
Fluorinated Merophosphinine and Phosphinine Dyes: Synthesis and Evaluation of UV-Visible Light Absorption Properties


Faculty of Molecular Chemistry and Materials Engineering, Kyoto Institute of Technology, Matsugasaki, Sakyo-ku, Kyoto 606-8585, Japan
*
Author to whom correspondence should be addressed.
Compounds 2023, 3(1), 153-168; https://doi.org/10.3390/compounds3010013
Submission received: 26 December 2022
/
Revised: 12 January 2023
/
Accepted: 19 January 2023
/
Published: 2 February 2023
(This article belongs to the Special Issue Feature Papers in Compounds (2022–2023))
Abstract
:Merophosphinine and phosphinine dyes were reported as phosphorus atom equivalents of merocyanine and cyanine dyes in the 1960s. Although these dyes are excellent sensitizers for enhancing photographic performance, their development has been considerably retarded because of difficulties in the synthetic process. Previously, while investigating the reactivity of fluoroalkenes with various nucleophiles, we developed fluorinated merophosphinine and phosphinine scaffolds in a single reaction step; however, we did not investigate their ultraviolet-visible (UV-vis) light absorption properties further. Therefore, in this study, we synthesized fluorinated merophosphinines and phosphinine derivatives using triphenylphosphonium ylides with various R substituents [R = H, C6H5, 4-MeOC6H4, 4-PhC6H4, or C6F5] and octafluorocyclopentene to study their absorption characteristics. Thermogravimetric analyses (TGA) indicated that several synthesized derivatives were thermally stable at temperatures above 241 °C. Additionally, when studied in various solvents, the fluorinated merophosphinine and phosphinine dyes exhibited well-defined maximum absorption wavelengths (λabs) in the 364–375 and 420–474 nm ranges, respectively. Thus, fluorinated merophosphinine and phosphinine derivatives may serve as promising thermally stable functional dyes for future technical applications.
1. Introduction
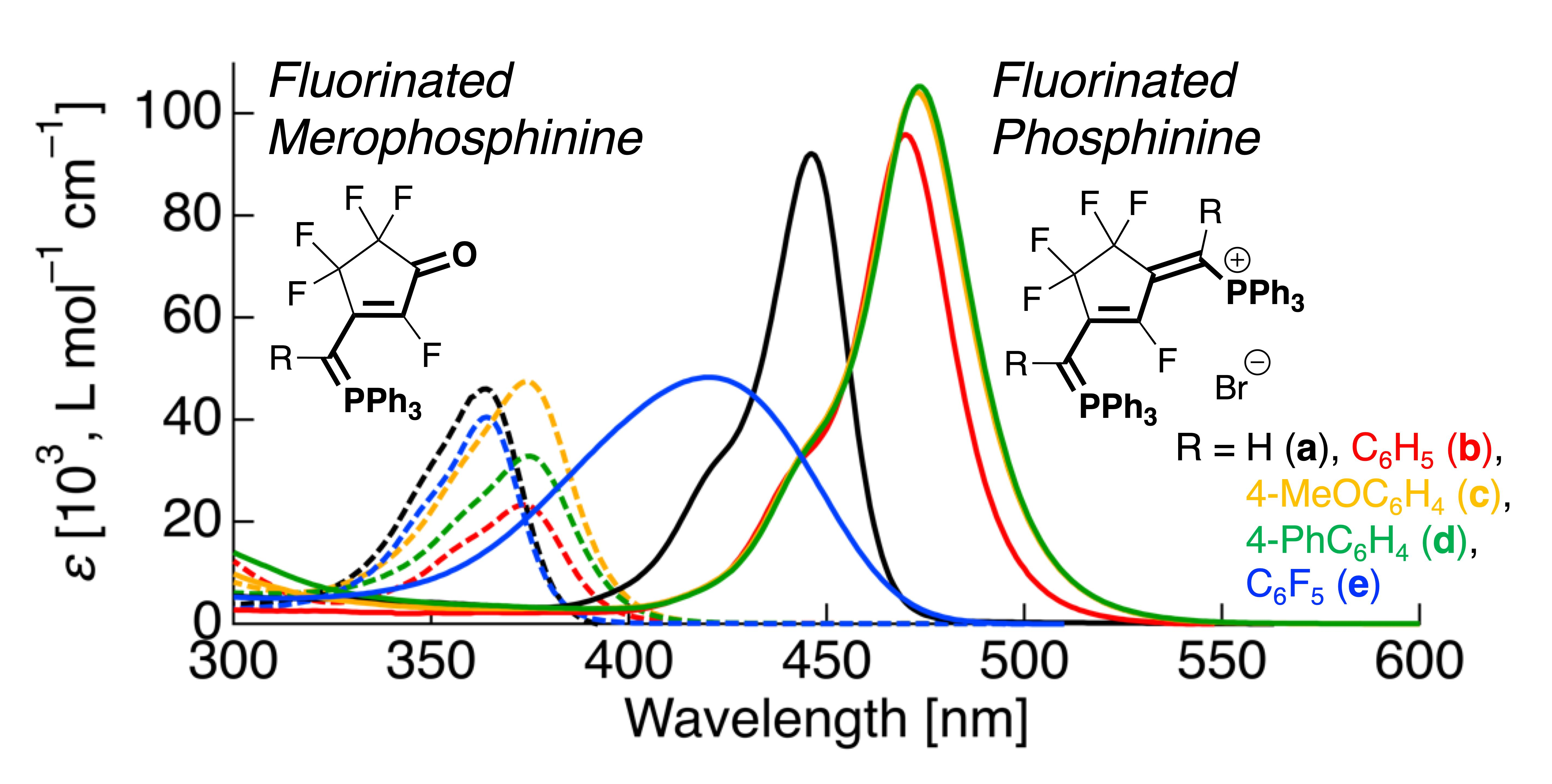
Organic dyes have attracted considerable interest because they absorb light and are key functional materials in electronics and medical diagnostics [1,2,3]. Among these dyes, merocyanine and cyanine dyes belong to the class of polymethine dyes. Merocyanine, which is composed of a –CH=CH– chain modified with an amino group (NR2) and an oxygen atom at each molecular terminal (Figure 1a), can be used as a dye sensitizer for photographic film or photovoltaic cells [4]. As a representative example of trimethine merocyanine, Michler’s ketone and merocyanine-540 (MC-540) have been reported to absorb light at 333 nm in hexane and at 574 nm, respectively, through π-π* transition [5,6]. Additionally, cyanines, which are modified with two identical NR2 groups at both molecular terminals of polyenes with bond alternations (Figure 1b), representatively showed absorption of visible light at a range of 550–740 nm through π-π* transition depending on the methine-linkage; as the methine-linkage is extended, the absorption wavelength shifts to the longer wavelength region [7,8]. Therefore, it also serves as a promising fluorescent dye for biomedical imaging [9,10,11].
Thus far, several structural analogs of merocyanine and cyanine dyes have been reported. For example, phosphorus-containing merophosphinine (Figure 1c) and phosphinine derivatives (Figure 1d) are structural analogs wherein the NR2 groups in the structures of merocyanine and cyanine are replaced by the PR3 groups. Although some absorption characteristics for pentamethine phosphinine have been reported so far (absorption wavelength: 664–749 nm range in DMSO) [12], little attention has been received to research the photophysical properties and electronic transitions of other derivatives. These dyes are excellent sensitizers that enhance photosensitivity with excellent photographic performances [13,14]. However, since Depoorter et al. reported merophosphinine and phosphinine derivatives, the development of dyes based on their structural analogs has been hindered because of difficulties in the synthetic process.
In previous studies, we have developed reactions using fluoroalkenes as starting materials to selectively synthesize various fluorine-containing organic compounds [15,16,17,18,19,20]. While investigating the reaction of octafluorocyclopentene (2) with various carbon nucleophiles, we found that the reaction of this with phosphonium ylide (1a), prepared from methylphosphonium bromide, selectively produced the PR3-terminated fluorinated π-conjugated ketone 3a (CCDC number: 635725) and the π-conjugated phosphonium salt 4aA (CCDC number: 635724), as shown in Figure 2a [18].
In the previous study, the structural features of compounds 3 and 4 were not identified; however, we have recently confirmed them to be fluorinated merophosphinine and phosphinine derivatives, respectively. Motivated by the potential of these compounds as photosensitizing dyes we, therefore, investigated the thermal stabilities and UV-vis absorption properties of several derivatives of 3 and 4 incorporating various aromatic substituents and different counter anions (Figure 2b).
2. Materials and Methods
2.1. General Methods
All reactions were performed using dried glassware and magnetic stir bars. All chemicals were of reagent grade and purified in the usual manner before use. Column chromatography was carried out using Wakogel® 60 N (38–100 μm) as the stationary phase, and thin-layer chromatography (TLC) analyses were performed on silica gel TLC plates (Merck, Silica gel 60F254). 1H and 13C nuclear magnetic resonance (NMR) spectra were obtained in chloroform-d (CDCl3) using an AVANCE III 400 NMR spectrometer (1H: 400 MHz and 13C: 100 MHz) (Bruker Corporation, Billerica, MA, USA), and chemical shifts were reported in parts per million (ppm) using the residual proton in the NMR solvent. 19F NMR (376 MHz) spectra were obtained using a Bruker AVANCE III 400 NMR spectrometer in CDCl3, and either hexafluorobenzene (δF = −163 ppm) or benzotrifluoride (δF = −63 ppm) was used as an internal standard. Infrared (IR) spectra were recorded using the KBr method with an FT/IR-4100 type A spectrometer (JASCO Corporation, Tokyo, Japan); all spectra were reported in wavenumbers (cm–1). High-resolution mass spectrometry (HRMS) was conducted using a JEOL JMS-700MS spectrometer (JASCO Corporation, Tokyo, Japan) employing the fast atom bombardment (FAB) method. HRMS data for fluorinated phosphinines, namely 4bA–4eA and 4aB–4aE, are reported for the mass number of anionic structures. Detailed information for the NMR spectrum, Quantum chemical calculation, and Car-tesian coordinate can be found in the Supplementary Materials section.
2.2. Typical Synthetic Procedure for Obtaining 2,4,4,5,5-pentafluoro-3-{1-phenyl-1-(triphenyl-λ5-phosphoniylidene)methyl}-2-cyclopenten-1-one (3b)
A 50 mL two-necked round-bottomed flask, equipped with a magnetic stir bar, was filled with a solution of (phenylmethyl)triphenylphosphonium bromide (0.95 g, 2.2 mmol) in tetrahydrofuran (THF, 10 mL). The solution was then cooled to −78 °C. A 1.6 mol·L−1 solution of n-butyllithium in hexane (1.38 mL, 2.2 mmol) was slowly added via a syringe and the resulting mixture was stirred at −78 °C for 0.5 h. Subsequently, a solution of octafluorocyclopentene (0.212 g, 1.0 mmol) in dichloromethane (CH2Cl2, 4 mL) was added to the main solution via syringe at −78 °C, followed by heating and stirring at 25 °C for 2 h. The reaction mixture was then poured into saturated aqueous NH4Cl (30 mL), followed by extraction of the crude product with CH2Cl2 (20 mL × 5). The organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure using a rotary evaporator. Column chromatography was carried out using a mixed solvent of hexane and ethyl acetate (v/v = 1/2) as the eluent, followed by recrystallization from a 1/1 (v/v) mixed solvent system of CH2Cl2 as a good solvent and hexane as a poor solvent via slow evaporation technique at ambient temperature, to obtain 3b in 11% isolated yield (62 mg, 0.11 mmol) as a purple solid. The compounds 3c–3e were also prepared using a similar procedure.
2.2.1. 2,4,4,5,5-Pentafluoro-3-{1-phenyl-1-(triphenyl-λ5-phosphoniylidene)methyl}-2-cyclopenten-1-one (3b)
Yield: 11% (purple solid); mp: 280–282 °C; 1H NMR (CDCl3): δ 6.96–7.02 (m, 5H), 7.38–7.53 (m, 12H), 7.57–7.66 (m, 3H); 13C NMR (CDCl3): δ 53.4, 110–114 (m, 2C for CF2CF2), 123.0 (d, J = 90.2 Hz), 127.4 (d, J = 2.2 Hz), 127.7 (d, J = 1.4 Hz), 129.3 (d, J = 13.2 Hz), 133.4 (d, J = 2.9 Hz), 133.6 (d, J = 9.6 Hz), 133.9 (d, J = 7.2 Hz), 134.3 (d, J = 4.3 Hz), 163.3 (d, J = 22.8 Hz); 19F NMR (CDCl3, C6F6): δ −110.79 (s, 1F), −110.81 (s, 1F), −125.01 (s, 1F), −125.03 (s, 1F), −148.75 to −148.82 (m, 1F); IR (KBr): ν 3057, 1666, 1290, 1198, 1092, 992, 807 cm−1; HRMS (FAB) m/z [M+H]+ calcd for C30H21F5OP: 523.1251; found: 523.1252.
2.2.2. 2,4,4,5,5-Pentafluoro-3-{1-(4-methoxyphenyl)-1-(triphenyl-λ5-phosphoniylidene)methyl}-2-cyclopenten-1-one (3c)
Yield: 19% (purple solid); mp: 306–308 °C; 1H NMR (CDCl3): δ 3.68 (s, 3H), 6.53 (d, J = 8.4 Hz, 2H), 6.85 (d, J = 8.4 Hz), 7.38–7.53 (m, 12H), 7.58–7.66 (m, 3H); 13C NMR (CDCl3): δ 55.1, 110–114 (m, 2C for CF2CF2), 113.1, 123.2 (d, J = 90.2 Hz), 124.9, 125.7, 127.0, 129.3 (d, J = 12.4 Hz), 133.4 (d, J = 2.2 Hz), 133.6 (d, J = 9.6 Hz), 135.3 (d, J = 4.3 Hz), 158.9; 19F NMR (CDCl3): δ −111.01 (s, 1F), −111.03 (s, 1F), −124.95 (s, 1F), −124.98 (s, 1F), −148.71 to −148.83 (m, 1F); IR (KBr): ν 3060, 2839, 1671, 1536, 1509, 1441, 1100, 998, 924 cm–1; HRMS (FAB) m/z [M+H]+ calcd for C31H23F5O2P: 553.1357; found: 553.1345.
2.2.3. 2,4,4,5,5-Pentafluoro-3-{1-(4-biphenyl)-1-(triphenyl-λ5-phosphoniylidene)methyl}-2-cyclopenten-1-one (3d)
Yield: 2% (purple solid); mp: 305–307 °C; 1H NMR (CDCl3): δ 7.02 (d, J = 8.0 Hz, 2H), 7.21 (d, J = 8.0 Hz, 2H), 7.36–7.55 (m, 16H), 7.58–7.72 (m, 4H); 13C NMR (CDCl3): δ 53.4, 110.0–114.0 (m, 2F for CF2CF2), 123.0 (d, J = 90.2 Hz), 126.3 (d, J = 2.2 Hz), 126.9, 127.4, 128.7, 129.3 (d, J = 12.4 Hz), 133.4 (d, J = 2.2 Hz), 133.6 (d, J = 9.6 Hz), 134.6 (d, J = 3.7 Hz), 140.1 (d, J = 2.9 Hz), 140.4, 163.6 (d, J = 41.8 Hz); 19F NMR (CDCl3): δ –110.67 (s, 1F), −110.69 (s, 1F), −124.96 (s, 1F), −124.99 (s, 1F), −148.66 to −148.85 (m, 1F); IR (KBr): ν 3058, 2849, 1675, 1539, 1294, 1113, 1014, 921 cm–1; HRMS (FAB) m/z [M+H]+ calcd for C36H25F5OP: 599.1564; found: 599.1562.
2.2.4. 2,4,4,5,5-Pentafluoro-3-{1-(2,3,4,5,6-pentafluorophenyl)-1-(triphenyl-λ5-phosphoniylidene)methyl}-2-cyclopenten-1-one (3e)
Yield: 26% (purple solid); mp: 282–284 °C; 1H NMR (CDCl3): δ 7.58–8.08 (m, 15H); 13C NMR (CDCl3): δ 53.4, 108.0–117.5 (m), 121.7–122.5 (m), 122.6–123.4 (m), 128.6–130.6 (m), 133.3 (d, J = 9.6 Hz), 133.7–134.2 (m), 165.8 (d, J = 11.8 Hz); 19F NMR (CDCl3, BTF): δ −113.78 to −113.92 (m, 2F), −124.17 (s, 1F), −124.20 (s, 1F), −134.97 (s, 1F), −135.02 (s, 1F), −147.13 to −147.32 (m, 1F), −152.72 (t, J = 20.7 Hz, 1F), −162.91 to −163.21 (m, 2F); IR (KBr): ν 3063, 1692, 1296, 1234, 1185, 1092, 872 cm–1; HRMS (FAB) m/z [M+H]+ calcd for C30H16F10OP: 613.0780; found: 613.0772.
2.3. Typical Synthetic Procedure of [2,4,4,5,5-pentafluoro-3-{1-phenyl-1-(triphenyl-λ5-phosphonilidene)methyl}-2-cyclopentenylidenemethyl]triphenylphosphonium Bromide (4bA)
A 50 mL two-necked round-bottomed flask, equipped with a magnetic stir bar, was filled with a solution of (phenylmethyl)triphenylphosphonium bromide (2.86 g, 6.6 mmol) in THF (10 mL), which was then cooled to −78 °C. A 1.6 mol·L−1 solution of n-butyllithium in hexane (1.38 mL, 2.2 mmol) was then added via a syringe, followed by stirring at −78 °C for 0.5 h. Subsequently, a solution of octafluorocyclopentene (2, 0.212 g, 1.0 mmol) in dichloromethane (CH2Cl2, 4 mL) was added via a syringe at −78 °C. After stirring at 25 °C for 2 h, the reaction mixture was poured into saturated aqueous NH4Cl (30 mL), followed by extraction of the crude product with CH2Cl2 (20 mL × 5). The organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure using a rotary evaporator. Column chromatography was carried out using a mixture of ethyl acetate and ethanol (v/v = 3/1) as the eluent, followed by recrystallization from CH2Cl2 and hexane (v/v = 1/1) via the slow evaporation technique at ambient temperature, to obtain 4bA in 24% isolated yield (0.23 g, 0.24 mmol) as yellow crystals. The compounds 4cA–4eA, incorporating different R substituents, and the compounds 4aB–4aE, complexed with different counter anions, were prepared using a similar procedure.
2.3.1. [2,4,4,5,5-Pentafluoro-3-{1-phenyl-1-(triphenyl-λ5-phosphonilidene)methyl}-2-cyclopentenylidenemethyl]triphenylphosphonium Bromide (4bA)
Yield: 24% (yellow solid); mp: 381–383 °C; 1H NMR (CDCl3): δ 6.81–6.88 (m, 4H), 6.91–6.98 (m, 4H), 6.99–7.05 (m, 2H), 7.07–7.24 (m, 10H), 7.38–7.49 (m, 13H), 7.61–7.69 (m, 7H); 13C NMR (CDCl3): δ 74.2 (d, J = 98.2 Hz), 112.0–119.0 (m, 2C for CF2CF2), 122.1 (d, J = 90.2 Hz), 127.4, 127.5, 129.4 (d, J = 12.5 Hz), 133.0 (d, J = 8.8 Hz), 133.6, 133.8, 135.8–136.6 (m), 140.0–143.0 (m); 19F NMR (CDCl3, C6F6): δ −107.61 (s, 2F), −107.63 (s, 2F), −128.10 to −128.33 (m, 1F); IR (KBr): ν 3052, 1701, 1478, 1266, 1180, 1098, 1017, 952 cm−1; HRMS (FAB) m/z [M+] calcd for C55H40F5P2: 857.2525; found: 857.2528.
2.3.2. [2,4,4,5,5-Pentafluoro-3-{1-(4-methoxyphenyl)-1-(triphenyl-λ5-phosphonilidene)methyl}-2-cyclopentenylidenemethyl]triphenylphosphonium Bromide (4cA)
Yield: 44% (yellow solid); mp: 190–192 °C; 1H NMR (CDCl3): δ 3.66 (s, 6H), 6.49 (d, J = 8.4 Hz, 4H), 6.73 (d, J = 8.4 Hz, 4H), 7.08–7.22 (m, 12H), 7.40–7.49 (m, 12H), 7.61–7.69 (m, 6H); 13C NMR (CDCl3): δ 55.1, 73.6 (d, J = 82.1 Hz), 112.0–119.0 (m, 2C for CF2CF2), 113.1, 122.3 (d, J = 91.6 Hz), 123.1, 124.4, 125.1 (d, J = 7.3 Hz), 129.6 (d, J = 12.4 Hz), 133.2 (d, J = 10.2 Hz), 133.7, 135.0, 135.8–136.6 (m), 140.0–143.0 (m); 19F NMR (CDCl3, C6F6): δ −109.20 (s, 2F), −109.23 (s, 2F), −129.21 to −129.42 (m, 1F); IR (KBr): ν 3055, 1603, 1506, 1246, 1173, 1104, 1018, 955, 805 cm−1; HRMS (FAB) m/z [M+] calcd for C57H44F5O2P2: 917.2737; found: 917.2730.
2.3.3. [2,4,4,5,5-Pentafluoro-3-{1-(4-biphenyl)-1-(triphenyl-λ5-phosphonilidene)methyl}-2-cyclopentenylidenemethyl]triphenylphosphonium Bromide (4dA)
Yield: 51% (yellow solid); mp: 217–218 °C; 1H NMR (CDCl3): δ 6.90 (d, J = 7.2 Hz, 4H), 7.14–7.24 (m, 12H), 7.36–7.49 (m, 24H), 7.62–7.70 (m, 8H); 13C NMR (CDCl3): δ 74.0 (d, J = 93.9 Hz), 111.0–118.0 (m, 2F for CF2CF2), 122.1 (d, J = 89.4 Hz), 126.2, 126.8, 127.4, 128.6, 129.6 (d, J = 12.5 Hz), 133.2 (d, J = 9.6 Hz), 133.8, 134.2, 136.6–136.6 (m), 140.1, 140.2; 19F NMR (CDCl3, C6F6): δ −108.78 (s, 2F), −108.79 (s, 2F), −129.25 to −129.45 (m, 1F); IR (KBr): ν 3055, 1508, 1436, 1343, 1284, 1215, 969, 875 cm–1; HRMS (FAB) m/z [M+] calcd for C67H48F5P2: 1009.3151; found: 1009.3141.
2.3.4. [2,4,4,5,5-Pentafluoro-3-{1-(2,3,4,5,6-pentafluorophenyl)-1-(triphenyl-λ5-phosphonilidene)methyl}-2-cyclopentenylidenemethyl]triphenylphosphonium Bromide (4eA)
Yield: 10% (yellow solid); mp: 218–220 °C; 1H NMR (CDCl3): δ 7.40–7.75 (m, 30H); 13C NMR (CDCl3): δ 91.4, 111.0–118.0 (m, 2F for CF2CF2), 124.5 (d, J = 111.1 Hz), 128.9 (d, J = 12.5 Hz), 132.9, 133.5 (d, J = 9.5 Hz), 135.0–138.9 (m, ArF), 142.0–145.3 (m, ArF), 147.2–149.8 (m, ArF); 19F NMR (CDCl3, BTF): δ −110.94 to −111.15 (m, 2F), −113.24 to −113.35 (m, 2F), −1334.72 to −134.96 (m, 2F), −135.60 to −135.81 (m, 1F), −139.74 to −140.06 (m, 2F), −155.16 (t, J = 19.2 Hz, 1F), −157.79 (t, J = 21.8 Hz, 1F), −164.10 to −164.28 (m, 2F), −164.34 to −164.58 (m, 2F); IR (KBr): ν 3061, 2931, 1723, 1284, 1215, 969, 875 cm–1; MS (FAB) m/z 181 (C6F5CH2+, 2.20), 349 (–C6F5CH2, 0.67), 806 (–3Ph, 0.96), 833 (–2Ph, 3.03).
2.3.5. [2,4,4,5,5-Pentafluoro-3-{(triphenyl-λ5-phosphonilidene)methyl}-2-cyclopentenylidenemethyl]triphenylphosphonium Iodide (4aB)
Yield: 54% (yellow solid); mp: 221–223 °C; 1H NMR (CDCl3): δ 4.24 (dd, J = 13.6, 2.0 Hz, 2H), 7.28–7.35 (m, 10H), 7.51–7.58 (m, 13H), 7.69–7.76 (m, 7H); 13C NMR (CDCl3): δ 53.4. 58.8 (d, J = 110.1 Hz), 111.0–117.0 (m, 2C for CF2CF2), 122.8 (d, J = 92.4 Hz), 129.9 (d, J = 13.2 Hz), 130.5 (d, J = 13.2 Hz), 132.5 (d, J = 10.4 Hz), 133.0 (d, J = 11.0 Hz), 134.1, 135.2 (d, J = 2.9 Hz), 138.3–139.1 (m), 140.2–142.8 (m); 19F NMR (CDCl3, C6F6): δ −114.64 (s, 2F), −114.66 (s, 2F), −137.26 to −137.38 (m, 1F); IR (KBr): ν 3050, 1507, 1402, 1295, 1229, 1101, 1023, 961 cm−1; HRMS (FAB) m/z [M+] calcd for C43H32F5P2: 705.1899; found: 705.1898.
2.3.6. [2,4,4,5,5-Pentafluoro-3-{(triphenyl-λ5-phosphonilidene)methyl}-2-cyclopentenylidenemethyl]triphenylphosphonium Tetrafluoroborate (4aC)
Yield: 32% (yellow solid); mp: 301–303 °C; 1H NMR (CDCl3): δ 4.23 (dd, J = 13.2, 2.0 Hz, 7.27–7.35 (m, 10H), 7.49–7.56 (m, 13H), 7.66–7.74 (m, 7H); 13C NMR (CDCl3): δ 58.8 (d, J = 110.0 Hz), 111.0–117.0 (m, 2C for CF2CF2), 122.7 (d, J = 91.3 Hz), 128.4 (d, J = 11.7 Hz), 129.8 (d, J = 12.5 Hz), 132.4 (d, J = 10.3 Hz), 134.1, 139.0–140.5 (m); 19F NMR (CDCl3, C6F6): δ −113.33 (s, 2F), −113.35 (s, 2F), −135.85 to −136.04 (m, 1F), −153.32 to −153.46 (m, 4F); IR (KBr): ν 3057, 1530, 1437, 1403, 1229, 1057, 963, 873 cm–1; HRMS (FAB) m/z [M+] calcd for C43H32F5P2: 705.1899; found: 705.1905.
2.3.7. [2,4,4,5,5-Pentafluoro-3-{(triphenyl-λ5-phosphonilidene)methyl}-2-cyclopentenylidenemethyl]triphenylphosphonium Trifluoromethanesulfonate (4aD)
Yield: 89% (yellow solid); mp: 238–240 °C; 1H NMR (CDCl3): δ 4.24 (dd, J = 13.2, 1.6 Hz, 2H), 7.27–7.35 (m, 10H), 7.48–7.56 (m, 13H), 7.67–7.73 (m, 7H); 13C NMR (CDCl3): δ 58.8 (d, J = 111.6), 111.0–118.0 (m, 2C for CF2CF2), 122.8 (d, J = 92.5 Hz), 129.9 (d, J = 13.2 Hz), 130.5 (d, J = 13.2 Hz), 132.5 (d, J = 10.4 Hz), 133.0 (d, J = 11.0 Hz), 134.1, 135.2, 139.4–140.2 (m), 145.8–148.5 (m); 19F NMR (CDCl3): δ −78.02 (s, 3F), −113.33, (s, 2F), −113.35 (s, 2F), −135.92 to −136.06 (m, 1F); IR (KBr): ν 3038, 1828, 1577, 1021, 961, 872 cm–1; HRMS (FAB) m/z [M+] calcd for C43H32F5P2: 705.1899; found: 705.1898.
2.3.8. [2,4,4,5,5-Pentafluoro-3-{(triphenyl-λ5-phosphonilidene)methyl}-2-cyclopentenylidenemethyl]triphenylphosphonium Perchlorate (4aE)
Yield: 20% (yellow solid); mp: >400 °C; 1H NMR (CDCl3): δ 4.24 (dd, J = 13.2, 1.6 Hz, 2H), 7.28–7.36 (m, 10H), 7.49–7.56 (m, 13H), 7.67–7.73 (m, 7H); 13C NMR (CDCl3): δ 58.8 (d, J = 113.0 Hz), 111.0–119.0 (m, 2C for CF2CF2), 122.8 (d, J = 92.5 Hz), 128.5 (d, J = 12.5 Hz), 129.9 (d, J = 12.5 Hz), 132.1 (d, J = 10.3 Hz), 132.5 (d, J = 11.1 Hz), 134.1, 134.8–136.2 (m), 142.0–147.0 (m); 19F NMR (CDCl3): δ −113.32 (s, 2F), −113.34 (s, 2F), −135.78 to −135.91 (m, 1F); IR (KBr): ν 3319, 1625, 1524, 1436, 1294, 1061, 873 cm−1; HRMS (FAB) m/z [M+] calcd for C43H32F5P2: 705.1899; found: 705.1898.
2.4. Quantum Chemical Calculation
All computations were performed using density functional theory (DFT) with the Gaussian 16 package (Rev. B.01) [21]. Geometry optimizations were executed using the M06-2X hybrid functional [22] and 6-31+G(d) basis set with a conductor-like polarizable continuum model (CPCM) [23] for CH2Cl2. Theoretical vertical transitions were calculated using a time-dependent DFT (TD-DFT) method at the same level of theory using the same solvation model.
2.5. Photophysical Properties
UV-vis absorption spectra were recorded using a JASCO V-750 absorption spectrometer (JASCO, Tokyo, Japan). The sample for each UV-vis absorption measurement was prepared by dissolving the solid sample in an organic solvent, e.g., CH2Cl2, and adjusting its concentration to 1.0 × 10–5 mol L–1.
2.6. Thermogravimetric Analyses
The thermogravimetric analyses (TGA) were conducted using a TGA-50 thermogravimetric analyzer (SHIMADZU, Kyoto, Japan) at a heating rate of 10 °C min–1 under a nitrogen atmosphere.
3. Results and Discussion
3.1. Synthesis
The synthesis of fluorinated merophosphinines and phosphinines was accomplished according to our previously reported procedure [18]. First, several triphenylphosphonium ylides (1, 2.2 equiv), prepared from various phosphonium salts [R = (a) H, (b) C6H5, (c) 4-MeOC6H4, (d) 4-PhC6H4, or (e) C6F5] and n-butyllithium, were treated with octafluorocyclopentene 2 to obtain the corresponding fluorinated merophosphinines 3b–3e after reaction termination. The use of 6.6 equiv of 1 under similar reaction conditions resulted in a further addition-elimination sequence, leading to the corresponding fluorinated phosphinine derivatives 4bA–4eA [R = (b) C6H5, (c) 4-MeOC6H4, (d) 4-PhC6H4, or (e) C6F5; counter anion (X) = (A) Br−] and 4aB–4aE [R = H; counter anion (X) = (B) I−, (C) , (D) OTf−, or (E) ]. The structures and chemical yields of all the fluorinated merophophinine and phosphinine derivatives are summarized in Table 1.
The addition-elimination and subsequent 1,4-HF elimination sequence of 2 with 2.2 equivalents of benzylphosphonium ylide 1b, prepared from benzyl bromide and n-butyllithium, provided the corresponding fluorinated merophosphinine 3b in 11% yield after hydrolysis. Another fluorinated merophosphinine, 3c, which incorporated a 4-methoxyphenyl group, was produced in 19% yield from the corresponding 4-methoxybenzyl bromide. The reaction of 2 with 4-phenylbenzylphosphinium ylide 1d was inefficient and provided the corresponding product 3d in 2% yield. Pentafluorobenzylphosphonium ylide 1e, prepared from pentafluorobenzyl bromide, also participated in the reaction, affording 3e in 26% yield. When 6.6 equiv of benzylphosphonium ylide 1b was used, the three-step addition-elimination/1,4-HF elimination/addition-elimination sequence proceeded to give the corresponding fluorinated phosphinine derivative 4bA in 23% yield. Two other benzylphosphonium ylides, 1c and 1d, participated in the reaction sequence to produce 4cA and 4dA in 44% and 51% yield, respectively. The reaction of 2 with 6.6 equiv of 1e was less efficient, thus giving the corresponding product 4eA in low yield. Fluorinated phosphine derivatives with different counter anions were also prepared from the corresponding phosphonium salts. The reaction of 2 with benzylphosphonium iodide-derived phosphonium ylide 1a, followed by hydrolysis, successfully produced the corresponding fluorinated phosphinine 4aB with an iodide ion in 54% yield. Other phosphonium ylides incorporating , TfO−, and counter anions also participated in the reaction to give the respective products 4aC-4aE in 20–89% yield. Additionally, all the afforded compounds were obtained in high purity using established chromatography and recrystallization methods, as indicated by spectroscopic analysis.
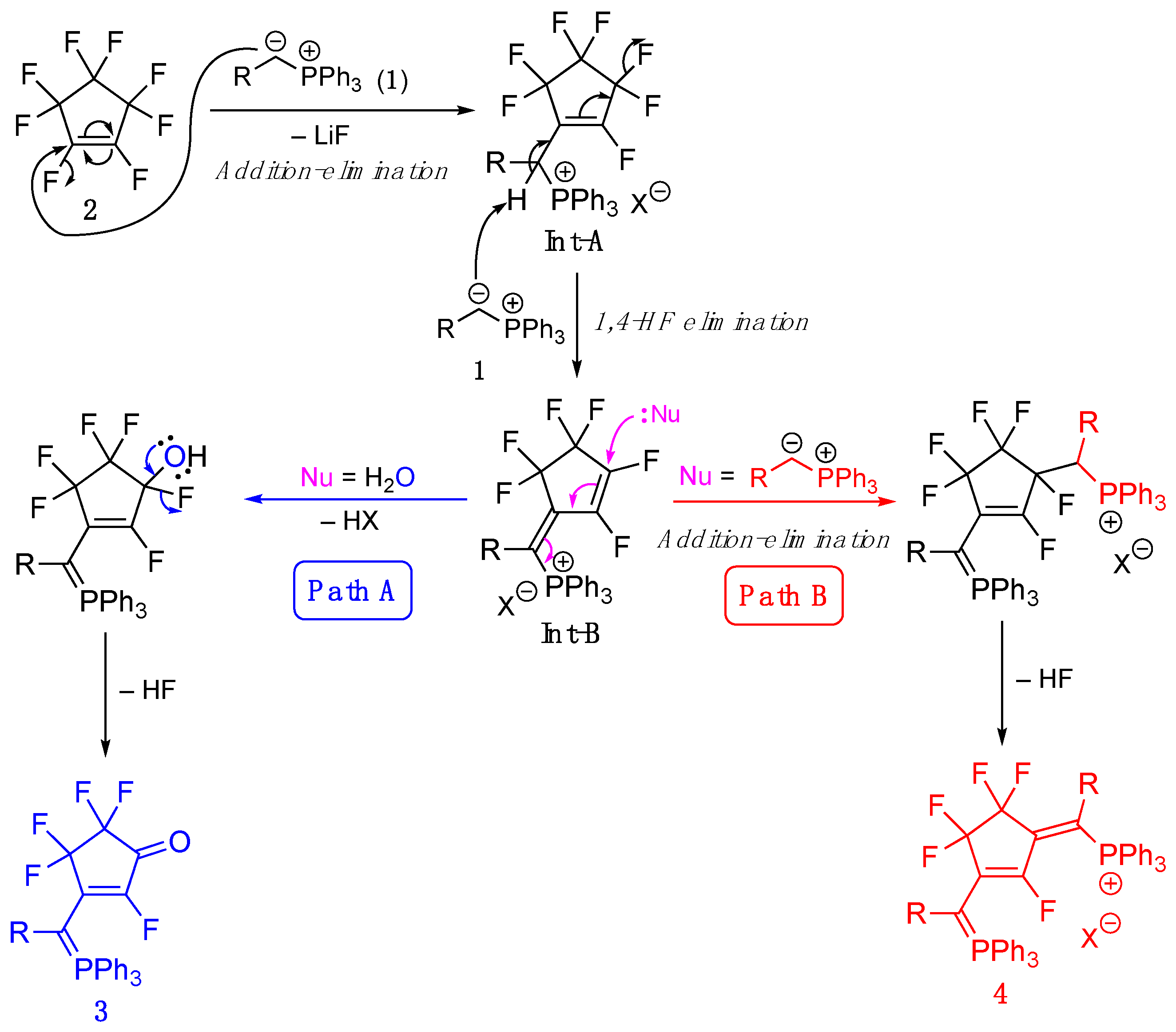
The proposed reaction mechanisms for the synthesis of 3 and 4 are shown in Figure 3. Nucleophilic addition-elimination of the phosphinium ylide 1 with 2 provides the corresponding monosubstituted intermediate (Int-A), which then undergoes 1,4-HF elimination to yield Int-B. If hydrolysis is performed, the fluorohydrine intermediate is produced via nucleophilic addition, which gives the corresponding merophosphininie 3 after HF elimination (Path A). Conversely, if no hydrolysis is carried out, an additional nucleophilic attack by the remaining phosphonium ylide, followed by HF elimination, affords the corresponding phosphinine 4 (Path B).
3.2. Thermal Stability
To evaluate the thermal stabilities of the synthesized fluorinated merophosphinine (3a–3e) and phosphinine (4aA–4eA) derivatives, we conducted TGA analysis on these compounds. Here, the temperature at which the sample weight decreased by 5% was defined as the thermal decomposition temperature (Tdec). Figure 4 shows the TGA thermograms of samples of the studied derivatives, which were conducted under a nitrogen atmosphere. The measured Tdec values are summarized in Table 2.
The Tdec of 3a, which incorporates a hydrogen atom as the R substituent, was 241 °C. Introducing an aromatic group as the R substituent increased the Tdec (Figure 4a); 3b, which incorporated a phenyl group as the R substituent, subsequently exhibited a Tdec of 295 °C, over 50 °C higher than that of 3a. A further increase in Tdec was observed in 3c, which incorporated an electron-rich aromatic backbone; this resulted in the highest Tdec (313 °C) of the studied compounds. Conversely, for 4-biphenyl-derived 3d, the thermal stability was reduced considerably. For 3e, which possessed an electron-deficient aromatic ring, thermal decomposition occurred at a mild Tdec of 304 °C. For the phosphinines, as shown in Figure 4b, the initial Tdec 4aA was equivalent to that of 3a (241 °C); however, once aromatic groups were incorporated as the R substituents, the observed Tdec increases were lower in magnitude than for the merophosphinines, attaining a maximum of 281 °C for 4aA. Notably, 4eA, which incorporated two electron-deficient pentafluorophenyl substituents, exhibited the lowest Tdec of the studied compounds (203 °C). These results show that fluorinated merophosphinine and phosphinine derivatives are generally stable above 200 °C. Furthermore, by appropriately selecting the R substituent, and therefore, the level of electron density donated to or withdrawn from the structural center of the derivatives, the thermal stability can be adjusted to a higher temperature range. This selectivity, therefore, imparts flexibility to these compounds for their application as functional dye materials.
3.3. UV-Vis Absorption Properties of Fluorinated Merophosphinines
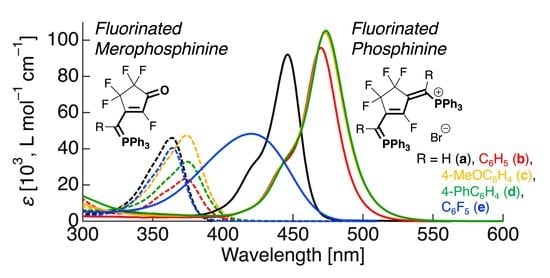
1.0 × 10–5 mol·L–1 solutions of the fluorinated merophosphinines 3a–3e in CH2Cl2 were used to investigate their UV-vis absorption properties. Figure 5 shows the resulting UV-vis absorption spectra and the representative molecular orbital distribution of 3a, which was calculated using quantum chemical calculations based on the compound’s observed electronic transitions. The photophysical data and the calculated transition energies are summarized in Table 3.
The 3a solution exhibited a pale purple color and a single absorption band with a maximum absorption wavelength (λabs) of approximately 364 nm (Figure 5a). To gain more information about the theoretical vertical transition in the UV-vis absorption process, the molecular orbital calculation was performed using Gaussian 16 by TD-DFT. The M06-2X/6-31+G(d) basis function was then used for the subsequent calculations of the vertical transition to the excited states. These calculations revealed that the electronic transition from the highest occupied molecular orbitals (HOMO) to the lowest unoccupied molecular orbital (LUMO) is involved in the excitation process. In 3a, the HOMO lobe is localized on the ethylidenecyclopentenone skeleton, while the LUMO is widely delocalized across the triphenylphosphane structure (Figure 5b). It is, therefore, reasonable to conclude that the absorption process occurs via a smooth π-π* transition because of the overlap of the wide π orbital lobes of these structures.
When a phenyl group was utilized as the R substituent, a redshift of 9 nm in the λabs of 3b (373 nm in CH2Cl2) was observed relative to that of 3a. From the TD-DFT calculation, 3b also exhibited a shift in the π-π* transition involving HOMO → LUMO; therefore, the observed redshift in the λabs of 3b can be attributed to a reduction in the HOMO-LUMO energy gap (ΔEH-L) by 0.16 eV, caused by the elongation of the π-conjugation length. Using 4-methoxyphenyl and 4-biphenyl groups as the R substituents in 3c and 3d, respectively, also reduced the ΔEH-L, resulting in redshifts of the λabs to 374 and 375 nm for 3c and 3d, respectively. However, when a pentafluorophenyl group was introduced as the R substituent, the λabs of 3e showed a considerable blueshift (364 nm) compared to that of 3b. As the theoretical calculation result also showed that the electronic transition in 3e occurs from HOMO → LUMO, it can be concluded that introducing fluorine atoms into the phenyl group raises the LUMO energy, thus widening the ΔEH-L in comparison to that of 3b.
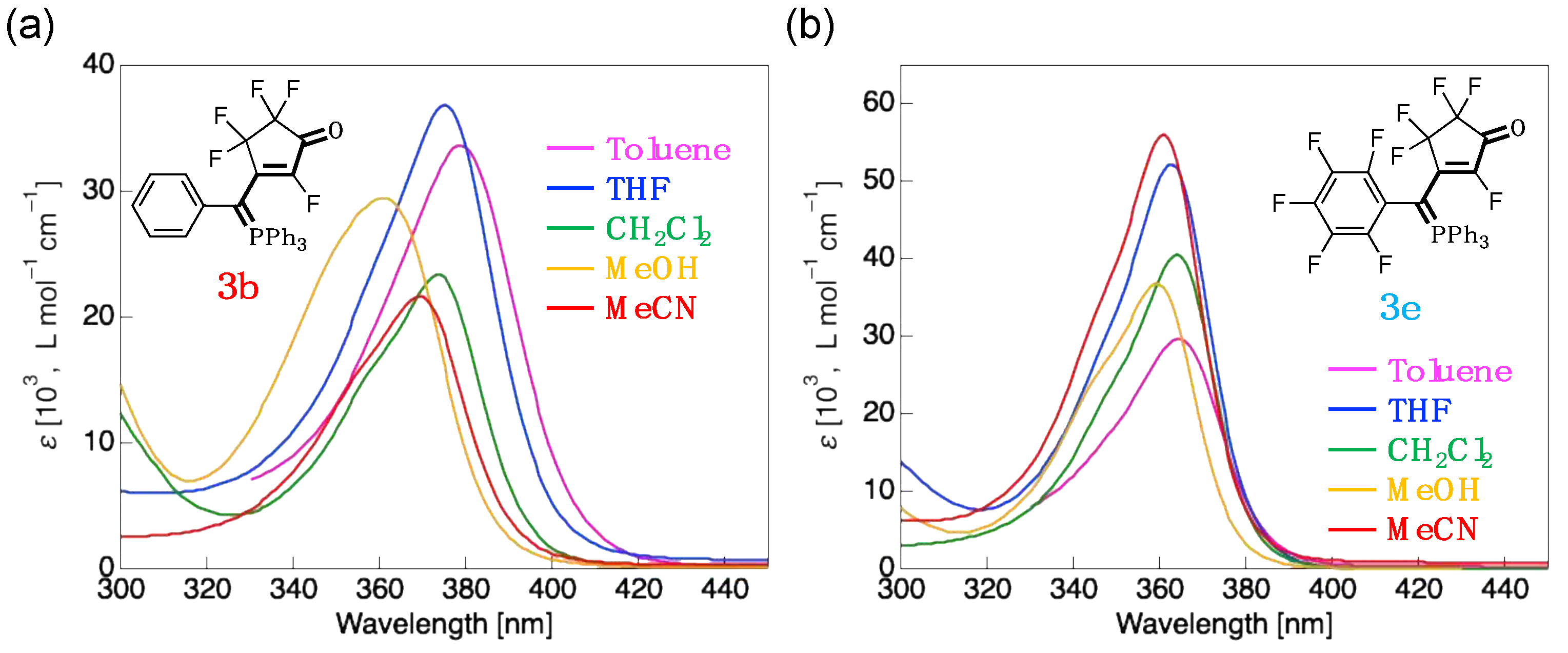
To investigate the difference in the absorption properties caused by the C6H5 and C6F5 groups, the effect of the solvent on the absorption spectra of 3b and 3e was studied. Figure 6 shows the absorption spectra of 3b and 3e measured in various solvents, including CH2Cl2, toluene, THF, MeOH, and MeCN. Photophysical data obtained in various solvents are also summarized in Table 3.
In less polar solvents, as shown in Figure 6a, the λabs of 3b exhibited redshifting compared to that observed in CH2Cl2; new absorption maxima were seen at 379 nm (z = 6 nm) in toluene and 375 nm (z = 2 nm) in THF. Conversely, the λabs gradually blueshifted with increasing solvent polarity, resulting in a minimum λabs of 361 nm in MeOH. The UV-vis absorption behavior of 3e in different solvents was observed to be somewhat similar to that of 3b (Figure 6b); the λabs was generally blueshifted as the solvent polarity increased, but the THF solution also exhibited a blueshift despite the reduced solvent polarity. From the above results, it was revealed that aromatic-substituted fluorinated merophosphinine derivatives, such as 3b and 3e, exhibit negative solvatochromism.
This negative solvatochromism can be rationally explained from theoretical calculations. The absorption bands for 3b and 3e can be assigned to the HOMO → LUMO transition, and the energies of these were observed to change with increasing solvent polarity. It was, therefore, shown that the HOMO energy decreased significantly in more polar solvents, thus increasing the ΔEH-L; this results in a blueshift in the absorption bands of 3b and 3e with increasing solvent polarity. This behavior indicates that the electronic structure of merocyanine 3 is highly dipolar [24,25].
3.4. UV-Vis Absorption Properties of Fluorinated Phosphinines
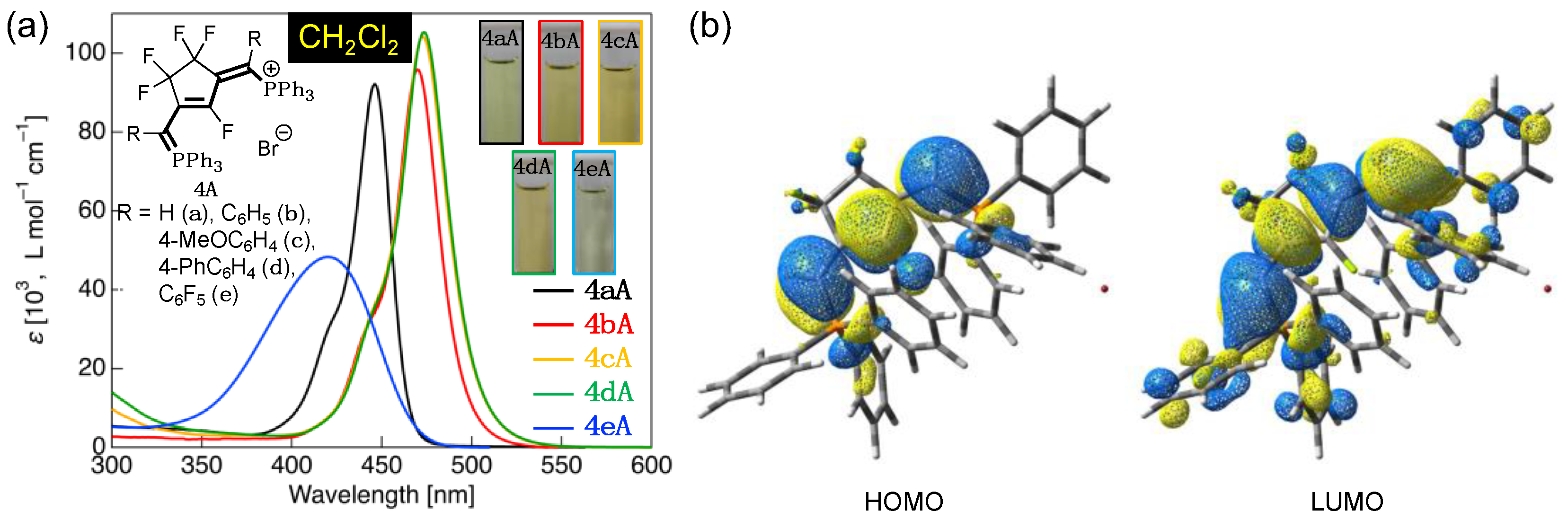
Subsequently, we evaluated the UV-vis absorption properties of the fluorinated phosphinine derivatives, 4aA–4eA, in CH2Cl2. UV-vis absorption spectra and the calculated HOMO/LUMO distribution in 4aA are shown in Figure 7, while the photophysical data for the compounds are listed in Table 4.
The H-substituted fluorinated phosphinine derivative 4aA exhibited two absorption features: a sharp absorption band with λabs at approximately 446 nm, and a shoulder peak at approximately 424 nm (Figure 7a). Compared to the previously observed absorption band of 3a, 4aA was considerably redshifted by approximately 82 nm. The theoretical calculations showed that the HOMO lobe of 4aA was localized over the π-conjugated structure of the phosphanylidene backbone to the phosphonium group. In contrast, the LUMO lobe was extended further to the triphenylphosphine site and was widely delocalized (Figure 7b). TD-DFT calculations further revealed that the theoretical vertical transition of 4aA was also a π-π* transition from HOMO to LUMO.
In 4bA, the λabs was redshifted by approximately 24 nm compared to that of 4aA. This can be attributed to the narrowing of the ΔEH-L owing to the extension of the π-conjugation length, which was induced by the introduction of a phenyl group as the R substituent. Introducing 4-methoxyphenyl or 4-biphenyl groups as the R substituents caused a further redshift, leading to λabs of 473 and 474 nm for 4cA and 4dA, respectively. This can be attributed to the following reasons: first, in 4cA, the introduction of an electron-donating methoxy group reduces the ΔEH-L by increasing the HOMO energy; second, in 4dA, the ΔEH-L is reduced by the extension of the π-conjugation length owing to the 4-biphenyl structure. In contrast, 4eA exhibited a broad absorption band with λabs at approximately 420 nm because of the decrease in HOMO energy caused by the electron-withdrawing effect of the pentafluorophenyl groups, thus increasing the ΔEH-L.
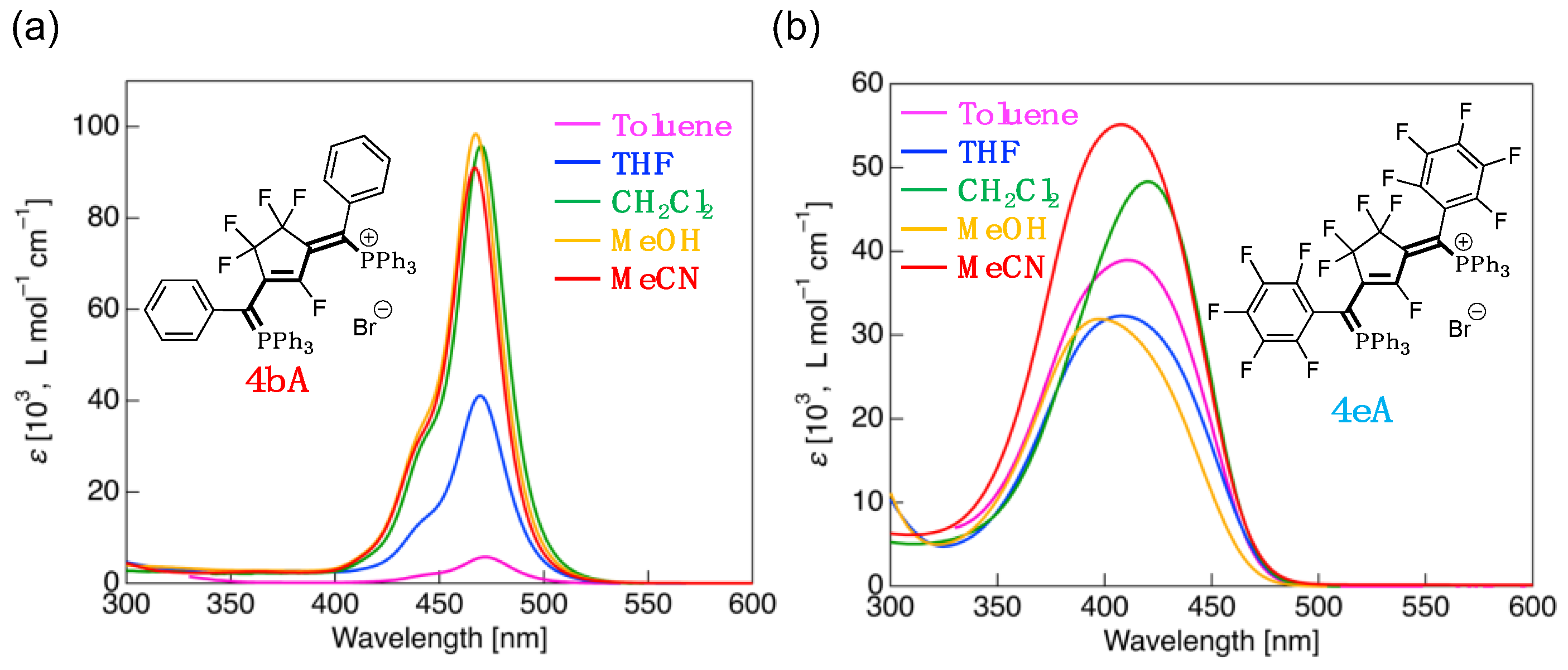
Subsequently, to investigate the solvent effect on the absorption properties, the UV-vis spectra of 4bA and 4eA were measured in various solvents (Figure 8). The photophysical data are summarized in Table 4.
4bA showed a negative solvatochromism similar to that of the fluorinated merophosphinine 3b (Figure 8a); in less polar solvents, such as toluene, the λabs was redshifted compared to that measured in CH2Cl2, while measuring in more polar solvents, such as MeOH, resulted in a maximum blueshift of 5 nm. As observed in the fluorinated merophosphinines, this is attributable to the decrease in ΔEH-L caused by the reduction in the HOMO energy in a more polar solvent. In the case of 4eA, as shown in Figure 8b, while the highest energy absorptions were observed in more polar solvents, the spectra measured in toluene and THF also exhibited blueshifts compared to that of CH2Cl2. When the theoretical transition in 4eA was calculated by TD-DFT using a toluene solvation model, it was found that, unlike the other solvents, the electronic transition involved HOMO-3 → LUMO; it is, therefore, considered that, for 4eA, the transition process differs depending on the solvent used.
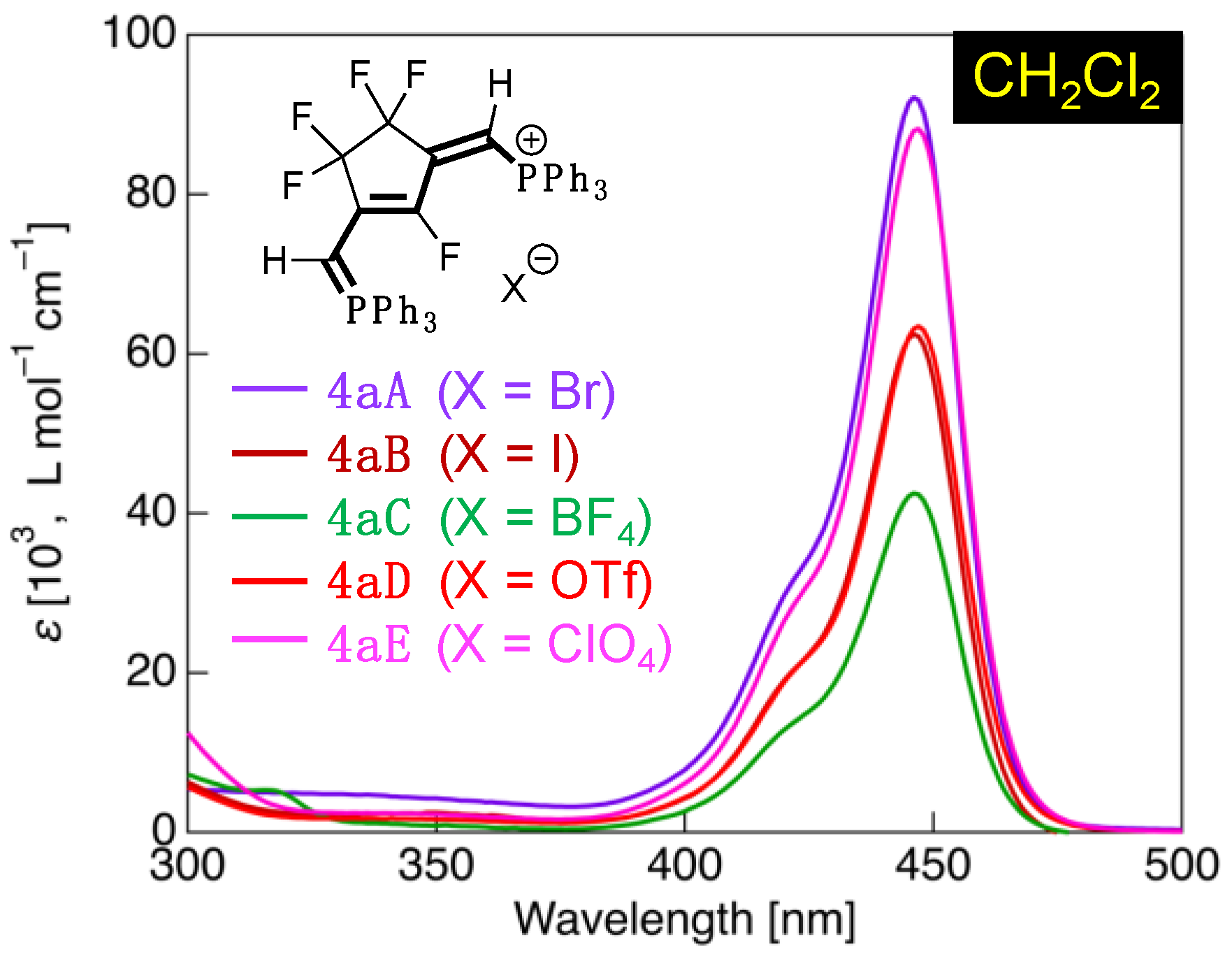
To confirm the effect of different counter anions on the UV-vis absorption characteristics of the fluorinated phosphinine derivatives, variants of 4aA (counter ion X = Br−) were prepared with X = I− (4aB), (4aC), OTf− (4aD), and (4aE) and measured in CH2Cl2 (Figure 9).
Here, all the prepared derivatives exhibited absorption bands with λabs at approximately 446 nm, as well as a shoulder peak at approximately 424 nm. The counter anion, therefore, does not affect the absorption properties of the fluorinated phosphinines. As mentioned previously, the theoretical vertical transition of 4aA calculated by the TD-SCF method was found to be a transition involving HOMO → LUMO. The HOMO and LUMO lobes are distributed over the π-conjugated sites of the phosphinine structure and do not interact with the counter ion; thus, the counter anion does not participate in the electronic transition.
4. Conclusions
In this study, we synthesized various fluorinated merophosphinines and phosphinine derivatives with different aromatic substituents and investigated their thermal stabilities and UV-vis absorption properties. The fluorinated merophosphinines were thermally stable up to 304 °C depending on the choice of the aromatic group employed. Similarly, the fluorinated phosphinine derivatives also showed enhanced thermal stabilities up to 281 °C; however, when a pentafluorophenyl group was used, the stability was reduced considerably. The fluorinated merophosphinine derivatives showed maximum absorption wavelengths in the 364–375 nm range in CH2Cl2 with a purple coloration. In contrast, the fluorinated phosphinine derivatives showed an absorption band with λabs in the 420–474 nm range with a yellow coloration. Both derivative classes showed negative solvatochromism; this can be attributed to a reduction in the HOMO energy and subsequent increase in the ΔEH-L when the solvent polarity is increased, as shown by theoretical calculations. It was also confirmed that the UV-vis absorption profile of the dyes is affected only by the organic structure and not by the counter anion. To date, only a few derivatives of merophosphinine and phosphinine dyes have been studied owing to the difficulty of accessing these structures; however, with the synthesis methods described in this study, novel fluorinated merophosphinine and phosphinine derivatives can now be readily produced which have tunable absorption properties and thermal stabilities. We anticipate that these findings will, therefore, inform the development of phosphinine dye chemistry and the design of new functional dyes for a variety of future technical applications. Our group is further investigating the development of fluorinated phosphinine molecules by further transformations using reported merophosphinine derivatives, such as the Wittig reaction, and their results will also be disclosed in the near future.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/compounds3010013/s1.
Author Contributions
Conceptualization, S.Y.; methodology, Y.T. and S.Y.; validation, Y.T. and S.Y.; investigation, Y.T. and S.Y.; resources, S.Y. and T.K.; data curation, Y.T., S.Y. and T.K.; writing—original draft preparation, Y.T. and S.Y.; writing—review and editing, Y.T., S.Y. and T.K.; visualization, S.Y.; supervision, S.Y.; project administration, S.Y.; All authors have read and agreed to the published version of the manuscript.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We would like to express our sincere gratitude to Nippon Zeon Corporation for providing the octafluorocyclopentene used as the starting material.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Mustroph, H.; Stollenwerk, M.; Bressau, V. Current developments in optical data storage with organic dyes. Angew. Chem. Int. Ed. 2006, 45, 2016–2035. [Google Scholar] [CrossRef] [PubMed]
- Liang, M.; Chen, J. Arylamine organic dyes for dye-sensitized solar cells. Chem. Soc. Rev. 2013, 42, 3453–3488. [Google Scholar] [CrossRef]
- Cai, Y.; Si, W.; Huang, W.; Chen, P.; Shao, J.; Dong, X. Organic dye based nanoparticles for cancer phototheranostics. Small 2018, 14, e1704247. [Google Scholar] [CrossRef] [PubMed]
- Kulinich, A.V.; Ishchenko, A.A.; Bulavko, G.V.; Davidenko, N.A. Effect of structure on the photovoltaic properties of merocyanine dyes in polymer films. Theor. Exp. Chem. 2018, 54, 178–185. [Google Scholar] [CrossRef]
- Reichardt, C. Solvatochromic dyes as solvent polarity indicators. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar] [CrossRef]
- Brooker, L.G.S.; Keyes, G.H.; Sprague, R.H.; van Dyke, R.H.; van Lare, E.; van Zandt, G.; White, F.L.; Cressman, H.W.J.; Dent, S.G., Jr. Color and constitution. X. 1 Absorption of the merocyanines 2. J. Am. Chem. Soc. 1951, 73, 5332–5350. [Google Scholar] [CrossRef]
- Matikonda, S.S.; Helmerich, D.A.; Meub, M.; Beliu, G.; Kollmannsberger, P.; Greer, A.; Sauer, M.; Schnermann, M.J. Defining the basis of cyanine phototruncation enables a new approach to single-molecule localization microscopy. ACS Cent. Sci. 2021, 7, 1144–1155. [Google Scholar] [CrossRef]
- Meguellati, K.; Spichty, M.; Ladame, S. Reversible synthesis and characterization of dynamic imino analogues of trimethine and pentamethine cyanine dyes. Org. Lett. 2009, 11, 1123–1126. [Google Scholar] [CrossRef]
- Gopika, G.S.; Prasad, P.M.H.; Lekshmi, A.G.; Lekshmypriya, S.; Sreesaila, S.; Arunima, C.; Kumar, M.S.; Anil, A.; Sreekumar, A.; Pillai, Z.S. Chemistry of cyanine dyes—A review. Mater. Today Proc. 2021, 46, 3102–3108. [Google Scholar] [CrossRef]
- Shindy, H.A. Fundamentals in the chemistry of cyanine dyes: A review. Dye. Pigment. 2017, 145, 505–513. [Google Scholar] [CrossRef]
- Tatikolov, A.S. Polymethine dyes as spectral-fluorescent probes for biomacromolecules. J. Photochem. Photobiol. C Photochem. Rev. 2012, 13, 55–90. [Google Scholar] [CrossRef]
- Weimei, L.; Zhenghua, Z.; Zhuguang, Y.; Mengzhen, H.; Bingkui, W. Novel laser dyes: Some bridged pentamethine phosphinines. Dye. Pigment. 1990, 14, 211–216. [Google Scholar] [CrossRef]
- Depoorter, H.; Nys, J.; van Dormael, A. New classes of phosphorus-containing dyes. Tetrahedron Lett. 1961, 2, 199–205. [Google Scholar] [CrossRef]
- Depoorter, H.; Nys, J.; van Dormael, A. Phosphorus containing polymethine dyes I. Methods of preparation and chemical properties. Bull. Soc. Chim. Belg. 1964, 73, 921–943. [Google Scholar] [CrossRef]
- Yamada, S.; Ishii, E.; Konno, T.; Ishihara, T. Preparation of perfluorocyclopentenylmetal species and their cross-coupling reaction with electrophiles—Remarkable accesses to versatile perfluorocyclopentene derivatives. Tetrahedron 2008, 64, 4215–4223. [Google Scholar] [CrossRef]
- Yamada, S.; Noma, M.; Hondo, K.; Konno, T.; Ishihara, T. Preparation and addition-elimination reactions of benzyl α,β,β-trifluoracrylate. A new stereoselective approach to (Z)-β-substituted α,β-difluoracrylates. J. Org. Chem. 2008, 73, 522–528. [Google Scholar] [CrossRef]
- Yamada, S.; Takahashi, T.; Konno, T.; Ishihara, T. A novel fluorine-metal exchange reaction of pentafluorocrotonate with organocuprate. Generation of β-metallated tetrafluorocrotonate and its cross-coupling reaction. Chem. Commun. 2007, 21, 3679–3681. [Google Scholar] [CrossRef]
- Yamada, S.; Ishii, E.; Konno, T.; Ishihara, T. Reaction of perfluorocyclopentene with various carbon nucleophiles—Heteroaromatic lithium reagents, enolate and phosphonium ylide. Org. Biomol. Chem. 2007, 5, 1442–1449. [Google Scholar] [CrossRef]
- Yamada, S.; Noma, M.; Konno, T.; Ishihara, T.; Yamanaka, H. Novel synthesis of (Z)-difluoroacrylates via a highly stereoselective addition-elimination reaction. Org. Lett. 2006, 8, 843–845. [Google Scholar] [CrossRef]
- Yamada, S.; Konno, T.; Ishihara, T.; Yamanaka, H. Reaction of octafluorocyclopentene with various carbon nucleophiles. J. Fluor. Chem. 2005, 126, 125–133. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Hohenstein, E.G.; Chill, S.T.; Sherrill, C.D. Assessment of the performance of the M05-2X and M06-2X exchange-correlation functionals for noncovalent interactions in biomolecules. J. Chem. Theory Comput. 2008, 4, 1996–2000. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Jensen, J.H. Improving the efficiency and convergence of geometry optimization with the polarizable continuum model: New energy gradients and molecular surface tessellation. J. Comput. Chem. 2004, 25, 1449–1462. [Google Scholar] [CrossRef] [PubMed]
- Kulinich, A.V.; Mikitenko, E.K.; Ishchenko, A.A. Scope of negative solvatochromism and solvatofluorochromism of merocyanines. Phys. Chem. Chem. Phys. 2016, 18, 3444–3453. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.; Choi, M.G.; Jeon, H.R.; Chang, S.-K. Negative solvatochromism of merocyanine dyes: Application as water content probes for organic solvents. Sens. Actuators B Chem. 2011, 157, 14–18. [Google Scholar] [CrossRef]
Figure 1.
Chemical structures and the corresponding resonance structures of: (a) merocyanine; (b) cyanine; (c) merophosphinine; (d) phosphinine.
Figure 1.
Chemical structures and the corresponding resonance structures of: (a) merocyanine; (b) cyanine; (c) merophosphinine; (d) phosphinine.

Figure 2.
(a) Scheme showing the previously investigated reaction of octafluorocyclopentene with triphenylphosphonium methylide [18]; (b) The chemical structures of the fluorinated merophosphinine (3) and phosphinine (4) derivatives synthesized in this study.
Figure 2.
(a) Scheme showing the previously investigated reaction of octafluorocyclopentene with triphenylphosphonium methylide [18]; (b) The chemical structures of the fluorinated merophosphinine (3) and phosphinine (4) derivatives synthesized in this study.

Figure 3.
The proposed reaction mechanisms for the synthesis of fluorinated merophosphinines 3 and phosphinines 4.
Figure 3.
The proposed reaction mechanisms for the synthesis of fluorinated merophosphinines 3 and phosphinines 4.

Figure 4.
TGA thermograms of the fluorinated derivatives under a nitrogen atmosphere (scan rate = 10 °C·min–1): (a) merophosphinines 3a–3e; (b) phosphinines 4aA–4eA.
Figure 4.
TGA thermograms of the fluorinated derivatives under a nitrogen atmosphere (scan rate = 10 °C·min–1): (a) merophosphinines 3a–3e; (b) phosphinines 4aA–4eA.

Figure 5.
(a) UV-vis absorption spectra and photographs (inset) of 3a–3e in CH2Cl2 (concentration = 1.0 × 10–5 mol·L–1, λex = 300–600 nm); (b) the HOMO and LUMO distribution in 3a as obtained from theoretical calculations.
Figure 5.
(a) UV-vis absorption spectra and photographs (inset) of 3a–3e in CH2Cl2 (concentration = 1.0 × 10–5 mol·L–1, λex = 300–600 nm); (b) the HOMO and LUMO distribution in 3a as obtained from theoretical calculations.

Figure 6.
UV-vis absorption spectra measured in various solvents for: (a) 3b (R = phenyl; C6H5); (b) 3e (R = pentafluorophenyl, C6F5).
Figure 6.
UV-vis absorption spectra measured in various solvents for: (a) 3b (R = phenyl; C6H5); (b) 3e (R = pentafluorophenyl, C6F5).

Figure 7.
(a) UV-vis absorption spectra and photographs (inset) of 4aA–4eA in CH2Cl2 (concentration = 1.0 × 10–5 mol·L–1); (b) the HOMO and LUMO distribution in 4aA as obtained from theoretical calculations.
Figure 7.
(a) UV-vis absorption spectra and photographs (inset) of 4aA–4eA in CH2Cl2 (concentration = 1.0 × 10–5 mol·L–1); (b) the HOMO and LUMO distribution in 4aA as obtained from theoretical calculations.

Figure 8.
UV-vis absorption spectra measured in various solvents for: (a) 4bA (R = phenyl, C6H5); (b) 4eA (R = pentaphenyl, C6F6).
Figure 8.
UV-vis absorption spectra measured in various solvents for: (a) 4bA (R = phenyl, C6H5); (b) 4eA (R = pentaphenyl, C6F6).

Figure 9.
UV-vis absorption spectrum of 4aA-4aE with various counter anions (X) in CH2Cl2 (concentration = 1.0 × 10–5 mol·L–1).
Figure 9.
UV-vis absorption spectrum of 4aA-4aE with various counter anions (X) in CH2Cl2 (concentration = 1.0 × 10–5 mol·L–1).


{kind=link}
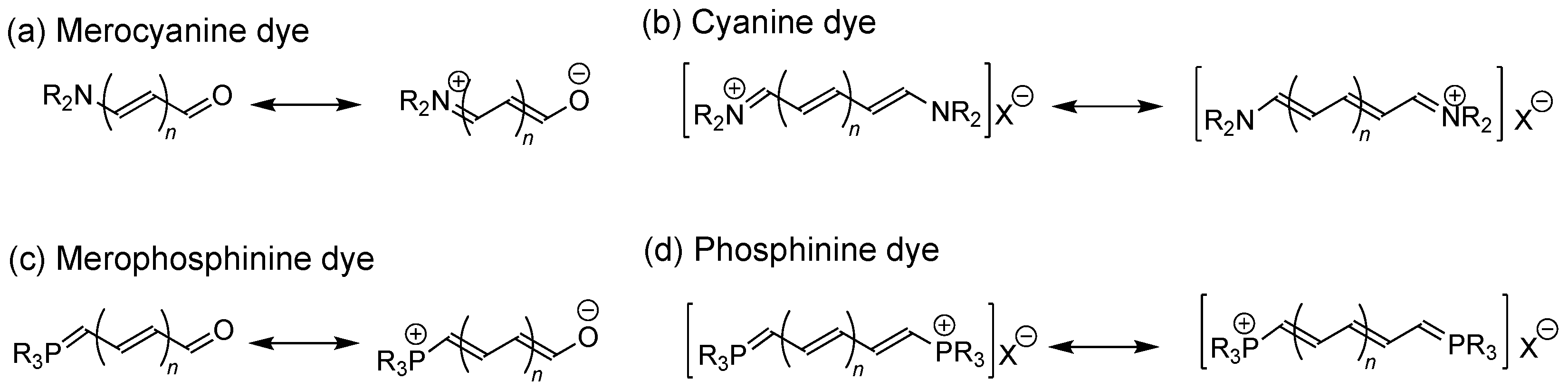{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Synthesis of the fluorinated merophosphinines (3) and phosphinines (4). (The values in parentheses are the isolated yields).
Table 1.
Synthesis of the fluorinated merophosphinines (3) and phosphinines (4). (The values in parentheses are the isolated yields).
 | |||
| Fluorinated Merophosphinines | |||
 |  |  |  |
| 3b (11%) | 3c (19%) | 3d (2%) | 3e (26%) |
| Fluorinated Phosphinines | |||
 |  |  |  |
| 4bA (23%) | 4cA (44%) | 4dA (51%) | 4eA (10%) |
 |  |  |  |
| 4aB (54%) | 4aC (32%) | 4aD (89%) | 4aE (20%) |
Table 2.
Thermal decomposition temperatures (Tdec) of the fluorinated merophosphinine (3a–3e) and phosphinine (4aA–4eA) derivatives.
Table 2.
Thermal decomposition temperatures (Tdec) of the fluorinated merophosphinine (3a–3e) and phosphinine (4aA–4eA) derivatives.
| Compound | Tdec [°C] 1 | Compound | Tdec [°C] 1 |
|---|---|---|---|
| 3a | 241 | 4aA | 241 |
| 3b | 295 | 4bA | 281 |
| 3c | 313 | 4cA | 270 |
| 3d | 217 | 4dA | 280 |
| 3e | 304 | 4eA | 203 |
1 Defined as the temperature at which the sample weight decreases by 5%.
Table 3.
Photophysical data of 3a–3e in numerous solvents and the corresponding calculated theoretical transitions.
Table 3.
Photophysical data of 3a–3e in numerous solvents and the corresponding calculated theoretical transitions.
| Compound | Solvent | λabs [nm] 1 (ε [L·mol–1·cm–1]) | Theoretical Electronic Transition and the Corresponding Orbital Energies [eV] 2 | ΔEH-L [eV] 3 |
|---|---|---|---|---|
| 3a | CH2Cl2 | 364 (46,000) | HOMO (–7.02) → LUMO (–1.08) | 5.94 (137) |
| 3b | CH2Cl2 | 373 (23,300) | HOMO (–6.87) → LUMO (–1.09) | 5.78 (133) |
| Toluene | 379 (33,600) | HOMO (–6.71) → LUMO (–1.04) | 5.67 (131) | |
| THF | 375 (36,900) | HOMO (–6.85) → LUMO (–1.08) | 5.77 (133) | |
| MeOH | 361 (29,400) | HOMO (–6.92) → LUMO (–1.11) | 5.81 (134) | |
| MeCN | 369 (21,600) | HOMO (–6.92) → LUMO (–1.11) | 5.81 (134) | |
| 3c | CH2Cl2 | 374 (47,500) | HOMO (–6.82) → LUMO (–1.07) | 5.75 (132) |
| 3d | CH2Cl2 | 375 (32,800) | HOMO (–6.85) → LUMO (–1.11) | 5.74 (132) |
| 3e | CH2Cl2 | 364 (40,500) | HOMO (–7.17) → LUMO (–1.22) | 5.95 (137) |
| Toluene | 364 (29,600) | HOMO (–7.06) → LUMO (–1.19) | 5.87 (135) | |
| THF | 363 (52,200) | HOMO (–7.16) → LUMO (–1.22) | 5.94 (137) | |
| MeOH | 359 (36,700) | HOMO (–7.21) → LUMO (–1.24) | 5.97 (138) | |
| MeCN | 361 (56,000) | HOMO (–7.21) → LUMO (–1.24) | 5.97 (138) |
1 Concentration: 1.0 × 10–5 mol·L–1. 2 Values were calculated using a time-dependent-self-consistent field (TD-SCF) method at the M06-2X/6-31+G(d) with a CPCM solvation model. Values in parentheses are the corresponding orbital energy (eV). 3 Energy gap between HOMO and LUMO. Values in parentheses indicate the ΔEH-L in kcal·mol–1 unit.
Table 4.
Photophysical data of 4aA–4eA 3e in numerous solvents and the corresponding calculated theoretical transitions.
Table 4.
Photophysical data of 4aA–4eA 3e in numerous solvents and the corresponding calculated theoretical transitions.
| Compound | Solvent | λabs [nm] 1 (ε [L mol–1 cm–1]) | Theoretical Transition and Orbital Energies [eV] 2 | ΔEH-L [eV] 3 |
|---|---|---|---|---|
| 4aA | CH2Cl2 | 424sh (33,600), 446 (92,100) | HOMO (–6.74) → LUMO (–1.53) | 5.21 (120) |
| 4bA | CH2Cl2 | 445sh (33,400), 470 (95,900) | HOMO (–6.43) → LUMO (–1.49) | 4.94 (114) |
| Toluene | 444sh (1900), 472 (5800) | HOMO (–6.40) → LUMO (–1.46) | 4.94 (114) | |
| THF | 444sh (14,600), 469 (41,000) | HOMO (–6.43) → LUMO (–1.49) | 4.94 (114) | |
| MeOH | 442sh (34,900), 467 (98,400) | HOMO (–6.44) → LUMO (–1.50) | 4.95 (114) | |
| MeCN | 442sh (33,000), 467 (91,100) | HOMO (–6.44) → LUMO (–1.50) | 4.95 (114) | |
| 4cA | CH2Cl2 | 444sh (33,800), 473 (104,200) | – 4 | – 4 |
| 4dA | CH2Cl2 | 444sh (33,200), 474 (105,300) | – 4 | – 4 |
| 4eA | CH2Cl2 | 420 (48,300) | HOMO (–6.93) → LUMO (–1.80) | 5.13 (118) |
| Toluene | 410 (38,900) | HOMO–3 (–6.95) → LUMO (–1.82) | 5.13 (118) | |
| THF | 408 (32,300) | HOMO (–6.93) → LUMO (–1.80) | 5.13 (118) | |
| MeOH | 397 (31,900) | HOMO (–6.92) → LUMO (–1.79) | 5.13 (118) | |
| MeCN | 408 (55,100) | HOMO (–6.92) → LUMO (–1.79) | 5.13 (118) |
1 Concentration: 1.0 × 10–5 mol·L–1. 2 Values were calculated using a time-dependent-self-consistent field (TD-SCF) method at the M06-2X/6-31+G(d) with a CPCM solvation model. Values in parentheses are the corresponding orbital energy (eV). 3 Energy gap between HOMO and LUMO. Values in parentheses indicates the ΔEH-L in kcal mol–1 unit. 4 Values were not obtained due to the high calculation cost. sh = shoulder peak.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Yamada, S.; Takahashi, Y.; Konno, T. Fluorinated Merophosphinine and Phosphinine Dyes: Synthesis and Evaluation of UV-Visible Light Absorption Properties. Compounds 2023, 3, 153-168. https://doi.org/10.3390/compounds3010013
AMA Style
Yamada S, Takahashi Y, Konno T. Fluorinated Merophosphinine and Phosphinine Dyes: Synthesis and Evaluation of UV-Visible Light Absorption Properties. Compounds. 2023; 3(1):153-168. https://doi.org/10.3390/compounds3010013
Chicago/Turabian StyleYamada, Shigeyuki, Yusuke Takahashi, and Tsutomu Konno. 2023. "Fluorinated Merophosphinine and Phosphinine Dyes: Synthesis and Evaluation of UV-Visible Light Absorption Properties" Compounds 3, no. 1: 153-168. https://doi.org/10.3390/compounds3010013