
A Genome-Wide Association Study of Dementia Using the Electronic Medical Record



Abstract
:1. Introduction
2. Materials and Methods
2.1. Database
2.2. Patient Cohort
2.3. Quality Control
2.4. Statistical Method
3. Results
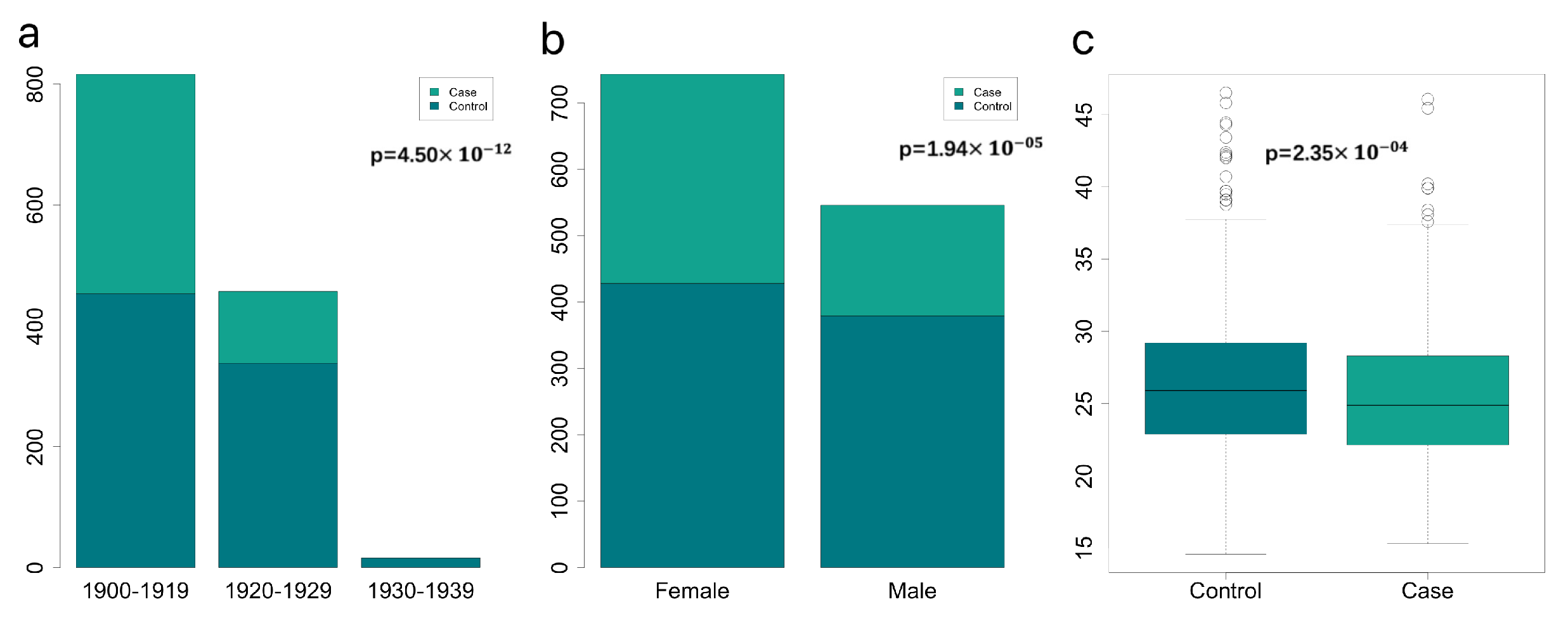
3.1. Important Non-Genomics Factors Affecting Risk of Dementia
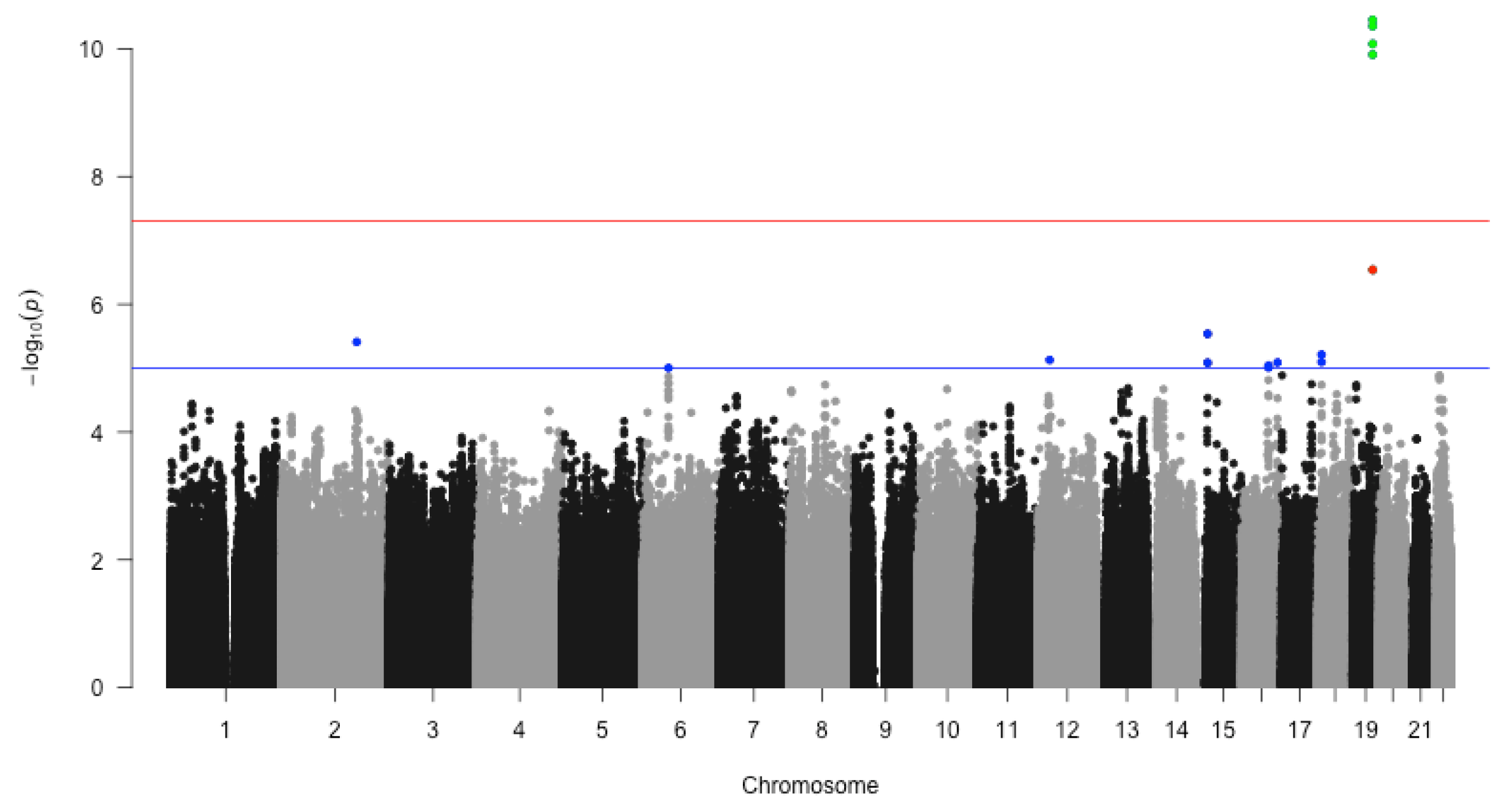
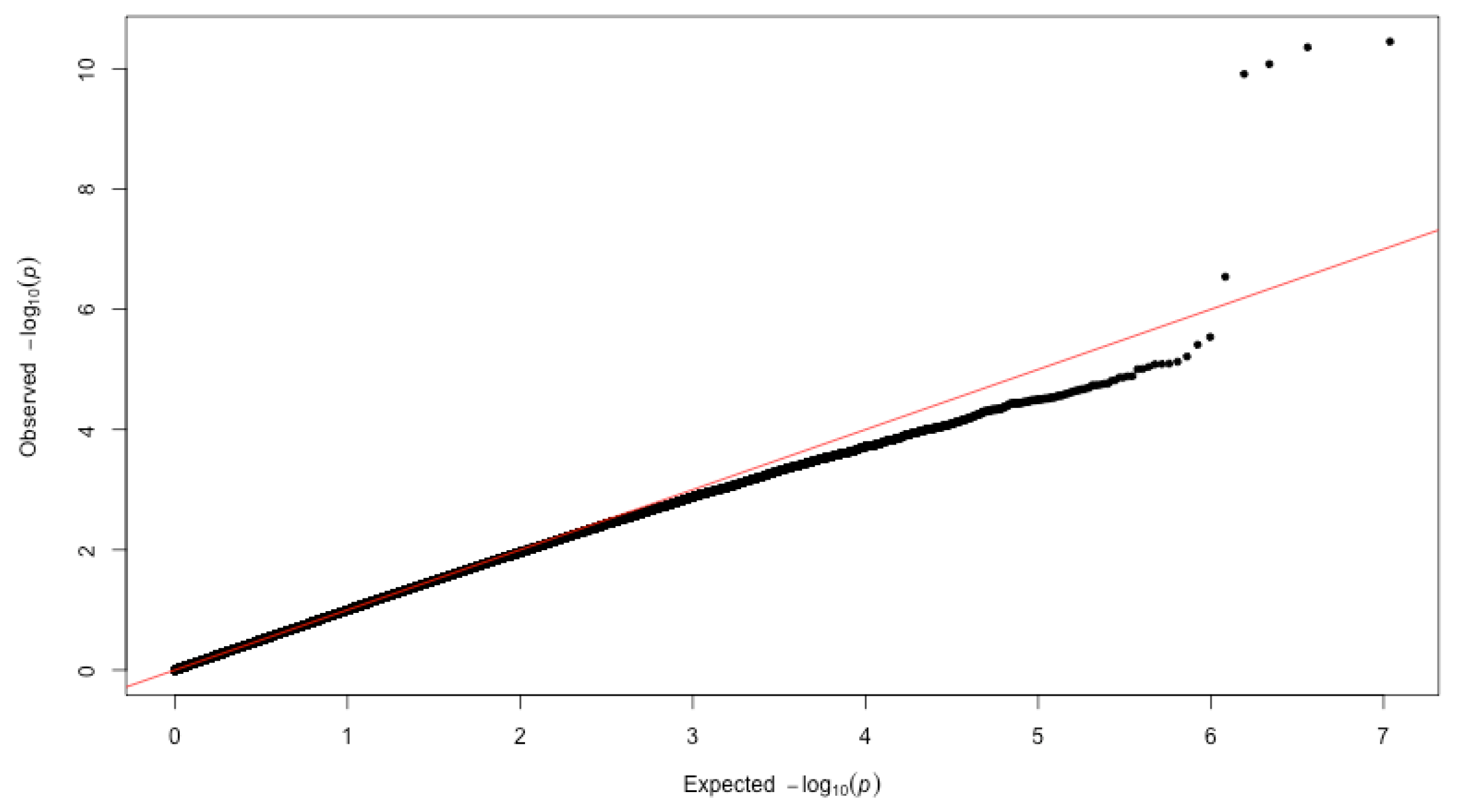
3.2. GWAS of Dementia
4. Discussions and Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| eMERGE | electronic Medical Records and Genomics |
| Q-Q plot | Quantile-Quantile plot |
| BMI | Body Mass Index |
| SNP | Single Nucleotide Polymorphism |
References
- Fu, W.Y.; Ip, N.Y. The role of genetic risk factors of Alzheimer’s disease in synaptic dysfunction. Semin. Cell Dev. Biol. 2023, 139, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.; Chauhan, A. Bystanders or not? Microglia and lymphocytes in aging and stroke. Neural Regen. Res. 2023, 18, 1397. [Google Scholar] [PubMed]
- Ayenigbara, I.O. Preventive Measures against the Development of Dementia in Old Age. Korean J. Fam. Med. 2022, 43, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Migliore, L.; Coppedè, F. Gene–environment interactions in Alzheimer disease: The emerging role of epigenetics. Nat. Rev. Neurol. 2022, 18, 643–660. [Google Scholar] [CrossRef]
- Wightman, D.P.; Jansen, I.E.; Savage, J.E.; Shadrin, A.A.; Bahrami, S.; Holland, D.; Rongve, A.; Børte, S.; Winsvold, B.S.; Drange, O.K.; et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 2021, 53, 1276–1282. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Grau, S.; Rojas, I.D.; Hernández, I.; Quintela, I.; Montrreal, L.; Alegret, M.; Hernández-Olasagarre, B.; Madrid, L.; González-Perez, A.; Maroñas, O.; et al. Genome-wide association analysis of dementia and its clinical endophenotypes reveal novel loci associated with Alzheimer’s disease and three causality networks: The GR@ACE project. Alzheimers Dement. 2019, 15, 1333–1347. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Schmechel, D.; Pericak-Vance, M.; Enghild, J.; Salvesen, G.S.; Roses, A.D. Apolipoprotein E: High-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 1977–1981. [Google Scholar] [CrossRef] [Green Version]
- Cervantes, S.; Samaranch, L.; Vidal-Taboada, J.M.; Lamet, I.; Bullido, M.J.; Frank-García, A.; Coria, F.; Lleó, A.; Clarimón, J.; Lorenzo, E.; et al. Genetic variation in APOE cluster region and Alzheimer’s disease risk. Neurobiol. Aging 2011, 32, 2107.e7. [Google Scholar] [CrossRef]
- Mishra, S.; Knupp, A.; Szabo, M.P.; Williams, C.A.; Kinoshita, C.; Hailey, D.W.; Wang, Y.; Andersen, O.M.; Young, J.E. The Alzheimer’s gene SORL1 is a regulator of endosomal traffic and recycling in human neurons. Cell. Mol. Life Sci. 2022, 79, 162. [Google Scholar] [CrossRef]
- Harold, D.; Abraham, R.; Hollingworth, P.; Sims, R.; Gerrish, A.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; Dowzell, K.; Williams, A.; et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1088–1093. [Google Scholar] [CrossRef]
- Roeder, K.; Wasserman, L. Genome-Wide Significance Levels and Weighted Hypothesis Testing. Stat. Sci. 2009, 24, 398–413. [Google Scholar] [CrossRef] [Green Version]
- Krohn, L.; Heilbron, K.; Blauwendraat, C.; Reynolds, R.H.; Yu, E.; Senkevich, K.; Rudakou, U.; Estiar, M.A.; Gustavsson, E.K.; Brolin, K.; et al. Genome-wide association study of REM sleep behavior disorder identifies polygenic risk and brain expression effects. Nat. Commun. 2022, 13, 7496. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Xing, L.; Su, J.; Zhang, X.; Qiu, W. Model-based clustering for identifying disease-associated SNPs in case-control genome-wide association studies. Sci. Rep. 2019, 9, 13686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magrangeas, F.; Kuiper, R.; Avet-Loiseau, H.; Gouraud, W.; Guérin-Charbonnel, C.; Ferrer, L.; Aussem, A.; Elghazel, H.; Suhard, J.; Sakissian, H.D.; et al. A Genome-Wide Association Study Identifies a Novel Locus for Bortezomib-Induced Peripheral Neuropathy in European Patients with Multiple Myeloma. Clin. Cancer Res. 2016, 22, 4350–4355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, G.; Liu, W.; Cheng, C.; Wilson, C.L.; Neale, G.; Yang, J.J.; Ness, K.K.; Robison, L.L.; Hudson, M.M.; Srivastava, D.K. Evaluation of a two-step iterative resampling procedure for internal validation of genome-wide association studies. J. Hum. Genet. 2015, 60, 729–738. [Google Scholar] [CrossRef] [Green Version]
- McCarty, C.A.; Chisholm, R.L.; Chute, C.G.; Kullo, I.J.; Jarvik, G.P.; Larson, E.B.; Li, R.; Masys, D.R.; Ritchie, M.D.; Roden, D.M.; et al. The eMERGE Network: A consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med. Genom. 2011, 4, 13. [Google Scholar] [CrossRef] [Green Version]
- Gottesman, O.; Kuivaniemi, H.; Tromp, G.; Faucett, W.A.; Li, R.; Manolio, T.A.; Sanderson, S.C.; Kannry, J.; Zinberg, R.; Basford, M.A.; et al. The Electronic Medical Records and Genomics (eMERGE) Network: Past, present and future. Genet. Med. 2013, 15, 761–771. [Google Scholar] [CrossRef] [Green Version]
- Nelson, M.R.; Marnellos, G.; Kammerer, S.; Hoyal, C.R.; Shi, M.M.; Cantor, C.R.; Braun, A. Large-Scale Validation of Single Nucleotide Polymorphisms in Gene Regions. Genome Res. 2004, 14, 1664–1668. [Google Scholar] [CrossRef] [Green Version]
- Marees, A.T.; Kluiver, H.d.; Stringer, S.; Vorspan, F.; Curis, E.; Marie-Claire, C.; Derks, E.M. A tutorial on conducting genome-wide association studies: Quality control and statistical analysis. Int. J. Methods Psychiatr. Res. 2018, 27, e1608. [Google Scholar] [CrossRef] [Green Version]
- Turner, S.; Armstrong, L.L.; Bradford, Y.; Carlson, C.S.; Crawford, D.C.; Crenshaw, A.T.; Andrade, M.; Doheny, K.F.; Haines, J.L.; Hayes, G.; et al. Quality Control Procedures for Genome-Wide Association Studies. Curr. Protoc. Hum. Genet. 2011, 68, 1.19.1–1.19.18. [Google Scholar] [CrossRef]
- Lahti, J.; Tuominen, S.; Yang, Q.; Pergola, G.; Ahmad, S.; Amin, N.; Armstrong, N.J.; Beiser, A.; Bey, K.; Bis, J.C.; et al. Genome-wide meta-analyses reveal novel loci for verbal short-term memory and learning. Mol. Psychiatry 2022, 27, 4419–4431. [Google Scholar] [CrossRef]
- Huang, Y.S.; Fang, T.H.; Kung, B.; Chen, C.H. Two Genetic Mechanisms in Two Siblings with Intellectual Disability, Autism Spectrum Disorder and Psychosis. J. Pers. Med. 2022, 12, 1013. [Google Scholar] [CrossRef]
- Mo, X.; Liu, M.; Gong, J.; Mei, Y.; Chen, H.; Mo, H.; Yang, X.; Li, J. PTPRM Is Critical for Synapse Formation Regulated by Zinc Ion. Front. Mol. Neurosci. 2022, 15, 822458. [Google Scholar] [CrossRef] [PubMed]
- Larivière, L.; Plaschka, C.; Seizl, M.; Petrotchenko, E.V.; Wenzeck, L.; Borchers, C.H.; Cramer, P. Model of the Mediator middle module based on protein cross-linking. Nucleic Acids Res. 2013, 41, 9266–9273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stephan, Y.; Sutin, A.R.; Luchetti, M.; Terracciano, A. Subjective age and risk of incident dementia: Evidence from the National Health and Aging Trends survey. J. Psychiatr. Res. 2018, 100, 1–4. [Google Scholar] [CrossRef] [Green Version]
- Mielke, M.M. Sex and Gender Differences in Alzheimer’s Disease Dementia. Psychiatr. Times 2018, 35, 14–17. [Google Scholar]
- Eruysal, E.; Ravdin, L.; Zhang, C.; Kamel, H.; Iadecola, C.; Ishii, M. Sexually Dimorphic Association of Circulating Plasminogen Activator Inhibitor-1 Levels and Body Mass Index with Cerebrospinal Fluid Biomarkers of Alzheimer’s Pathology in Preclinical Alzheimer’s Disease. J. Alzheimers Dis. 2022, 91, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gale, S.A.; Acar, D.; Daffner, K.R. Dementia. Am. J. Med. 2018, 131, 1161–1169. [Google Scholar] [CrossRef] [PubMed]
- Uffelmann, E.; Huang, Q.Q.; Munung, N.S.; Vries, J.d.; Okada, Y.; Martin, A.R.; Martin, H.C.; Lappalainen, T.; Posthuma, D. Genome-wide association studies. Nat. Rev. Methods Prim. 2021, 1, 59. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Characteristic | Case (N = 578) | Control (N = 855) | p-Value |
|---|---|---|---|
| Gender (%) | |||
| Female | 346 (59.86) | 438 (51.22) | |
| Male | 232 (40.14) | 417 (48.77) | |
| Birth year (%) | < | ||
| Birth in 1900–1919 | 451 (78.03) | 486 (56.84) | |
| Birth in 1920–1929 | 126 (21.80) | 352 (41.16) | |
| Birth in 1930–1939 | 1 (0.17) | 17 (1.99) | |
| BMI (Mean ± SD) | 25.51 (±4.62) | 26.43 (±4.73) | |
| Smoke Status (%) | 0.46 | ||
| Never | 72 (43.11) | 223 (37.99) | |
| Current | 9 (5.39) | 39 (6.64) | |
| Past | 86 (54.50) | 325 (55.37) | |
| STC (Mean ± SD) | 232.41 (±42.37) | 229.47 (±43.10) | 0.26 |
| H-PSA (Mean ± SD) | 4.17 (±6.18) | 3.63 (±4.43) | 0.53 |
| Race (%) | 0.34 | ||
| Black or African American | 31 (5.37) | 32 (3.74) | |
| Unknown | 11 (1.90) | 10 (1.17) | |
| American Indian or Alaska Native | 2 (0.35) | 1 (0.12) | |
| Asian | 11 (1.90) | 19 (2.22) | |
| White | 523 (90.48) | 793 (92.70) |
| SNPID | CHR | p-Value | FDR | BP | GENE | |
|---|---|---|---|---|---|---|
| 1 | rs11556505 | 19 | 45396144 | TOMM40 * | ||
| 2 | rs2075650 | 19 | 45,395,619 | TOMM40 * | ||
| 3 | rs34404554 | 19 | 45,395,909 | TOMM40 * | ||
| 4 | rs71352238 | 19 | 45,394,336 | TOMM40 * | ||
| 5 | rs34095326 | 19 | 45,395,844 | TOMM40 * | ||
| 6 | rs72689267 | 15 | 26,117,761 | ATP10A-DT - | ||
| 7 | rs668168 | 18 | 8,392,719 | PTPRM - | ||
| 8 | rs144822097 | 12 | 27,183,821 | MED21 | ||
| 9 | rs670305 | 18 | 8,392,750 | PTPRM - | ||
| 10 | rs7178765 | 15 | 26,121,173 | ATP10A-DT - | ||
| 11 | rs4785108 | 16 | 60,446,014 | LOC101927605 | ||
| 12 | rs9888985 | 16 | 60,427,440 | LOC101927605 | ||
| 13 | rs10458022 | 6 | 58,308,335 | LOC101927293 |
| Gene | Function | Reference | |
|---|---|---|---|
| 1 | MED21 | an enzyme in humans | [24] |
| 2 | ATP10A-DT | it can affect neurodevelopmental disorders | [22] |
| 3 | PTPRM | it involved in the formation of synapses regulated by zinc ions, which is related to the transmission of information in the brain. | [23] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, X.; Dong, Y.; Xing, L.; Zhang, X. A Genome-Wide Association Study of Dementia Using the Electronic Medical Record. BioMedInformatics 2023, 3, 141-149. https://doi.org/10.3390/biomedinformatics3010010
Cao X, Dong Y, Xing L, Zhang X. A Genome-Wide Association Study of Dementia Using the Electronic Medical Record. BioMedInformatics. 2023; 3(1):141-149. https://doi.org/10.3390/biomedinformatics3010010
Chicago/Turabian StyleCao, Xiaowen, Yao Dong, Li Xing, and Xuekui Zhang. 2023. "A Genome-Wide Association Study of Dementia Using the Electronic Medical Record" BioMedInformatics 3, no. 1: 141-149. https://doi.org/10.3390/biomedinformatics3010010