
The True Nature of the Energy Calibration for Nuclear Resonant Vibrational Spectroscopy: A Time-Based Conversion


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
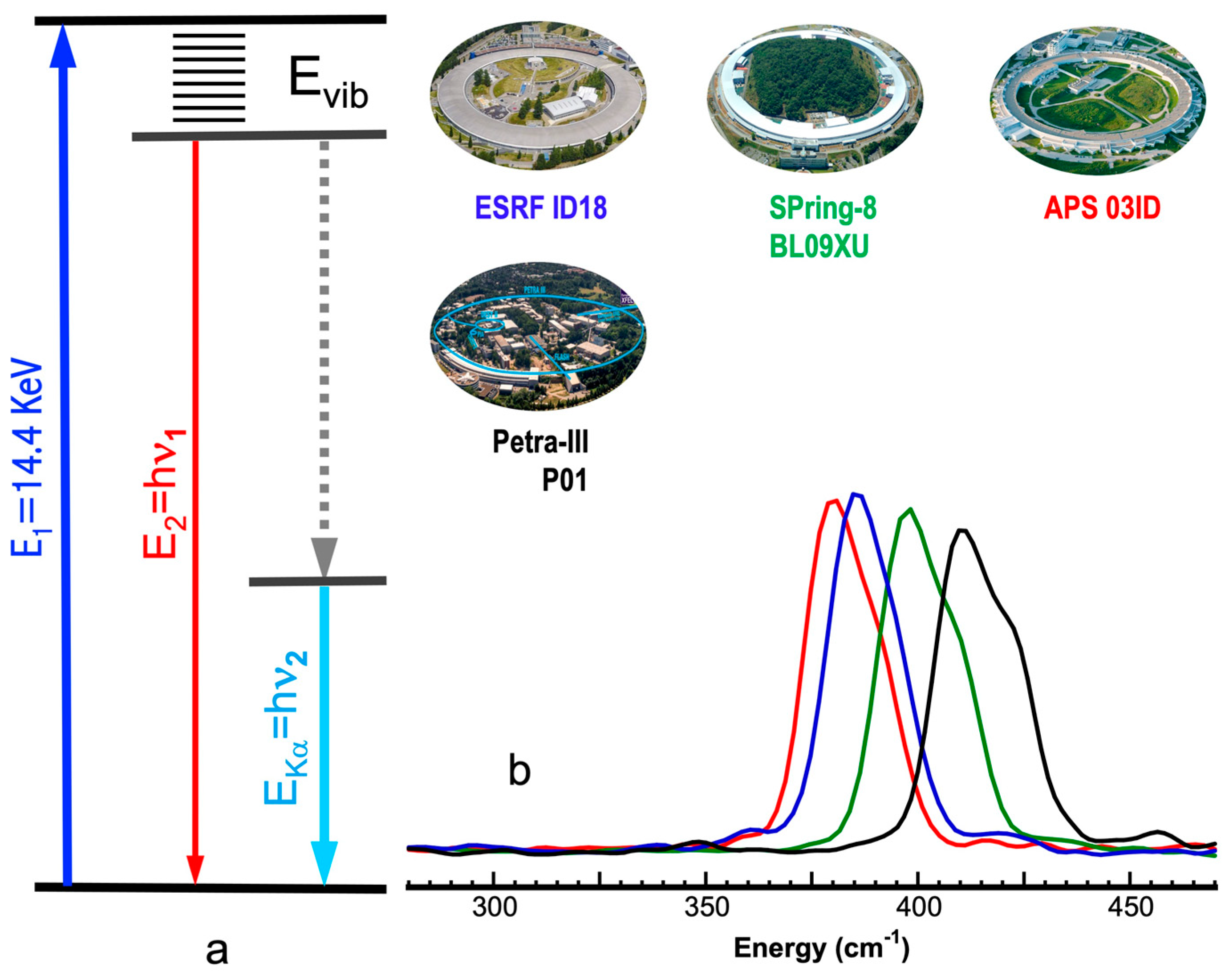
1.1. Nuclear Resonant Vibrational Spectroscopy
1.2. Energy Calibration in NRVS
1.3. Using In-Situ Energy Calibrations
2. Experimental Aspects
3. Results and Discussions
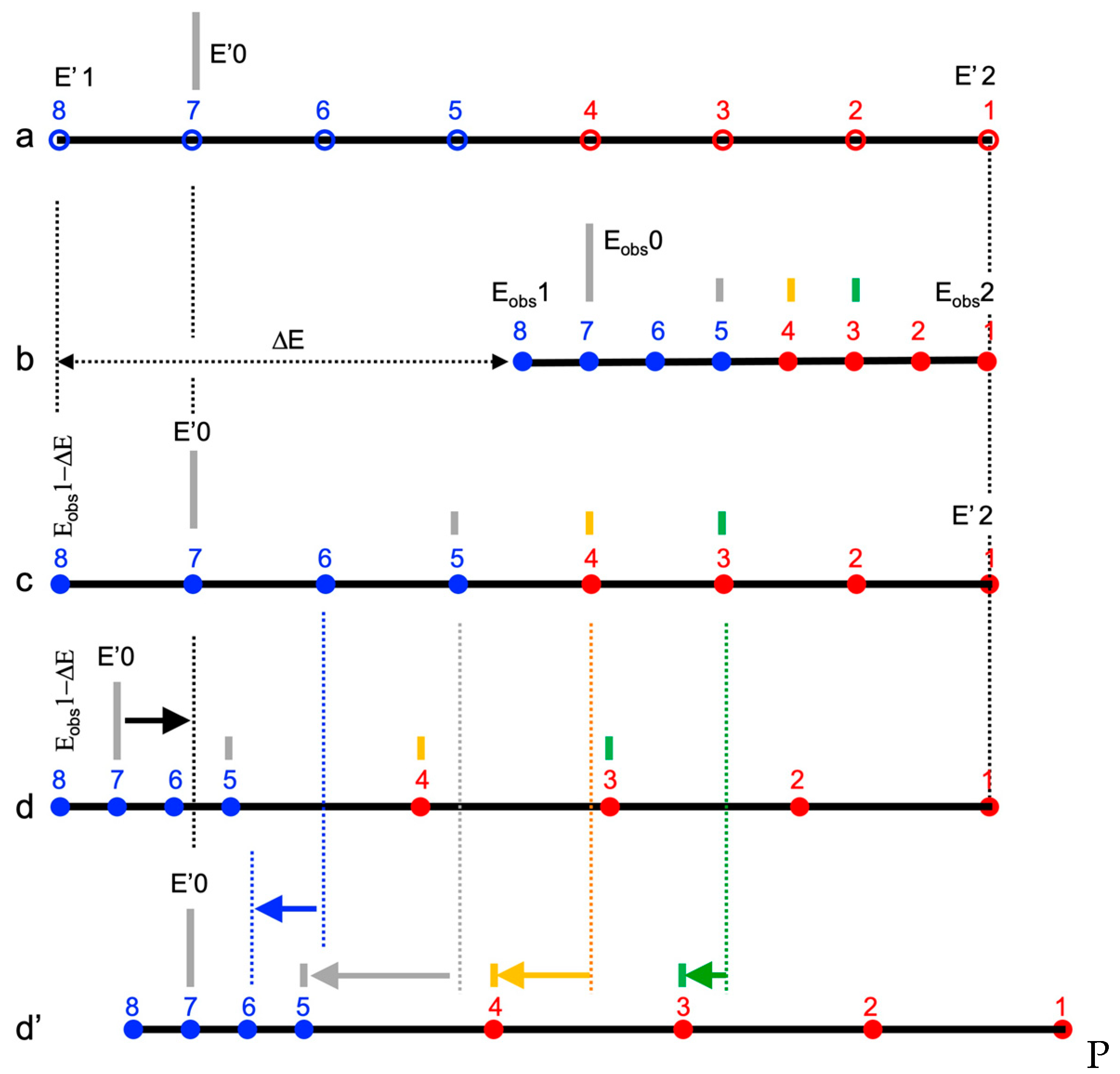
3.1. Understanding Time-Scaled Calibrations
= Eobs⋅{1 + ΔEi/(E2obs − E1obs)} − {E2obs⋅ΔEi/(E2obs − E1obs)}
3.2. Comparing Two Calibration Procedures
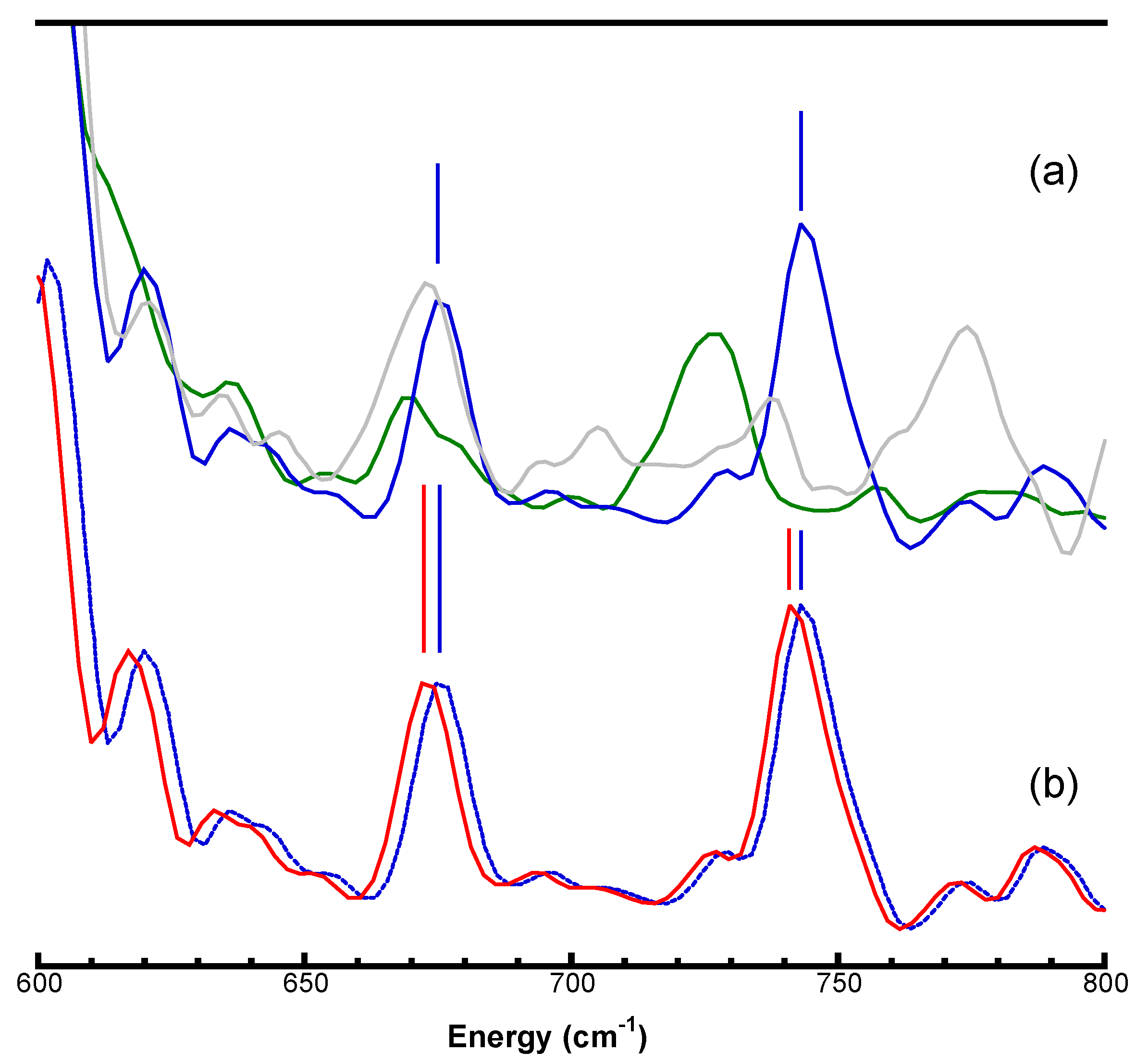
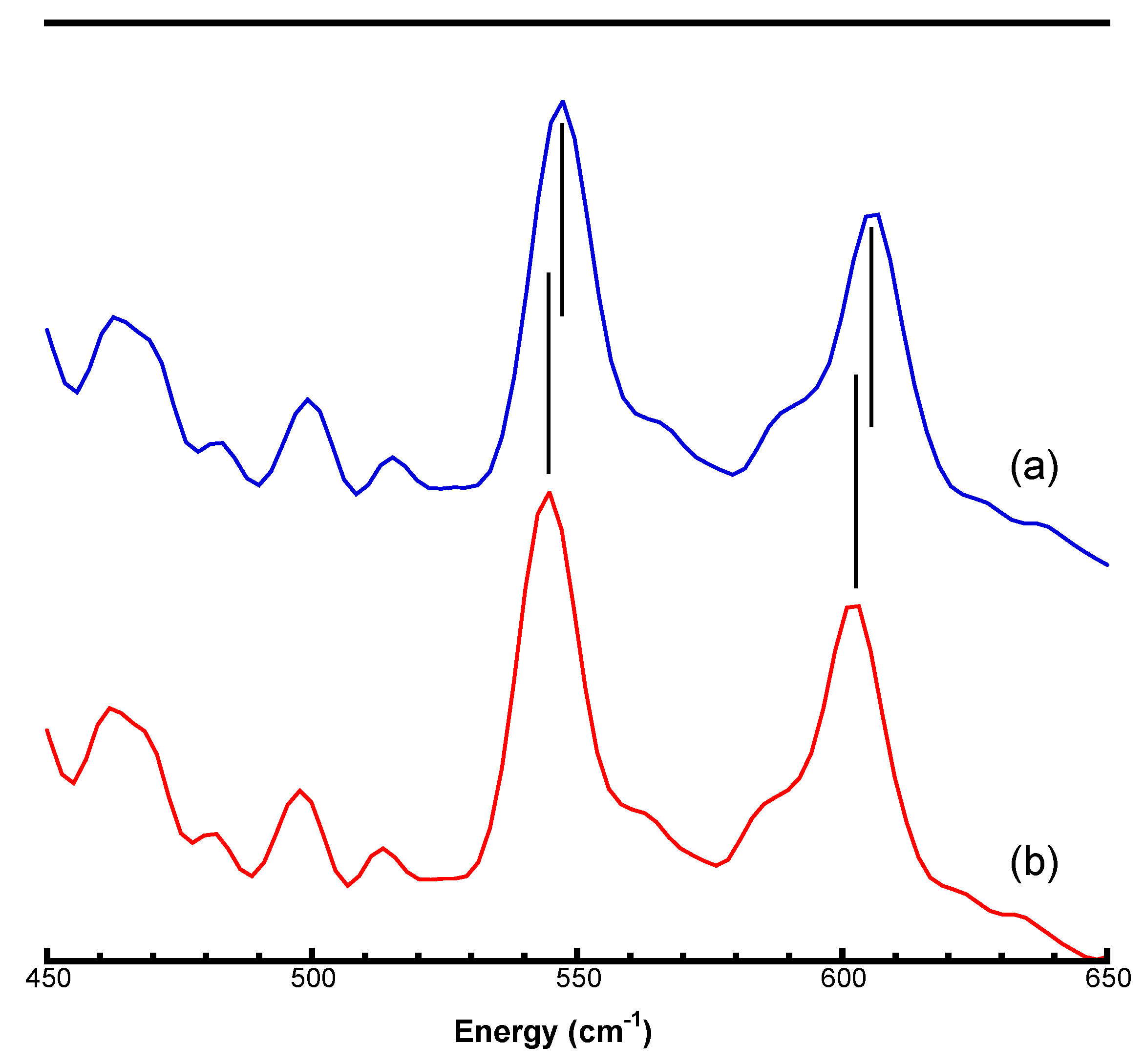
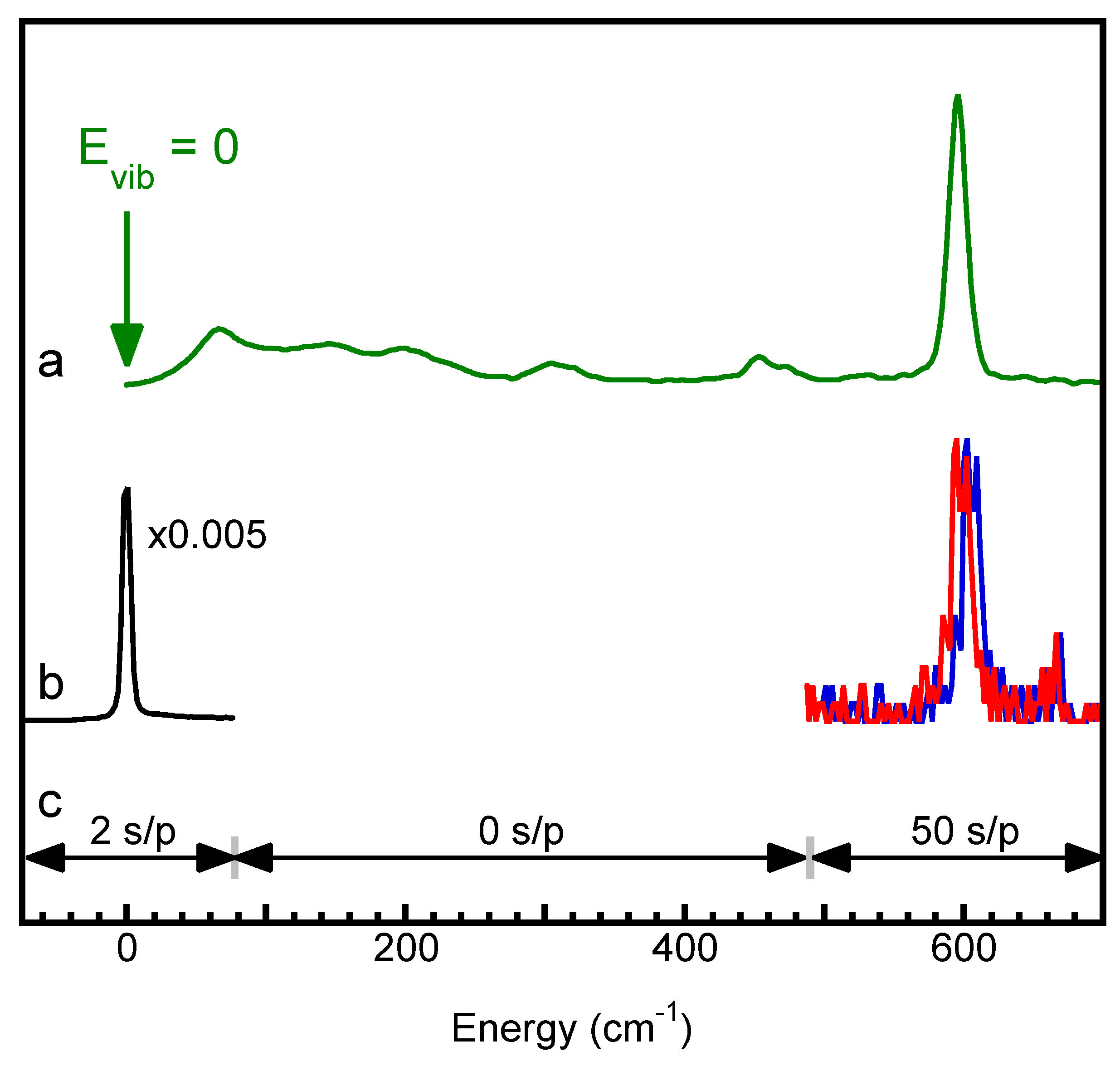
3.3. Re-Calibrating Published PVDOS
= [E*real/α − (Σtk/Ttot)⋅(ΔE)]⋅α/{1 + ΔE/(E2*obs − E1*obs)}
3.4. Examples of Re-Calibrated NRVS
3.5. Dealing with Jump Scans
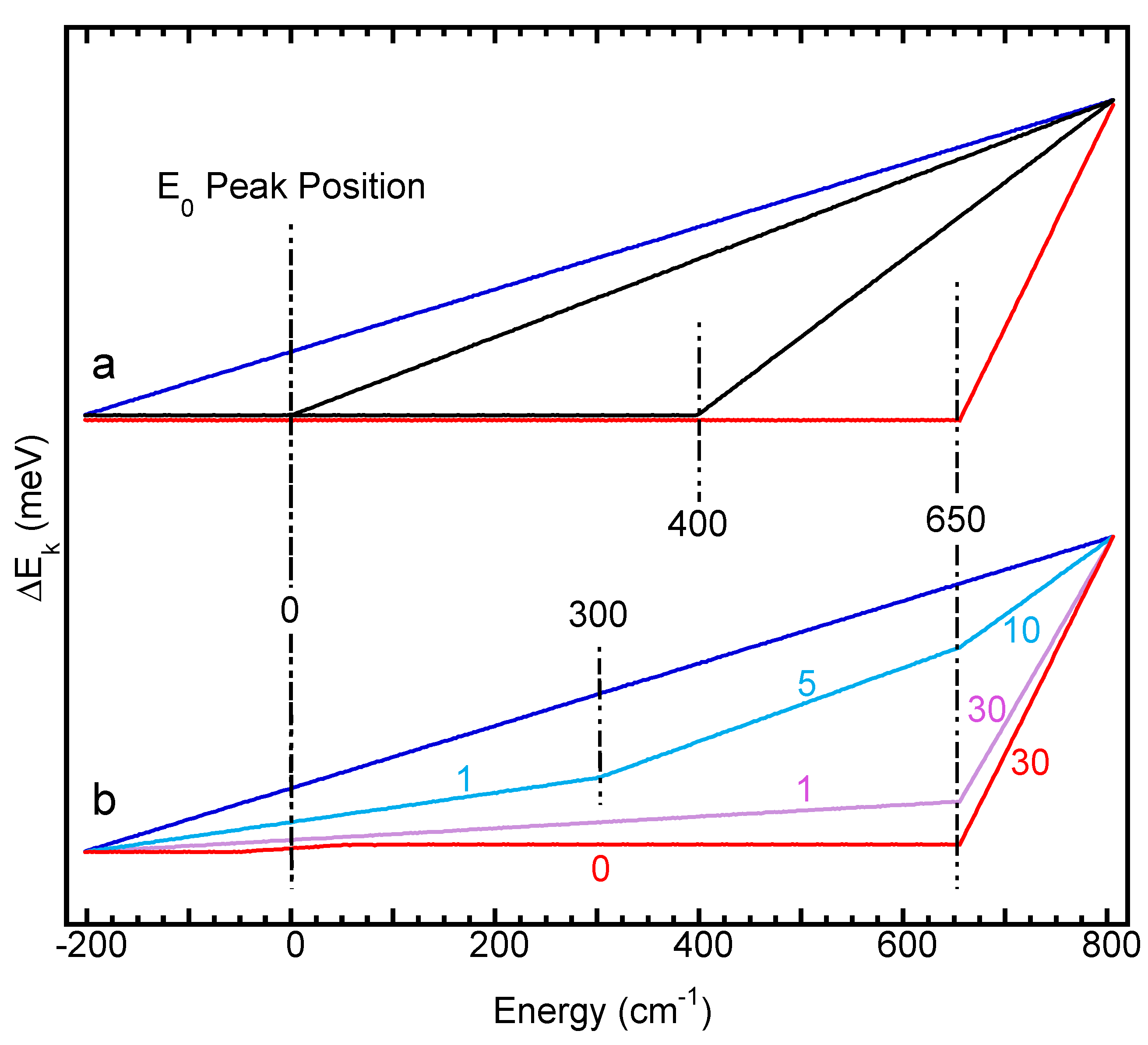
3.6. Further Discussions
- (1)
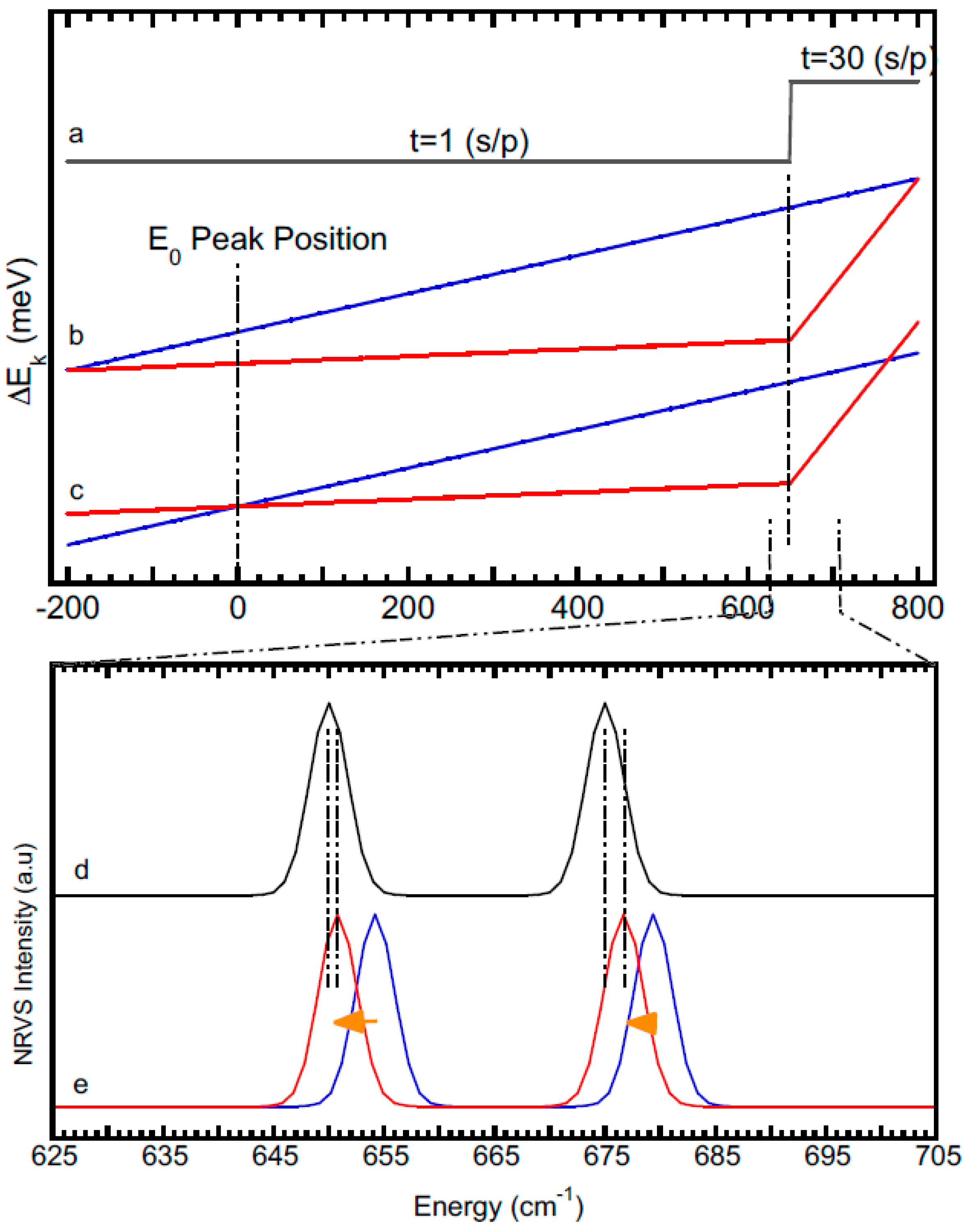
- it is obvious that the wider the scanning region (vs. the skipped region) [(800 → 650 cm−1) → (800 → 400 cm−1) → (800 → 0 cm−1)], the closer its energy distribution curve to the case of an even NRVS scan. For example, the left black curve is the closest one to the blue curve in Figure 8a;
- (2)
- the time ratio of the two adjacent scanning regions dictates the difference between the energies calculated via the Equation (1) and those calculated via the Equation (2): for example, the 5:10 [Figure 8b, light blue] provides a much closer result to the even time scan (blue) than the 1:30 (purple) or 30:0 (red) does. This leads to the concept that a multiple section scan that changes the scanning time at a gradual pace is closer to the even time scan. In practical measurements, it is better to start with an even time scan or a sectional scan but with multiple and stepwise changes in its scanning time parameters. When the major features are all well resolved and calibrated, some extremely weak features need to be probed with heavy counting on one region, such as the 30/1 or the 30/0 s/p scans discussed in Figure 8a. It becomes necessary to use the time-based energy calibration procedure (2) for any sectional scans but it is especially necessary for the ones with large time steps, such as the 30/1 or 30/0 ones;
- (3)
- no matter which calibration procedure is to be used and no matter what the scanning parameters, no point should have an energy drift amount greater than the total energy drift per scan (ΔEi). Therefore, the final factor for controlling the possible energy calibration errors is to scan NRVS with a lesser ΔEi value. For example, NRVS experimentalists need to avoid the moments right after each hutch opening or so and wait for the beam to be stabilized. For further reduction of ΔEi values, NRVS users also have the option to use less time for each scan and take more scans to average.

4. Summary
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Seto, M.; Yoda, Y.; Kikuta, S.; Zhang, X.W.; Ando, M. Observation of Nuclear Resonant Scattering Accompanied by Phonon Excitation Using Synchrotron Radiation. Phys. Rev. Lett. 1995, 74, 3828–3831. [Google Scholar] [CrossRef]
- Sturhahn, W.; Toellner, T.S.; Alp, E.E.; Zhang, X.; Ando, M.; Yoda, Y.; Kikuta, S.; Seto, M.; Kimball, C.W.; Dabrowski, B. Phonon Density of States Measured by Inelastic Nuclear Resonant Scattering. Phys. Rev. Lett. 1995, 74, 3832–3835. [Google Scholar] [CrossRef] [Green Version]
- Yoda, Y.; Yabashi, M.; Izumi, K.; Zhang, X.W.; Kishimoto, S.; Kitao, S.; Seto, M.; Mitsui, T.; Harami, T.; Imai, Y.; et al. Nuclear resonant scattering beamline at SPring-8. Nucl. Inst. Meth. A 2001, 467, 715–718. [Google Scholar] [CrossRef]
- Yoda, Y.; Imai, Y.; Kobayashi, H.; Goto, S.; Takeshita, K.; Seto, M. Upgrade of the nuclear resonant scattering beamline, BL09XU in SPring-8. Hyperfine Interact. 2012, 206, 83. [Google Scholar] [CrossRef]
- Wang, H.; Yoda, Y.; Dong, W.; Huang, S.D. Energy calibration issues in nuclear resonant vibrational spectroscopy: Observing small spectral shifts and making fast calibrations. J. Synchrotron Radiat. 2013, 20, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Alp, E.; Yoda, Y.; Cramer, S. A Practical Guide for Nuclear Resonance Vibrational Spectroscopy (NRVS) of Biochemical Samples and Model Compounds. In Metalloproteins; Fontecilla-Camps, J.C., Nicolet, Y., Eds.; Humana Press: Totowa, NJ, USA, 2014; Volume 1122, pp. 125–137. [Google Scholar]
- Yoda, Y.; Okada, K.; Wang, H.; Cramer, S.P.; Seto, M. High-resolution monochromator for iron nuclear resonance vibrational spectroscopy of biological samples. Jap. J. App. Phys. 2016, 55, 122401. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Braun, A.; Cramer, S.P.; Gee, L.B.; Yoda, Y. Nuclear Resonance Vibrational Spectroscopy: A Modern Tool to Pinpoint Site-Specific Cooperative Processes. Crystals 2021, 11, 909. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, L.; Wang, H. Machine learning concept in de-spiking process for nuclear resonant vibrational spectra—Automation using no external parameter. Vib. Spectrosc. 2022, 119, 103352. [Google Scholar] [CrossRef]
- Wang, H.; Yoda, Y.; Ogata, H.; Tanaka, Y.; Lubitz, W. A strenuous experimental journey searching for spectroscopic evidence of a bridging nickel-iron-hydride in [NiFe] hydrogenase. J. Synchrotron Radiat. 2015, 22, 1334–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Salzberg, A.P.; Weiner, B.R. Laser ablation of aluminum at 193, 248, and 351 nm. Appl. Phys. Lett. 1991, 59, 935–937. [Google Scholar] [CrossRef]
- He, M.; Wang, H.; Weiner, B.R. Production and laser-induced fluorescence spectrum of aluminum sulfide. Chem. Phys. Lett. 1993, 204, 563–566. [Google Scholar] [CrossRef]
- Wang, H.; Chen, X.; Weiner, B.R. Laser photodissociation dynamics of thionyl chloride: Concerted and stepwise cleavage of S-Cl bonds. J. Phys. Chem. 1993, 97, 12260–12268. [Google Scholar] [CrossRef]
- Chen, X.; Asmar, F.; Wang, H.; Weiner, B.R. Nascent sulfur monoxide (X3.SIGMA.-) vibrational distributions from the photodissociation of sulfur dioxide, sulfonyl chloride, and dimethylsulfoxide at 193 nm. J. Phys. Chem. 1991, 95, 6415–6417. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, H.; Xiao, Y.; Vogt, S.; Thauer, R.K.; Shima, S.; Volkers, P.I.; Rauchfuss, T.B.; Pelmenschikov, V.; Case, D.A.; et al. Characterization of the Fe site in iron-sulfur cluster-free hydrogenase (Hmd) and of a model compound via nuclear resonance vibrational spectroscopy (NRVS). Inorg. Chem. 2008, 47, 3969–3977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.C.; Xiao, Y.; Wang, H.; George, S.J.; Coucouvanis, D.; Koutmos, M.; Sturhahn, W.; Alp, E.E.; Zhao, J.; Cramer, S.P. Normal-Mode Analysis of FeCl4- and Fe2S2Cl42- via Vibrational Mössbauer, Resonance Raman, and FT-IR Spectroscopies. Inorg. Chem. 2005, 44, 5562–5570. [Google Scholar] [CrossRef]
- Xiao, Y.; Wang, H.; George, S.J.; Smith, M.C.; Adams, M.W.; Jenney, F.E., Jr.; Sturhahn, W.; Alp, E.E.; Zhao, J.; Yoda, Y.; et al. Normal mode analysis of Pyrococcus furiosus rubredoxin via nuclear resonance vibrational spectroscopy (NRVS) and resonance raman spectroscopy. J. Am. Chem. Soc. 2005, 127, 14596–14606. [Google Scholar] [CrossRef]
- Xiao, Y.; Fisher, K.; Smith, M.C.; Newton, W.; Case, D.A.; George, S.J.; Wang, H.; Sturhahn, W.; Alp, E.E.; Zhao, J.; et al. How Nitrogenase Shakes—Initial Information about P-Cluster and FeMo-Cofactor Normal Modes from Nuclear Resonance Vibrational Spectroscopy (NRVS). J. Am. Chem. Soc. 2006, 128, 7608–7612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gee, L.B.; Pelmenschikov, V.; Wang, H.; Mishra, N.; Liu, Y.-C.; Yoda, Y.; Tamasaku, K.; Chiang, M.-H.; Cramer, S.P. Vibrational characterization of a diiron bridging hydride complex—A model for hydrogen catalysis. Chem. Sci. 2020, 11, 5487–5493. [Google Scholar] [CrossRef] [PubMed]
- Gee, L.B.; Wang, H.; Cramer, S.P. Chapter Fourteen—NRVS for Fe in Biology: Experiment and Basic Interpretation. In Methods in Enzymology; David, S.S., Ed.; Academic Press: Cambridge, MA, USA, 2018; Volume 599, pp. 409–425. [Google Scholar]
- Guo, Y.; Brecht, E.; Aznavour, K.; Nix, J.C.; Xiao, Y.; Wang, H.; George, S.J.; Bau, R.; Keable, S.; Peters, J.W.; et al. Nuclear resonance vibrational spectroscopy (NRVS) of rubredoxin and MoFe protein crystals. Hyperfine Interact. 2012, 222, 77–90. [Google Scholar] [CrossRef]
- Ogata, H.; Kramer, T.; Wang, H.; Schilter, D.; Pelmenschikov, V.; van Gastel, M.; Neese, F.; Rauchfuss, T.B.; Gee, L.B.; Scott, A.D.; et al. Hydride bridge in [NiFe]-hydrogenase observed by nuclear resonance vibrational spectroscopy. Nat. Commun. 2015, 6, 7890. [Google Scholar] [CrossRef] [Green Version]
- Kamali, S.; Wang, H.; Mitra, D.; Ogata, H.; Lubitz, W.; Manor, B.C.; Rauchfuss, T.B.; Byrne, D.; Bonnefoy, V.; Jenney, F.E.; et al. Observation of the Fe-CN and Fe-CO Vibrations in the Active Site of [NiFe] Hydrogenase by Nuclear Resonance Vibrational Spectroscopy. Angew. Chem. Int. Ed. 2013, 52, 724–728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tinberg, C.E.; Tonzetich, Z.J.; Wang, H.; Do, L.H.; Yoda, Y.; Cramer, S.P.; Lippard, S.J. Characterization of Iron Dinitrosyl Species Formed in the Reaction of Nitric Oxide with a Biological Rieske Center. J. Am. Chem. Soc. 2010, 132, 18168–18176. [Google Scholar] [CrossRef] [PubMed]
- Do, L.H.; Wang, H.; Tinberg, C.E.; Dowty, E.; Yoda, Y.; Cramer, S.P.; Lippard, S.J. Characterization of a synthetic peroxodiiron(III) protein model complex by nuclear resonance vibrational spectroscopy. Chem. Commun. 2011, 47, 10945–10947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbert-Wilson, R.; Siebel, J.F.; Adamska-Venkatesh, A.; Pham, C.C.; Reijerse, E.; Wang, H.; Cramer, S.P.; Lubitz, W.; Rauchfuss, T.B. Spectroscopic Investigations of [FeFe] Hydrogenase Maturated with [57Fe2(adt)(CN)2(CO)4]2−. J. Am. Chem. Soc. 2015, 137, 8998–9005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelmenschikov, V.; Birrell, J.A.; Pham, C.C.; Mishra, N.; Wang, H.X.; Sommer, C.; Reijerse, E.; Richers, C.P.; Tamasaku, K.; Yoda, Y.; et al. Reaction Coordinate Leading to H-2 Production in [FeFe]-Hydrogenase Identified by Nuclear Resonance Vibrational Spectroscopy and Density Functional Theory. J. Am. Chem. Soc. 2017, 139, 16894–16902. [Google Scholar] [CrossRef] [PubMed]
- Pham, C.C.; Mulder, D.W.; Pelmenschikov, V.; King, P.W.; Ratzloff, M.W.; Wang, H.; Mishra, N.; Alp, E.E.; Zhao, J.; Hu, M.Y.; et al. Terminal Hydride Species in [FeFe]-Hydrogenases are Vibrationally Coupled to the Active Site Environment. Ang. Chem. 2018, 57, 10605–10609. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Huang, S.D.; Yan, L.; Hu, M.Y.; Zhao, J.; Alp, E.E.; Yoda, Y.; Petersen, C.M.; Thompson, M.K. Europium-151 and iron-57 nuclear resonant vibrational spectroscopy of naturally abundant KEu(iii)Fe(ii)(CN)6 and Eu(iii)Fe(iii)(CN)6 complexes. Dalton Trans. 2022. [Google Scholar] [CrossRef] [PubMed]
- Wittkamp, F.; Mishra, N.; Wang, H.; Wille, H.-C.; Steinbrügge, R.; Kaupp, M.; Cramer, S.P.; Apfel, U.-P.; Pelmenschikov, V. Insights from 125Te and 57Fe nuclear resonance vibrational spectroscopy: A [4Fe–4Te] cluster from two points of view. Chem. Sci. 2019, 10, 7535–7541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pelmenschikov, V.; Birrell, J.A.; Gee, L.B.; Richers, C.P.; Reijerse, E.J.; Wang, H.; Arragain, S.; Mishra, N.; Yoda, Y.; Matsuura, H.; et al. Vibrational Perturbation of the [FeFe] Hydrogenase H-Cluster Revealed by 13C2H-ADT Labeling. J. Am. Chem. Soc. 2021, 143, 8237–8243. [Google Scholar] [CrossRef]
- Gu, W.W.; Jacquamet, L.; Patil, D.S.; Wang, H.X.; Evans, D.J.; Smith, M.C.; Millar, M.; Koch, S.; Eichhorn, D.M.; Latimer, M.; et al. Refinement of the nickel site structure in Desulfovibrio gigas hydrogenase using range-extended EXAFS spectroscopy. J. Inorg. Biochem. 2003, 93, 41–51. [Google Scholar] [CrossRef]
- DeBeer George, S.; Metz, M.; Szilagyi, R.K.; Wang, H.; Cramer, S.P.; Lu, Y.; Tolman, W.B.; Hedman, B.; Hodgson, K.O.; Solomon, E.I. A Quantitative Description of the Ground-State Wave Function of CuA by X-ray Absorption Spectroscopy: Comparison to Plastocyanin and Relevance to Electron Transfer. J. Am. Chem. Soc. 2001, 123, 5757–5767. [Google Scholar] [CrossRef] [PubMed]
- O’Dowd, B.; Williams, S.; Wang, H.X.; No, J.H.; Rao, G.D.; Wang, W.X.; McCammon, J.A.; Cramer, S.P.; Oldfield, E. Spectroscopic and Computational Investigations of Ligand Binding to IspH: Discovery of Non-diphosphate Inhibitors. Chembiochem 2017, 18, 914–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funk, T.; Deb, A.; George, S.J.; Wang, H.; Cramer, S.P. X-ray magnetic circular dichroism—A high energy probe of magnetic properties. Coord. Chem. Rev. 2005, 249, 3–30. [Google Scholar] [CrossRef]
- Wang, H.X.; Ralston, C.Y.; Patil, D.S.; Jones, R.M.; Gu, W.; Verhagen, M.; Adams, M.; Ge, P.; Riordan, C.; Marganian, C.A.; et al. Nickel L-edge soft X-ray spectroscopy of nickel-iron hydrogenases and model compounds—Evidence for high-spin nickel(II) in the active enzyme. J. Am. Chem. Soc. 2000, 122, 10544–10552. [Google Scholar] [CrossRef]
- Wang, H.X.; Ge, P.H.; Riordan, C.G.; Brooker, S.; Woomer, C.G.; Collins, T.; Melendres, C.A.; Graudejus, O.; Bartlett, N.; Cramer, S.P. Integrated X-ray L absorption spectra. Counting holes in Ni complexes. J. Phys. Chem. B 1998, 102, 8343–8346. [Google Scholar] [CrossRef]
- Wang, H.; Friedrich, S.; Li, L.; Mao, Z.; Ge, P.; Balasubramanian, M.; Patil, D.S. L-edge sum rule analysis on 3d transition metal sites: From d10 to d0 and towards application to extremely dilute metallo-enzymes. Phys. Chem. Chem. Phys. 2018, 20, 8166–8176. [Google Scholar] [CrossRef] [PubMed]
- Pierce and Rod. Accuracy and Precision. Available online: https://www.mathsisfun.com/accuracy-precision.html (accessed on 1 June 2022).
- Wang, J.; Yoda, Y.; Wang, H. Tracking energy scale variations from scan to scan in nuclear resonant vibrational spectroscop y: In situ correction using zero-energy position drifts ΔEi rather than making in situ calibration measurements. Rev. Sci. Instrum. 2022, 93, 095101. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.Y.; Sturhahn, W. High-energy-resolution X-ray monochromator calibration using the detailed-balance principle. J. Synchrotron Radiat. 2012, 19, 602–608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sturhahn, W. Nuclear resonant spectroscopy. J. Phys. Condens. Matter 2004, 16, S497–S530. [Google Scholar] [CrossRef]
- Sturhahn, W. CONUSS and PHOENIX: Evaluation of nuclear resonant scattering data. Hyperfine Interact. 2000, 125, 149–172. [Google Scholar] [CrossRef]
- Watanabe, H.; Yamada, N.; Okaji, M. Linear Thermal Expansion Coefficient of Silicon from 293 to 1000 K. Int. J. Thermophys. 2004, 25, 221–236. [Google Scholar] [CrossRef]
- Lumen-Physics. Heat and Heat Transfer Methods. Available online: https://courses.lumenlearning.com/physics/chapter/14-5-conduction/ (accessed on 1 June 2022).
- Resnick, R.; Halliday, D.; Walker, J. Fundamentals of Physics Extended; Welly: New York, NY, USA, 2016. [Google Scholar]
- Morra, S. Fantastic [FeFe]-Hydrogenases and Where to Find Them. Front. Microbiol. 2022, 13, 853626. [Google Scholar] [CrossRef] [PubMed]
- Chiang, M.-H.; Pelmenschikov, V.; Gee, L.B.; Liu, Y.-C.; Hsieh, C.-C.; Wang, H.; Yoda, Y.; Matsuura, H.; Li, L.; Cramer, S.P. High-Frequency Fe–H and Fe–H2 Modes in a trans-Fe(η2-H2)(H) Complex: A Speed Record for Nuclear Resonance Vibrational Spectroscopy. Inorg. Chem. 2021, 60, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Chumakov, A.I.; Rüffer, R.; Leupold, O.; Sergueev, I. Insight to Dynamics of Molecules with Nuclear Inelastic Scattering. Struct. Chem. 2003, 14, 109–119. [Google Scholar] [CrossRef]
- Shokouhimehr, M.; Soehnlen, E.S.; Hao, J.; Griswold, M.; Flask, C.; Fan, X.; Basilion, J.P.; Basu, S.; Huang, S.D. Dual purpose Prussian blue nanoparticles for cellular imaging and drug delivery: A new generation of T-1-weighted MRI contrast and small molecule delivery agents. J. Mater. Chem. 2010, 20, 5251–5259. [Google Scholar] [CrossRef]
- Kandanapitiye, M.S.; Valley, B.; Yang, L.D.; Fry, A.M.; Woodward, P.M.; Huang, S.D. Gallium Analogue of Soluble Prussian Blue KGa Fe(CN)(6) center dot nH(2)O: Synthesis, Characterization, and Potential Biomedical Applications. Inorg. Chem. 2013, 52, 2790–2792. [Google Scholar] [CrossRef] [PubMed]
- Seto, M.; Kitao, S.; Kobayashi, Y.; Haruki, R.; Mitsui, T.; Yoda, Y.; Zhang, X.W.; Kishimoto, S.; Maeda, Y. Nuclear Resonant Inelastic and Forward Scattering of Synchrotron Radiation by 40K. Hyperfine Interact. 2002, 141, 99–108. [Google Scholar] [CrossRef]
- Kobayashi, H.; Yoda, Y.; Shirakawa, M.; Ochiai, A. 151Eu Nuclear Resonant Inelastic Scattering of Eu4As3 around Charge Ordering Temperature. J. Phys. Soc. Jpn. 2006, 75, 034602. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Yoda, Y.; Wang, J. The True Nature of the Energy Calibration for Nuclear Resonant Vibrational Spectroscopy: A Time-Based Conversion. Physchem 2022, 2, 369-388. https://doi.org/10.3390/physchem2040027
Wang H, Yoda Y, Wang J. The True Nature of the Energy Calibration for Nuclear Resonant Vibrational Spectroscopy: A Time-Based Conversion. Physchem. 2022; 2(4):369-388. https://doi.org/10.3390/physchem2040027
Chicago/Turabian StyleWang, Hongxin, Yoshitaka Yoda, and Jessie Wang. 2022. "The True Nature of the Energy Calibration for Nuclear Resonant Vibrational Spectroscopy: A Time-Based Conversion" Physchem 2, no. 4: 369-388. https://doi.org/10.3390/physchem2040027