


Evaluation of DNA Methylation-Based Age-Prediction Models from Saliva and Buccal Swab Samples Using Pyrosequencing Data


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Design and Sample Collection
2.2. Workflow Validation
2.3. DNA Extraction
2.4. Bisulfite Conversion
2.5. Converted DNA PCR
2.6. Purification and Pyrosequencing
2.7. Statistical Analysis
3. Results
3.1. Trueness
3.2. Sensitivity
3.3. Repeatability
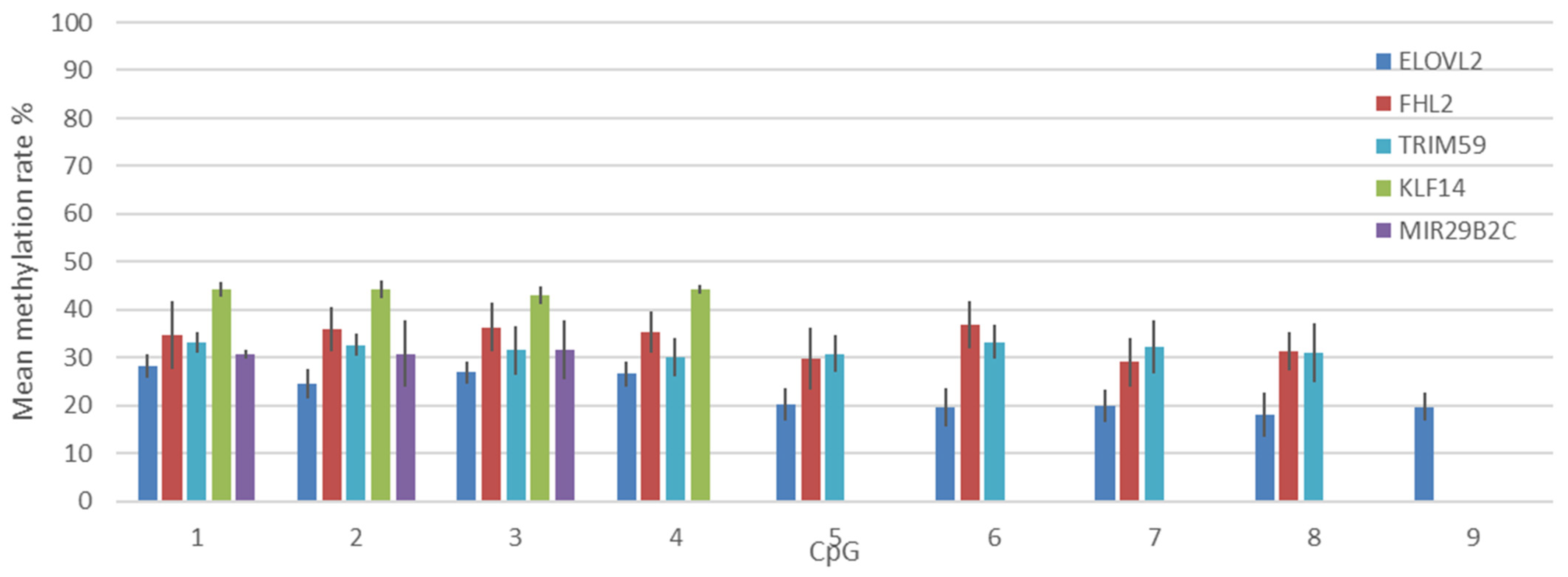
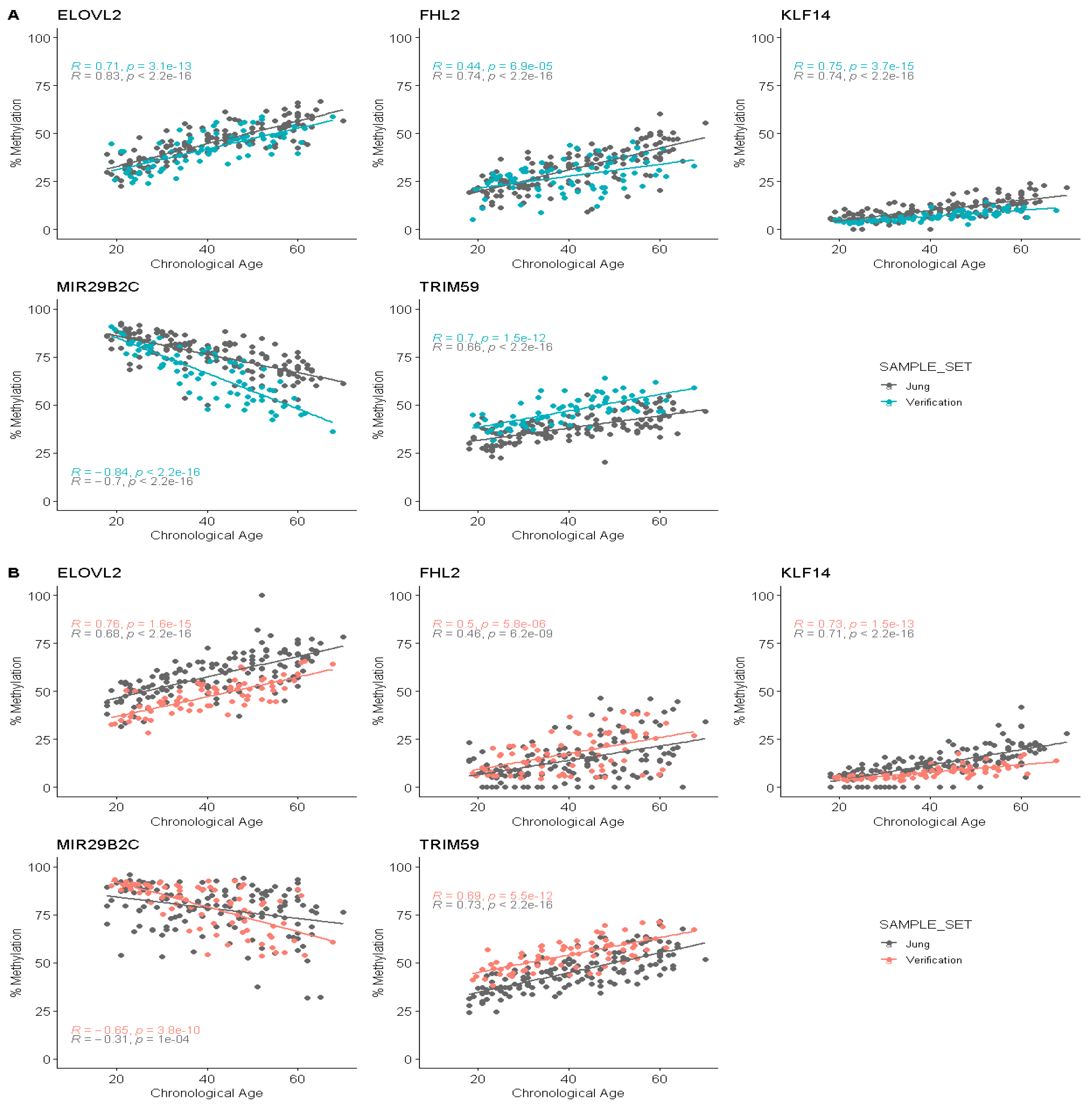
3.4. Correlation between Age and Methylation at Five CpG Sites Using Pyrosequencing
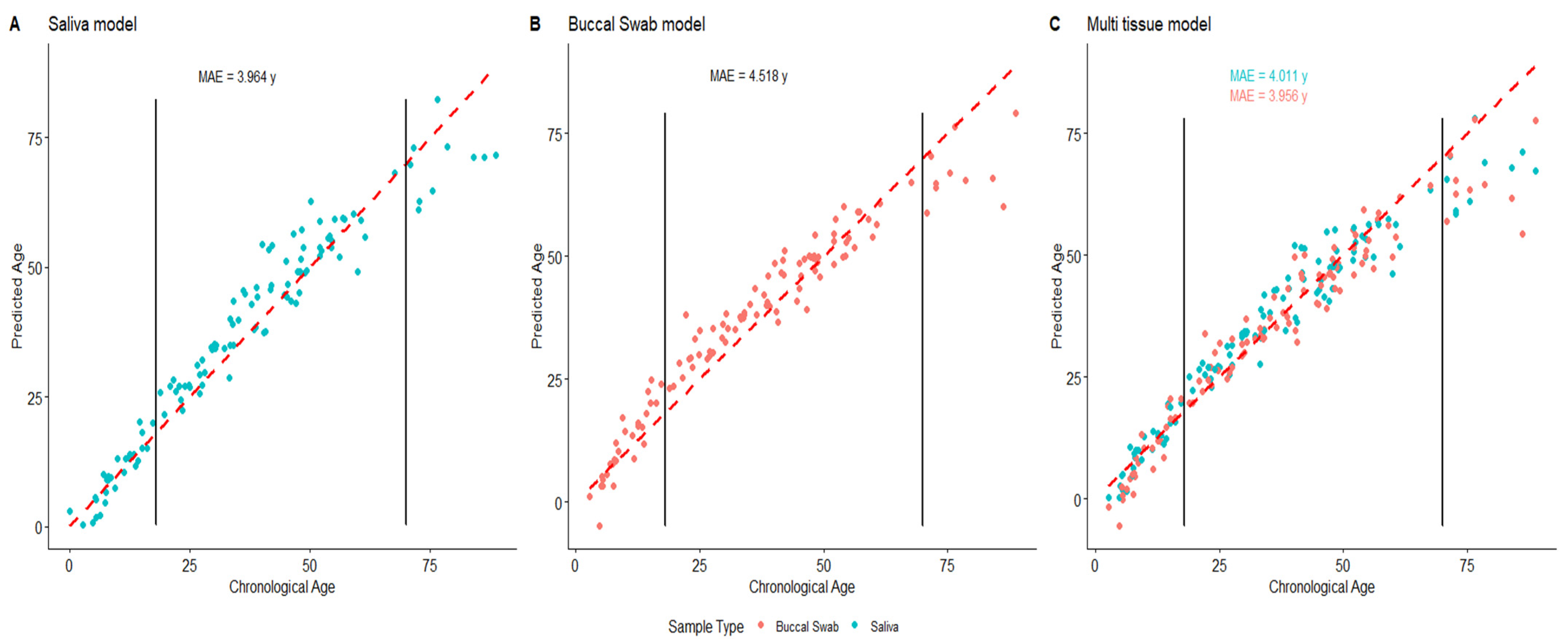
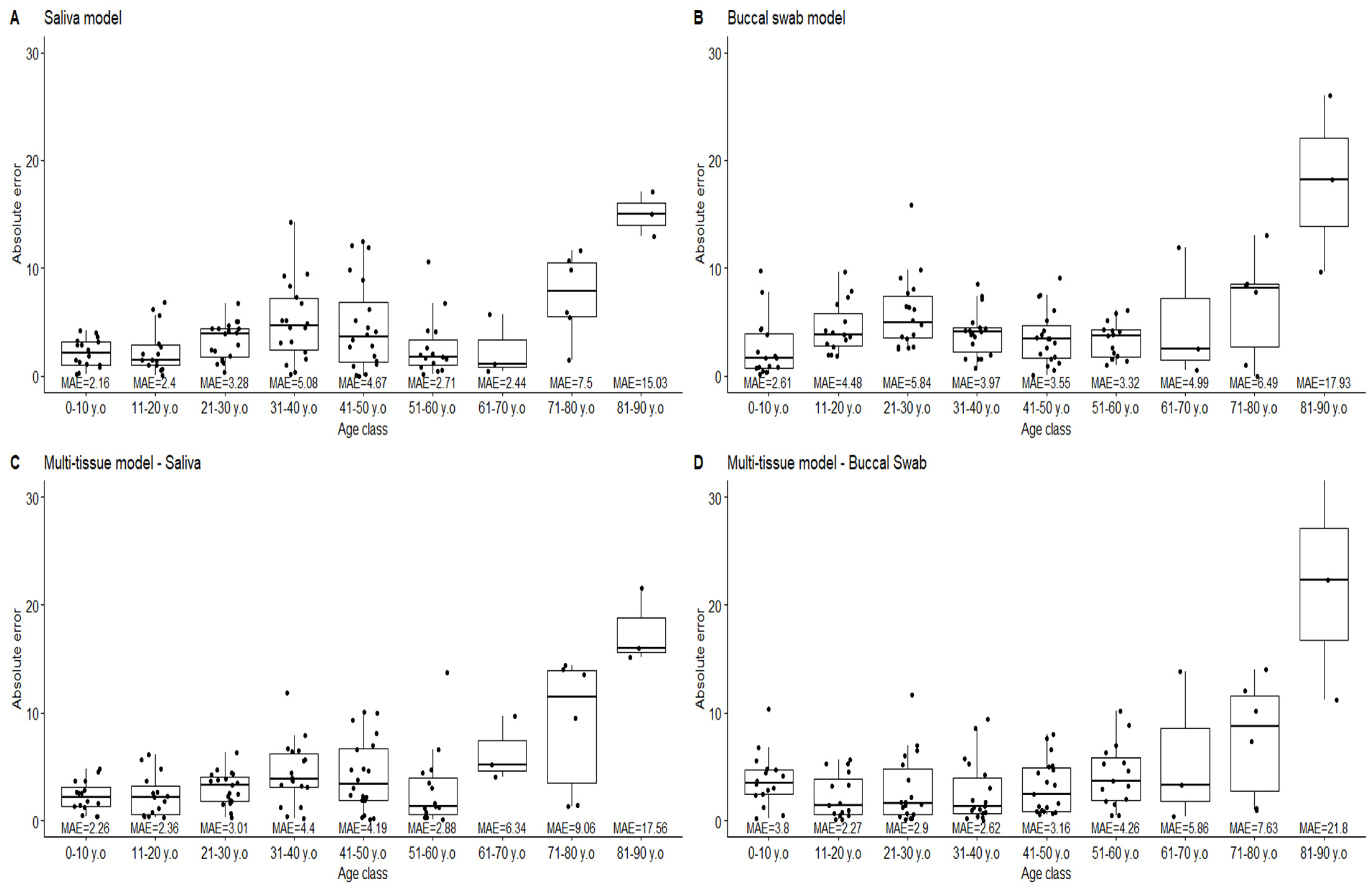
3.5. Age Prediction on Saliva and Buccal Swab Samples Using Tissue-Specific and Multitissue Models
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Parson, W. Age Estimation with DNA: From Forensic DNA Fingerprinting to Forensic (Epi)Genomics: A Mini-Review. Gerontology 2018, 64, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Thiburce, N.; Nolot, F.; Pussiau, A.; Guyomarc’h, P.; Mazevet, M. Forensic Science and “The Duty of Memory”: The Face of Verdun. J. Forensic Identif. 2020, 70, 1–15. [Google Scholar]
- Vidaki, A.; Kayser, M. Recent progress, methods and perspectives in forensic epigenetics. Forensic Sci. Int. Genet. 2018, 37, 180–195. [Google Scholar] [CrossRef]
- Kader, F.; Ghai, M. DNA methylation and application in forensic sciences. Forensic Sci. Int. 2015, 249, 255–265. [Google Scholar] [CrossRef]
- Zubakov, D.; Liu, F.; Kokmeijer, I.; Choi, Y.; van Meurs, J.; van IJcken, W.; Uitterlinden, A.; Hofman, A.; Broer, L.; van Duijn, C.; et al. Human age estimation from blood using mRNA, DNA methylation, DNA rearrangement, and telomere length. Forensic Sci. Int. Genet. 2016, 24, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naue, J.; Hoefsloot, H.; Kloosterman, A.; Verschure, P. Forensic DNA methylation profiling from minimal traces: How low can we go? Forensic Sci. Int. Genet. 2018, 33, 17–23. [Google Scholar] [CrossRef]
- Eipel, M.; Mayer, F.; Arent, T.; Ferreira, M.; Birkhofer, C.; Gerstenmaier, U.; Costa, I.; Ritz-Timme, S.; Wagner, W. Epigenetic age predictions based on buccal swabs are more precise in combination with cell type-specific DNA methylation signatures. Aging 2016, 8, 1034–1048. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.R.; Jung, S.-E.; Lee, S.E.; Shin, K.-J.; Yang, W.H.; Lee, H.Y. DNA methylation-based age prediction from saliva: High age predictability by combination of 7 CpG markers. Forensic Sci. Int. Genet. 2017, 29, 118–125. [Google Scholar] [CrossRef]
- Jung, S.-E.; Lim, S.M.; Hong, S.R.; Lee, E.H.; Shin, K.-J.; Lee, H.Y. DNA methylation of the ELOVL2, FHL2, KLF14, C1orf132/MIR29B2C, and TRIM59 genes for age prediction from blood, saliva, and buccal swab samples. Forensic Sci. Int. Genet. 2019, 38, 1–8. [Google Scholar] [CrossRef]
- Cho, S.; Jung, S.-E.; Hong, S.R.; Lee, E.H.; Lee, J.H.; Lee, S.D.; Lee, H.Y. Independent validation of DNA-based approaches for age prediction in blood. Forensic Sci. Int. Genet. 2017, 29, 250–256. [Google Scholar] [CrossRef] [PubMed]
- Zbieć-Piekarska, R.; Spólnicka, M.; Kupiec, T.; Parys-Proszek, A.; Makowska, Ż.; Pałeczka, A.; Kucharczyk, K.; Płoski, R.; Branicki, W. Development of a forensically useful age prediction method based on DNA methylation analysis. Forensic Sci. Int. Genet. 2015, 17, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.R.; Shin, K.-J.; Jung, S.-E.; Lee, E.H.; Lee, H.Y. Platform-independent models for age prediction using DNA methylation data. Forensic Sci. Int. Genet. 2019, 38, 39–47. [Google Scholar] [CrossRef]
- Kint, S.; De Spiegelaere, W.; De Kesel, J.; Vandekerckhove, L.; Van Criekinge, W. Evaluation of bisulfite kits for DNA methylation profiling in terms of DNA fragmentation and DNA recovery using digital PCR. PLoS ONE 2018, 13, e0199091. [Google Scholar] [CrossRef] [Green Version]
- Tost, J.; Gut, I.G. DNA methylation analysis by pyrosequencing. Nat. Protoc. 2007, 2, 2265–2275. [Google Scholar] [CrossRef] [PubMed]
- Candiloro, I.L.M.; Mikeska, T.; Dobrovic, A. Assessing alternative base substitutions at primer CpG sites to optimise unbiased PCR amplification of methylated sequences. Clin. Epigenet. 2017, 9, 31. [Google Scholar] [CrossRef]
- Wojdacz, T.K.; Borgbo, T.; Hansen, L.L. Primer design versus PCR bias in methylation independent PCR amplifications. Epigenetics 2009, 4, 231–234. [Google Scholar] [CrossRef] [Green Version]
- Shen, L.; Guo, Y.; Chen, X.; Ahmed, S.; Issa, J.-P.J. Optimizing annealing temperature overcomes bias in bisulfite PCR methylation analysis. BioTechniques 2007, 42, 48–58. [Google Scholar] [CrossRef]
- Warnecke, P.M.; Stirzaker, C.; Melki, J.R.; Millar, D.S.; Paul, C.L.; Clark, S.J. Detection and measurement of PCR bias in quantitative methylation analysis of bisulphite-treated DNA. Nucleic Acids Res. 1997, 25, 4422–4426. [Google Scholar] [CrossRef] [Green Version]
- Heidegger, A.; Xavier, C.; Niederstätter, H.; de la Puente, M.; Pośpiech, E.; Pisarek, A.; Kayser, M.; Branicki, W.; Parson, W. Development and optimization of the VISAGE basic prototype tool for forensic age estimation. Forensic Sci. Int. Genet. 2020, 48, 102322. [Google Scholar] [CrossRef]
- Schwender, K.; Holländer, O.; Klopfleisch, S.; Eveslage, M.; Danzer, M.F.; Pfeiffer, H.; Vennemann, M. Development of two age estimation models for buccal swab samples based on 3 CpG sites analyzed with pyrosequencing and minisequencing. Forensic Sci. Int. Genet. 2021, 53, 102521. [Google Scholar] [CrossRef] [PubMed]
- Ciccarone, F.; Tagliatesta, S.; Caiafa, P.; Zampieri, M. DNA methylation dynamics in aging: How far are we from understanding the mechanisms? Mech. Ageing Dev. 2018, 174, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.W.K.; Pausova, Z. Cigarette smoking and DNA methylation. Front. Genet. 2013, 4, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woźniak, A.; Heidegger, A.; Piniewska-Róg, D.; Pośpiech, E.; Xavier, C.; Pisarek, A.; Kartasińska, E.; Boroń, M.; Freire-Aradas, A.; Wojtas, M.; et al. Development of the VISAGE enhanced tool and statistical models for epigenetic age estimation in blood, buccal cells and bones. Aging 2021, 13, 6459–6484. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | CpG position (GRCh38) | Primers (5′-->3′) F: Forward R: Reverse S: Sequencing B: Biotin | Ref. | Strand | Amp. Size (bp) | [MgCl2] (mM) | Sequence to analyze (5′-->3′): Y, R: Analyzed CpG Y, R: Other CpGs T or A: Conversion Control |
| ELOVL2 | Chr6: 11044628 | F: AGGGGAGTAGGGTAAGTGAGG R: B-AACCATTTCCCCCTAATATATACTTCA S: GGGAGGAGATTTGTAGGTTT | (2) | - | 303 | 2.5 | AGTYGGYGTYGGTTTYGYGYGGYGGTTTAAYGTTTAYGGAGTTTTAG |
| FHL2 | Chr2: 105399282 | F: GGGTTTTGGGAGTATAGTAGT R: B-AAAATAACCCCCTCCTCC S: GTTTTGGGAGTATAGTAGTTAT | (1) | + | 191 | 1.5 | TYGGGAGYGTYGTTTTYGGYGTGGGTTTTYGGGYGYGAGTTT |
| KLF14 | Chr7: 130734355 | F: B-AGGTTGTTGTAATTTAGAAGTTT R: ATATTTAACAACCTCAAAAATTATCTTATC S: TTAACAACCTCAAAAATTATCTTATCTCC | (1) | + | 114 | 2.5 | RCRTTCTTTCTTCTACCRACRAACCAAATAATAATAACAAAAC |
| MIR29B2C | Chr1: 207823681 | F: B-GGGTTAYGTTATTAAGTTTTGAAG R: TAAAACCAAATTCTAAAACATTC S: AAACCAAAATTTAAATCTAC | (1) | + | 116 | 2.5 | RCAAACRACRATAAATAATCC |
| TRIM59 | Chr3: 160450189 | F: TATGGTATAGGTGGTTTGGGGGAGA R: B- ATAAAAAACACTACCCTCCACAACATAAC S: TTGGGGGAGAGGTTG | (2) | + | 146 | 2.5 | GGTTTGGYGYGGGAYGAGGYGAAGYGTYGGTGGTYGAYGGTTTTT |
| Age Class | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Model | Interval | 0–10 n = 16 | 11–20 n = 15 | 21–30 n = 19 | 31–40 n = 18 | 41–50 n = 20 | 51–60 n = 15 | 61–70 n = 3 | 71–80 n = 6 | 81–90 n = 3 |
| Saliva | ±5 y | 100% | 80% | 84% | 56% | 65% | 87% | 67% | 17% | 0% |
| ±10 y | 100% | 100% | 100% | 94% | 85% | 93% | 100% | 67% | 0% | |
| Buccal swab | ±5 y | 88% | 67% | 50% | 83% | 74% | 80% | 67% | 33% | 0% |
| ±10 y | 100% | 100% | 94% | 100% | 100% | 100% | 67% | 83% | 33% | |
| Multi-tissue-Saliva | ±5 y | 100% | 87% | 95% | 61% | 70% | 87% | 33% | 33% | 0% |
| ±10 y | 100% | 100% | 100% | 94% | 90% | 93% | 100% | 50% | 0% | |
| Multi-tissue-Buccal swab | ±5 y | 81% | 80% | 72% | 78% | 74% | 60% | 67% | 33% | 0% |
| ±10 y | 94% | 100% | 94% | 100% | 100% | 93% | 67% | 50% | 0% | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Poussard, A.; Curci, J.-Y.; Siatka, C.; Hermitte, F.; Pussiau, A.; Singla-Sanchez, H.; Hubac, S. Evaluation of DNA Methylation-Based Age-Prediction Models from Saliva and Buccal Swab Samples Using Pyrosequencing Data. Forensic Sci. 2023, 3, 192-204. https://doi.org/10.3390/forensicsci3020015
Poussard A, Curci J-Y, Siatka C, Hermitte F, Pussiau A, Singla-Sanchez H, Hubac S. Evaluation of DNA Methylation-Based Age-Prediction Models from Saliva and Buccal Swab Samples Using Pyrosequencing Data. Forensic Sciences. 2023; 3(2):192-204. https://doi.org/10.3390/forensicsci3020015
Chicago/Turabian StylePoussard, Alexandre, Jean-Yves Curci, Christian Siatka, Francis Hermitte, Amaury Pussiau, Hélène Singla-Sanchez, and Sylvain Hubac. 2023. "Evaluation of DNA Methylation-Based Age-Prediction Models from Saliva and Buccal Swab Samples Using Pyrosequencing Data" Forensic Sciences 3, no. 2: 192-204. https://doi.org/10.3390/forensicsci3020015