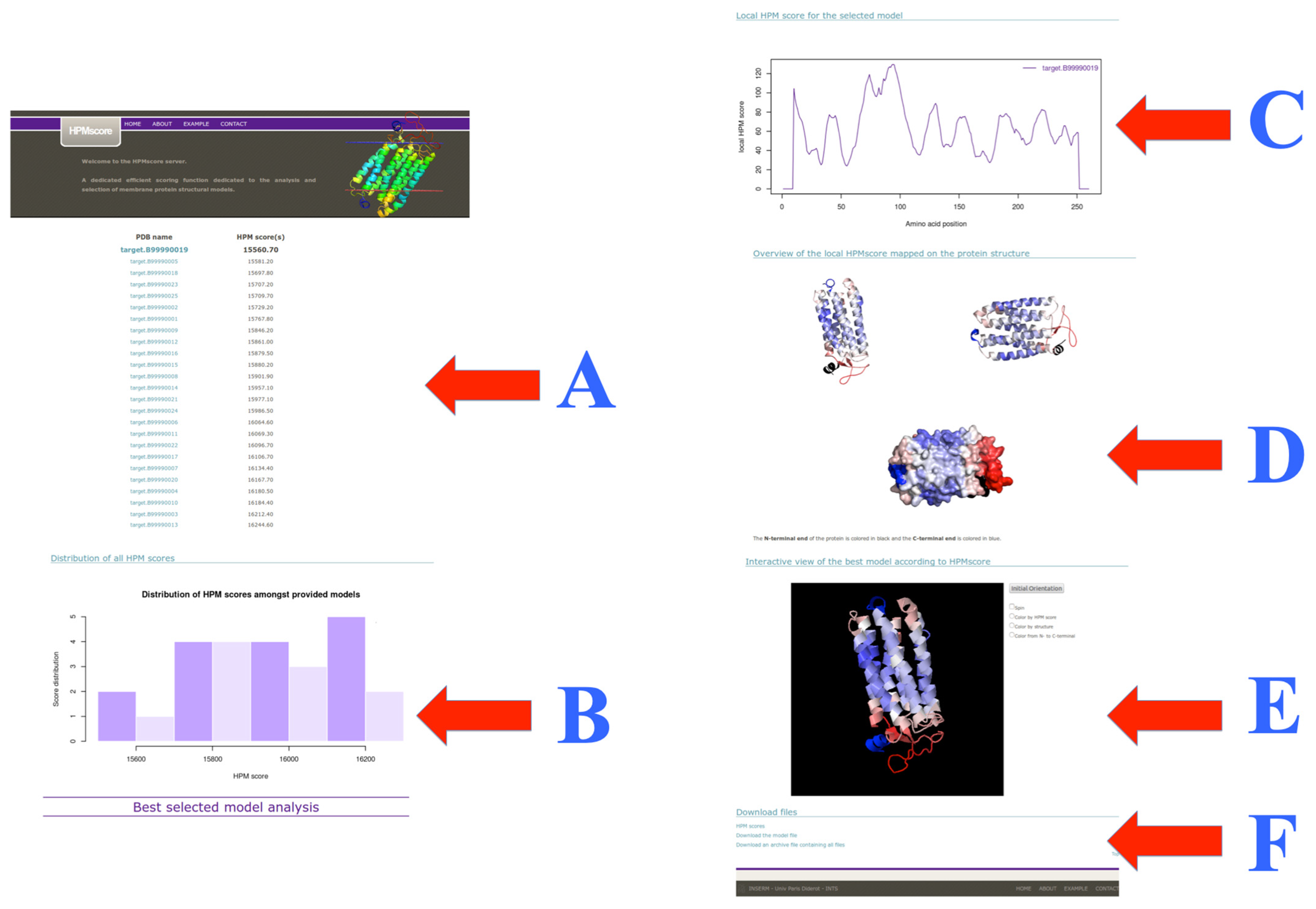
3.4. Web Server Usage and Example
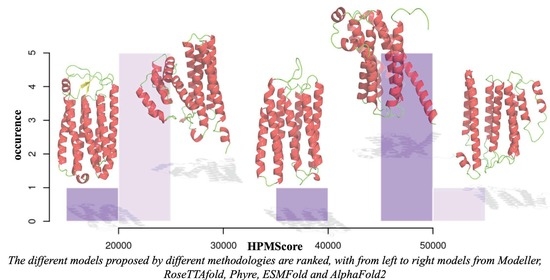
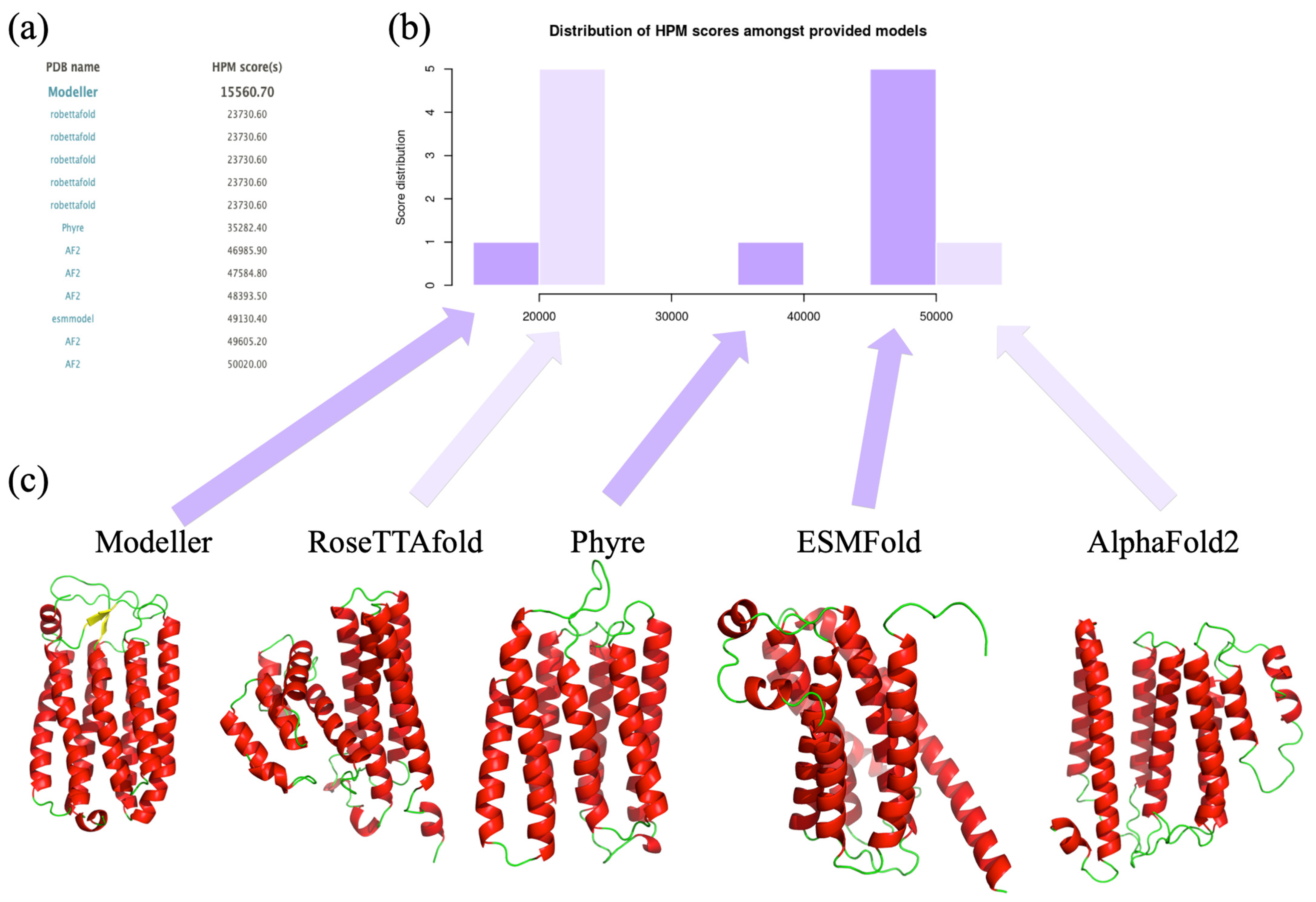
3.5. Use with Structural Models Coming from Different Approaches
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

References
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobson, L.; Reményi, I.; Tusnády, G.E. The human transmembrane proteome. Biol. Direct 2015, 10, 31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaucha, J.; Heinzinger, M.; Kulandaisamy, A.; Kataka, E.; Salvádor, Ó.L.; Popov, P.; Rost, B.; Gromiha, M.M.; Zhorov, B.S.; Frishman, D. Mutations in transmembrane proteins: Diseases, evolutionary insights, prediction and comparison with globular proteins. Brief. Bioinform. 2020, 22, bbaa132. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Chen, Y.; Pu, F.; Sun, P.; He, F.; Zhang, L.; Li, Y.; Ma, Z.; Wang, H. Understanding membrane protein drug targets in computational perspective. Curr. Drug Targets 2019, 20, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Varga, J.; Dobson, L.; Reményi, I.; Tusnády, G.E. Tstmp: Target selection for structural genomics of human transmembrane proteins. Nucleic Acids Res. 2017, 45, D325–D330. [Google Scholar] [CrossRef] [Green Version]
- Latek, D.; Trzaskowski, B.; Niewieczerza, S.; Miszta, P.; Mynarczyk, K.; Dębiński, A.; Puławski, W.; Yuan, S.; Sztyler, A.; Orze, U.; et al. Modeling of membrane proteins. In Computational Methods to Study the Structure and Dynamics of Biomolecules and Biomolecular Processes; Liwo, A., Ed.; Springer: Berlin/Heidelberg, Germany, 2018; pp. 371–451. [Google Scholar]
- Almeida, J.G.; Preto, A.J.; Koukos, P.I.; Bonvin, A.; Moreira, I.S. Membrane proteins structures: A review on computational modeling tools. Biochim. Biophys. Acta. Biomembr. 2017, 1859, 2021–2039. [Google Scholar] [CrossRef] [PubMed]
- Dobson, L.; Szekeres, L.I.; Gerdán, C.; Langó, T.; Zeke, A.; Tusnády, G.E. Tmalphafold database: Membrane localization and evaluation of alphafold2 predicted alpha-helical transmembrane protein structures. Nucleic Acids Res. 2022, 51, D517–D522. [Google Scholar] [CrossRef]
- Lazaridis, T.; Karplus, M. Discrimination of the native from misfolded protein models with an energy function including implicit solvation. J. Mol. Biol. 1999, 288, 477–487. [Google Scholar] [CrossRef] [Green Version]
- Felts, A.K.; Gallicchio, E.; Wallqvist, A.; Levy, R.M. Distinguishing native conformations of proteins from decoys with an effective free energy estimator based on the opls all-atom force field and the surface generalized born solvent model. Proteins 2002, 48, 404–422. [Google Scholar] [CrossRef] [PubMed]
- Dutagaci, B.; Wittayanarakul, K.; Mori, T.; Feig, M.A.-O. Discrimination of native-like states of membrane proteins with implicit membrane-based scoring functions. J. Chem. Comput. 2017, 13, 3049–3059. [Google Scholar] [CrossRef]
- Postic, G.; Janel, N.; Tufféry, P.; Moroy, G. An information gain-based approach for evaluating protein structure models. Comput. Struct. Biotechnol. J. 2020, 18, 2228–2236. [Google Scholar] [CrossRef]
- Shen, M.Y.; Sali, A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [Green Version]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Webb, B.; Sali, A. Protein structure modeling with modeller. Methods Mol. Biol. 2021, 2199, 239–255. [Google Scholar] [PubMed]
- Sippl, M.J. Recognition of errors in three-dimensional structures of proteins. Proteins 1993, 17, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Wiederstein, M.; Sippl, M.J. Prosa-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Eisenberg, D.; Lüthy, R.; Bowie, J.U. Verify3d: Assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997, 277, 396–404. [Google Scholar] [CrossRef]
- Lüthy, R.; Bowie, J.U.; Eisenberg, D. Assessment of protein models with three-dimensional profiles. Nature 1992, 356, 83–85. [Google Scholar] [CrossRef]
- Benkert, P.; Biasini, M.; Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 2011, 27, 343–350. [Google Scholar] [CrossRef] [Green Version]
- Kortemme, T.; Morozov, A.V.; Baker, D. An orientation-dependent hydrogen bonding potential improves prediction of specificity and structure for proteins and protein-protein complexes. J. Mol. Biol. 2003, 326, 1239–1259. [Google Scholar] [CrossRef] [Green Version]
- Shin, W.H.; Kang, X.; Zhang, J.; Kihara, D. Prediction of local quality of protein structure models considering spatial neighbors in graphical models. Sci. Rep. 2017, 7, 40629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tosatto, S.C. The victor/frst function for model quality estimation. J. Comput. Biol. A J. Comput. Mol. Cell Biol. 2005, 12, 1316–1327. [Google Scholar] [CrossRef]
- Conover, M.; Staples, M.; Si, D.; Sun, M.; Cao, R. Angularqa: Protein model quality assessment with lstm networks. Comput. Math. Biophys 2019, 7, 1–9. [Google Scholar] [CrossRef]
- Uziela, K.; Shu, N.; Wallner, B.; Elofsson, A. Proq3: Improved model quality assessments using rosetta energy terms. Sci. Rep. 2016, 6, 33509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, R.; Bhattacharya, D.; Hou, J.; Cheng, J. Deepqa: Improving the estimation of single protein model quality with deep belief networks. BMC Bioinform. 2016, 17, 495. [Google Scholar] [CrossRef] [Green Version]
- Studer, G.; Rempfer, C.; Waterhouse, A.M.; Gumienny, R.; Haas, J.; Schwede, T. Qmeandisco-distance constraints applied on model quality estimation. Bioinformatics 2020, 36, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Stern, H.A. Scoring function accuracy for membrane protein structure prediction. Proteins 2007, 68, 67–75. [Google Scholar] [CrossRef]
- Heim, A.J.; Li, Z. Developing a high-quality scoring function for membrane protein structures based on specific inter-residue interactions. J. Comput.-Aided Mol. Des. 2012, 26, 301–309. [Google Scholar] [CrossRef] [Green Version]
- Ray, A.; Lindahl, E.; Wallner, B. Model quality assessment for membrane proteins. Bioinformatics 2010, 26, 3067–3074. [Google Scholar] [CrossRef] [Green Version]
- Wallner, B. Proqm-resample: Improved model quality assessment for membrane proteins by limited conformational sampling. Bioinformatics 2014, 30, 2221–2223. [Google Scholar] [CrossRef] [Green Version]
- Nugent, T.; Jones, D.T. Membrane protein orientation and refinement using a knowledge-based statistical potential. BMC Bioinform. 2013, 14, 276. [Google Scholar] [CrossRef] [Green Version]
- Postic, G.; Ghouzam, Y.; Gelly, J.C. An empirical energy function for structural assessment of protein transmembrane domains. Biochimie 2015, 115, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Postic, G.; Ghouzam, Y.; Guiraud, V.; Gelly, J.C. Membrane positioning for high- and low-resolution protein structures through a binary classification approach. Protein Eng. Des. Sel. PEDS 2016, 29, 87–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studer, G.; Biasini, M.; Schwede, T. Assessing the local structural quality of transmembrane protein models using statistical potentials (qmeanbrane). Bioinformatics 2014, 30, i505–i511. [Google Scholar] [CrossRef] [Green Version]
- Barth, P.; Schonbrun, J.; Baker, D. Toward high-resolution prediction and design of transmembrane helical protein structures. Proc. Natl. Acad. Sci. USA 2007, 104, 15682–15687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alford, R.F.; Koehler Leman, J.; Weitzner, B.D.; Duran, A.M.; Tilley, D.C.; Elazar, A.; Gray, J.J. An integrated framework advancing membrane protein modeling and design. PLoS Comput. Biol. 2015, 11, e1004398. [Google Scholar] [CrossRef] [Green Version]
- Duran, A.M.; Meiler, J. Computational design of membrane proteins using rosettamembrane. Protein Sci. 2018, 27, 341–355. [Google Scholar] [CrossRef] [Green Version]
- Yarov-Yarovoy, V.; Schonbrun, J.; Baker, D. Multipass membrane protein structure prediction using rosetta. Proteins 2006, 62, 1010–1025. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with alphafold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Hegedűs, T.; Geisler, M.; Lukács, G.L.; Farkas, B. Ins and outs of alphafold2 transmembrane protein structure predictions. Cell. Mol. Life Sci. CMLS 2022, 79, 73. [Google Scholar] [CrossRef]
- Tunyasuvunakool, K.A.-O.; Adler, J.A.-O.; Wu, Z.; Green, T.A.-O.; Zielinski, M.; Žídek, A.; Bridgland, A.; Cowie, A.; Meyer, C.; Laydon, A.; et al. Highly accurate protein structure prediction for the human proteome. Nature 2021, 596, 590–596. [Google Scholar] [CrossRef] [PubMed]
- De Brevern, A.G. An agnostic analysis of the human alphafold2 proteome using local protein conformations. Biochimie 2023, 207, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Akdel, M.; Pires, D.E.V.; Pardo, E.P.; Jänes, J.; Zalevsky, A.O.; Mészáros, B.; Bryant, P.; Good, L.L.; Laskowski, R.A.; Pozzati, G.; et al. A structural biology community assessment of alphafold2 applications. Nat. Struct. Mol. Biol. 2022, 29, 1056–1067. [Google Scholar] [CrossRef] [PubMed]
- Esque, J.; Urbain, A.; Etchebest, C.; de Brevern, A.G. Sequence-structure relationship study in all-alpha transmembrane proteins using an unsupervised learning approach. Amino Acids 2015, 47, 2303–2322. [Google Scholar] [CrossRef]
- De Brevern, A.G.; Hazout, S. Hybrid protein model (hpm): A method to compact protein 3d-structure information and physicochemical properties. IEEE-Comp. Soc. (SPIRE 2000) 2000, S1, 49–54. [Google Scholar]
- De Brevern, A.G.; Hazout, S. ‘Hybrid protein model’ for optimally defining 3d protein structure fragments. Bioinformatics 2003, 19, 345–353. [Google Scholar] [CrossRef] [Green Version]
- Benros, C.; de Brevern, A.G.; Etchebest, C.; Hazout, S. Assessing a novel approach for predicting local 3d protein structures from sequence. Proteins: Struct. Funct. Bioinform. 2005, 62, 865–880. [Google Scholar] [CrossRef] [Green Version]
- Benros, C.; de Brevern, A.G.; Hazout, S. Analyzing the sequence–structure relationship of a library of local structural prototypes. J. Theor. Biol. 2009, 256, 215–226. [Google Scholar] [CrossRef] [Green Version]
- Bornot, A.; Etchebest, C.; de Brevern, A.G. A new prediction strategy for long local protein structures using an original description. Proteins 2009, 76, 570–587. [Google Scholar] [CrossRef] [Green Version]
- Bornot, A.; Etchebest, C.; de Brevern, A.G. Predicting protein flexibility through the prediction of local structures. Proteins 2011, 79, 839–852. [Google Scholar] [CrossRef] [Green Version]
- Narwani, T.J.; Etchebest, C.; Craveur, P.; Léonard, S.; Rebehmed, J.; Srinivasan, N.; Bornot, A.; Gelly, J.C.; de Brevern, A.G. In silico prediction of protein flexibility with local structure approach. Biochimie 2019, 165, 150–155. [Google Scholar] [CrossRef] [Green Version]
- De Brevern, A.G.; Bornot, A.; Craveur, P.; Etchebest, C.; Gelly, J.C. Predyflexy: Flexibility and local structure prediction from sequence. Nucleic Acids Res. 2012, 40, W317–W322. [Google Scholar] [CrossRef] [Green Version]
- De Brevern, A.G.; Etchebest, C.; Hazout, S. Bayesian probabilistic approach for predicting backbone structures in terms of protein blocks. Proteins 2000, 41, 271–287. [Google Scholar] [CrossRef]
- Joseph, A.P.; Agarwal, G.; Mahajan, S.; Gelly, J.C.; Swapna, L.S.; Offmann, B.; Cadet, F.; Bornot, A.; Tyagi, M.; Valadie, H.; et al. A short survey on protein blocks. Biophys. Rev. 2011, 2, 137–147. [Google Scholar] [CrossRef] [PubMed]
- Zemla, A.; Venclovas, C.; Fidelis, K.; Rost, B. A modified definition of sov, a segment-based measure for protein secondary structure prediction assessment. Proteins 1999, 34, 220–223. [Google Scholar] [CrossRef]
- Stamm, M.; Forrest, L.R. Structure alignment of membrane proteins: Accuracy of available tools and a consensus strategy. Proteins 2015, 83, 1720–1732. [Google Scholar] [CrossRef] [Green Version]
- Lomize, M.A.; Lomize, A.L.; Pogozheva, I.D.; Mosberg, H.I. Opm: Orientations of proteins in membranes database. Bioinformatics 2006, 22, 623–625. [Google Scholar] [CrossRef] [Green Version]
- Lomize, M.A.; Pogozheva, I.D.; Joo, H.; Mosberg, H.I.; Lomize, A.L. Opm database and ppm web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 2012, 40, D370–D376. [Google Scholar] [CrossRef] [PubMed]
- Sarti, E.; Aleksandrova, A.A.; Ganta, S.K.; Yavatkar, A.S.; Forrest, L.R. Encompass: An online database for analyzing structure and symmetry in membrane proteins. Nucleic Acids Res. 2019, 8, D315–D325. [Google Scholar] [CrossRef] [Green Version]
- BioPerl. 2020. Available online: https://github.com/bioperl/bioperl-live (accessed on 1 March 2023).
- Kohonen, T. Self-organized formation of topologically correct feature maps. Biol. Cybern 1982, 43, 59–69. [Google Scholar] [CrossRef]
- Kohonen, T. Self-Organizing Maps, 3rd ed.; Springer: Berlin/Heidelberg, Germany, 2001; p. 501. [Google Scholar]
- Delano, W.L. The Pymol Molecular Graphics System. 2002. Available online: http://www.pymol.org (accessed on 1 March 2023).
- Joseph, A.P.; Srinivasan, N.; de Brevern, A.G. Improvement of protein structure comparison using a structural alphabet. Biochimie 2011, 93, 1434–1445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, A.; Porter, C. ProFit Software. Available online: http://www.bioinf.org.uk/software/profit/ (accessed on 1 March 2023).
- Zhang, Y.; Skolnick, J. Tm-align: A protein structure alignment algorithm based on the tm-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef]
- Kelley, L.A.; Sternberg, M.J. Protein structure prediction on the web: A case study using the phyre server. Nat. Protoc. 2009, 4, 363–371. [Google Scholar] [CrossRef] [Green Version]
- Lin, Z.; Akin, H.; Rao, R.; Hie, B.; Zhu, Z.; Lu, W.; Smetanin, N.; Verkuil, R.; Kabeli, O.; Shmueli, Y.; et al. Evolutionary-scale prediction of atomic level protein structure with a language model. bioRxiv 2022. [Google Scholar] [CrossRef]
- Koehler Leman, J.; Ulmschneider, M.B.; Gray, J.J. Computational modeling of membrane proteins. Proteins 2015, 83, 1–24. [Google Scholar] [CrossRef] [Green Version]
- Rost, B.; Casadio, R.; Fariselli, P.; Sander, C. Transmembrane helices predicted at 95% accuracy. Protein Sci. 1995, 4, 521–533. [Google Scholar] [CrossRef] [Green Version]
- Bernhofer, M.; Dallago, C.; Karl, T.; Satagopam, V.; Heinzinger, M.; Littmann, M.; Olenyi, T.; Qiu, J.; Schütze, K.; Yachdav, G.; et al. Predictprotein-predicting protein structure and function for 29 years. Nucleic Acids Res. 2021, 49, W535–W540. [Google Scholar] [CrossRef]
- Buchan, D.W.A.; Jones, D.T. The psipred protein analysis workbench: 20 years on. Nucleic Acids Res. 2019, 47, W402–W407. [Google Scholar] [CrossRef] [Green Version]
- McGuffin, L.J.; Bryson, K.; Jones, D.T. The psipred protein structure prediction server. Bioinformatics 2000, 16, 404–405. [Google Scholar] [CrossRef] [Green Version]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 1999, 292, 195–202. [Google Scholar] [CrossRef] [Green Version]
- Cid, H.; Bunster, M.; Arriagada, E.; Campos, M. Prediction of secondary structure of proteins by means of hydrophobicity profiles. FEBS Lett. 1982, 150, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Hessa, T.; Kim, H.; Bihlmaier, K.; Lundin, C.; Boekel, J.; Andersson, H.; Nilsson, I.; White, S.H.; von Heijne, G. Recognition of transmembrane helices by the endoplasmic reticulum translocon. Nature 2005, 433, 377–381. [Google Scholar] [CrossRef]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry 1994, 33, 3038–3049. [Google Scholar] [CrossRef]
- Jones, D.T. Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics 2007, 23, 538–544. [Google Scholar] [CrossRef] [Green Version]
- Fariselli, P.; Casadio, R. Htp: A neural network-based method for predicting the topology of helical transmembrane domains in proteins. Comput. Appl. Biosci. CABIOS 1996, 12, 41–48. [Google Scholar] [CrossRef] [Green Version]
- Hirokawa, T.; Boon-Chieng, S.; Mitaku, S. Sosui: Classification and secondary structure prediction system for membrane proteins. Bioinformatics 1998, 14, 378–379. [Google Scholar] [CrossRef] [Green Version]
- Tusnády, G.E.; Simon, I. Principles governing amino acid composition of integral membrane proteins: Application to topology prediction. J. Mol. Biol. 1998, 283, 489–506. [Google Scholar] [CrossRef] [Green Version]
- Dosztányi, Z.; Magyar, C.; Tusnády, G.E.; Cserzo, M.; Fiser, A.; Simon, I. Servers for sequence-structure relationship analysis and prediction. Nucleic Acids Res. 2003, 31, 3359–3363. [Google Scholar] [CrossRef] [Green Version]
- Sonnhammer, E.L.; von Heijne, G.; Krogh, A. A hidden markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1998, 6, 175–182. [Google Scholar]
- Pasquier, C.; Promponas, V.J.; Palaios, G.A.; Hamodrakas, J.S.; Hamodrakas, S.J. A novel method for predicting transmembrane segments in proteins based on a statistical analysis of the swissprot database: The pred-tmr algorithm. Protein Eng. 1999, 12, 381–385. [Google Scholar] [CrossRef]
- Viklund, H.; Elofsson, A. Octopus: Improving topology prediction by two-track ann-based preference scores and an extended topological grammar. Bioinformatics 2008, 24, 1662–1668. [Google Scholar] [CrossRef] [Green Version]
- Bernsel, A.; Viklund, H.; Hennerdal, A.; Elofsson, A. Topcons: Consensus prediction of membrane protein topology. Nucleic Acids Res. 2009, 37, W465–W468. [Google Scholar] [CrossRef] [Green Version]
- Tsirigos, K.D.; Peters, C.; Shu, N.; Käll, L.; Elofsson, A. The topcons web server for consensus prediction of membrane protein topology and signal peptides. Nucleic Acids Res. 2015, 43, W401–W407. [Google Scholar] [CrossRef]
- Cao, B.; Porollo, A.; Adamczak, R.; Jarrell, M.; Meller, J. Enhanced recognition of protein transmembrane domains with prediction-based structural profiles. Bioinformatics 2006, 22, 303–309. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Mattick, J.S.; Teasdale, R.D. Svmtm: Support vector machines to predict transmembrane segments. J. Comput. Chem. 2004, 25, 632–636. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Zhang, C.; Liu, S.; Zhou, Y. Web-based toolkits for topology prediction of transmembrane helical proteins, fold recognition, structure and binding scoring, folding-kinetics analysis and comparative analysis of domain combinations. Nucleic Acids Res. 2005, 33, W193–W197. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.; Lee, B.; Jang, I.; Kim, S.; Bhak, J. Localizome: A server for identifying transmembrane topologies and tm helices of eukaryotic proteins utilizing domain information. Nucleic Acids Res. 2006, 34, W99–W103. [Google Scholar] [CrossRef]
- Yin, X.; Yang, J.; Xiao, F.; Yang, Y.; Shen, H.B. Membrain: An easy-to-use online webserver for transmembrane protein structure prediction. Nano-Micro Lett. 2018, 10, 2. [Google Scholar] [CrossRef] [Green Version]
- Hönigschmid, P.; Breimann, S.; Weigl, M.; Frishman, D. Allestm: Predicting multiple structural features of transmembrane proteins. BMC Bioinform. 2020, 21, 242. [Google Scholar] [CrossRef] [PubMed]
- Koehler Leman, J.; Mueller, B.K.; Gray, J.J. Expanding the toolkit for membrane protein modeling in rosetta. Bioinformatics 2017, 33, 754–756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernhofer, M.; Rost, B. Tmbed: Transmembrane proteins predicted through language model embeddings. BMC Bioinform. 2022, 23, 326. [Google Scholar] [CrossRef]
- Von Heijne, G. Membrane-protein topology. Nat. Rev. Mol. Cell Biol. 2006, 7, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Mendenhall, J.; Capra, J.A.; Meiler, J. A multitask deep-learning method for predicting membrane associations and secondary structures of proteins. J. Proteome Res. 2021, 20, 4089–4100. [Google Scholar] [CrossRef]
- Qu, J.; Yin, S.S.; Wang, H. Prediction of metal ion binding sites of transmembrane proteins. Comput. Math. Methods Med. 2021, 2021, 2327832. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. Swiss-model: Homology modelling of protein structures and complexes. Nucleic Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef] [Green Version]
- Ebejer, J.-P.; Hill, J.R.; Kelm, S.; Shi, J.; Deane, C.M. Memoir: Template-based structure prediction for membrane proteins. Nucleic Acids Res. 2013, 41, W379–W383. [Google Scholar] [CrossRef] [Green Version]
- Kelm, S.; Shi, J.; Deane, C.M. Medeller: Homology-based coordinate generation for membrane proteins. Bioinformatics 2010, 26, 2833–2840. [Google Scholar] [CrossRef] [Green Version]
- Kozma, D.; Tusnády, G.E. Tmfoldweb: A web server for predicting transmembrane protein fold class. Biol. Direct. 2017, 10, 54. [Google Scholar] [CrossRef] [Green Version]
- Kozma, D.; Tusnády, G.E. Tmfoldrec: A statistical potential-based transmembrane protein fold recognition tool. BMC Bioinform. 2015, 16, 201. [Google Scholar] [CrossRef] [Green Version]
- .Yarov-Yarovoy, V.; Baker, D.; Catterall, W.A. Voltage sensor conformations in the open and closed states in ROSETTA structural models of K(+) channels. Proc. Natl. Acad. Sci. USA 2006, 103, 7292–7297. [Google Scholar] [CrossRef] [Green Version]
- Benkert, P.; Künzli, M.; Schwede, T. Qmean server for protein model quality estimation. Nucleic Acids Res. 2009, 37, W510–W514. [Google Scholar] [CrossRef] [Green Version]
- Snider, C.; Jayasinghe, S.; Fau-Hristova, K.; Hristova, K.; Fau-White, S.H.; White, S.H. Mpex: A tool for exploring membrane proteins. Protein Sci. 2009, 18, 2624–2628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jayasinghe, S.; Hristova, K.; Fau-White, S.H.; White, S.H. Mptopo: A database of membrane protein topology. Protein Sci. 2001, 10, 455–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokrab, Y.; Stevens, T.J.; Mizuguchi, K. A structural dissection of amino acid substitutions in helical transmembrane proteins. Proteins 2010, 78, 2895–2907. [Google Scholar] [CrossRef] [PubMed]
- Olivella, M.; Gonzalez, A.; Pardo, L.; Deupi, X. Relation between sequence and structure in membrane proteins. Bioinformatics 2013, 29, 1589–1592. [Google Scholar] [CrossRef] [Green Version]
- Kabsch, W. A discussion of the solution for the best rotation to relate two sets of vectors. Acta Crystallogr. Sect. A 1978, 34, 827–828. [Google Scholar] [CrossRef] [Green Version]
- Del Alamo, D.; Govaerts, C.; McHaourab, H.S. Alphafold2 predicts the inward-facing conformation of the multidrug transporter lmrp. Proteins 2021, 89, 1226–1228. [Google Scholar] [CrossRef]
- Xiao, Q.; Xu, M.; Wang, W.; Wu, T.; Zhang, W.; Qin, W.; Sun, B. Utilization of alphafold2 to predict mfs protein conformations after selective mutation. Int. J. Mol. Sci. 2022, 23, 7235. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) | |||
| Scoring Method/Models Considered for Ranking | TOP 1 | TOP 5 | TOP 10 |
| DOPE | 40.1% | 44.0% | 45.5% |
| HPM | 46.9% | 48.4% | 48.4% |
| HPM and DOPE | 13.0% | 7.6% | 6.1% |
| (B) | |||
| Scoring Method/Models Considered for Ranking | TOP 1 | TOP 5 | TOP 10 |
| DOPE | 47.4% | 46.4% | 46.3% |
| HPM | 45.6% | 46.8% | 47.0% |
| HPM and DOPE | 7.0% | 6.8% | 6.7% |
| Scoring Method/% Sequence Identity Range | Poor Alignments (0–35%) | Average Alignments (35–75%) | Good Alignments (75–100%) |
|---|---|---|---|
| Sequence count | 15,786 | 10,102 | 3682 |
| DOPE | 43.8% | 35.5% | 36.8% |
| HPM | 42.6% | 51.8% | 52.6% |
| HPM and DOPE | 13.6% | 12.7% | 10.6% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Téletchéa, S.; Esque, J.; Urbain, A.; Etchebest, C.; de Brevern, A.G. Evaluation of Transmembrane Protein Structural Models Using HPMScore. BioMedInformatics 2023, 3, 306-326. https://doi.org/10.3390/biomedinformatics3020021
Téletchéa S, Esque J, Urbain A, Etchebest C, de Brevern AG. Evaluation of Transmembrane Protein Structural Models Using HPMScore. BioMedInformatics. 2023; 3(2):306-326. https://doi.org/10.3390/biomedinformatics3020021
Chicago/Turabian StyleTéletchéa, Stéphane, Jérémy Esque, Aurélie Urbain, Catherine Etchebest, and Alexandre G. de Brevern. 2023. "Evaluation of Transmembrane Protein Structural Models Using HPMScore" BioMedInformatics 3, no. 2: 306-326. https://doi.org/10.3390/biomedinformatics3020021