
Comparing Polyphosphate and Orthophosphate Treatments of Solution-Precipitated Aragonite Powders



Abstract
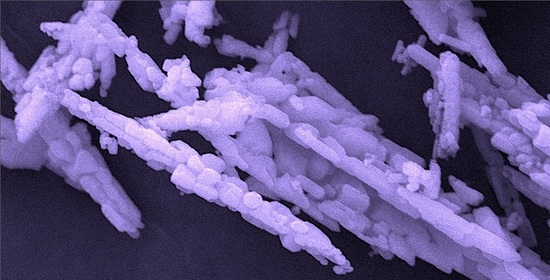
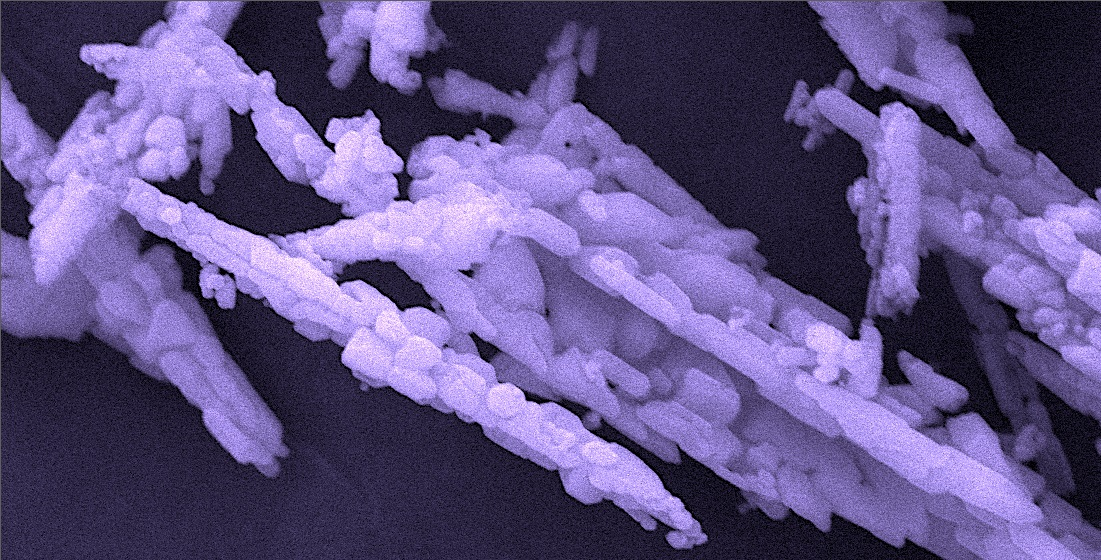
:
1. Introduction
2. Materials and Methods
2.1. Precipitation
2.2. Annealing
2.3. Aqueous Treatments
2.4. Characterization
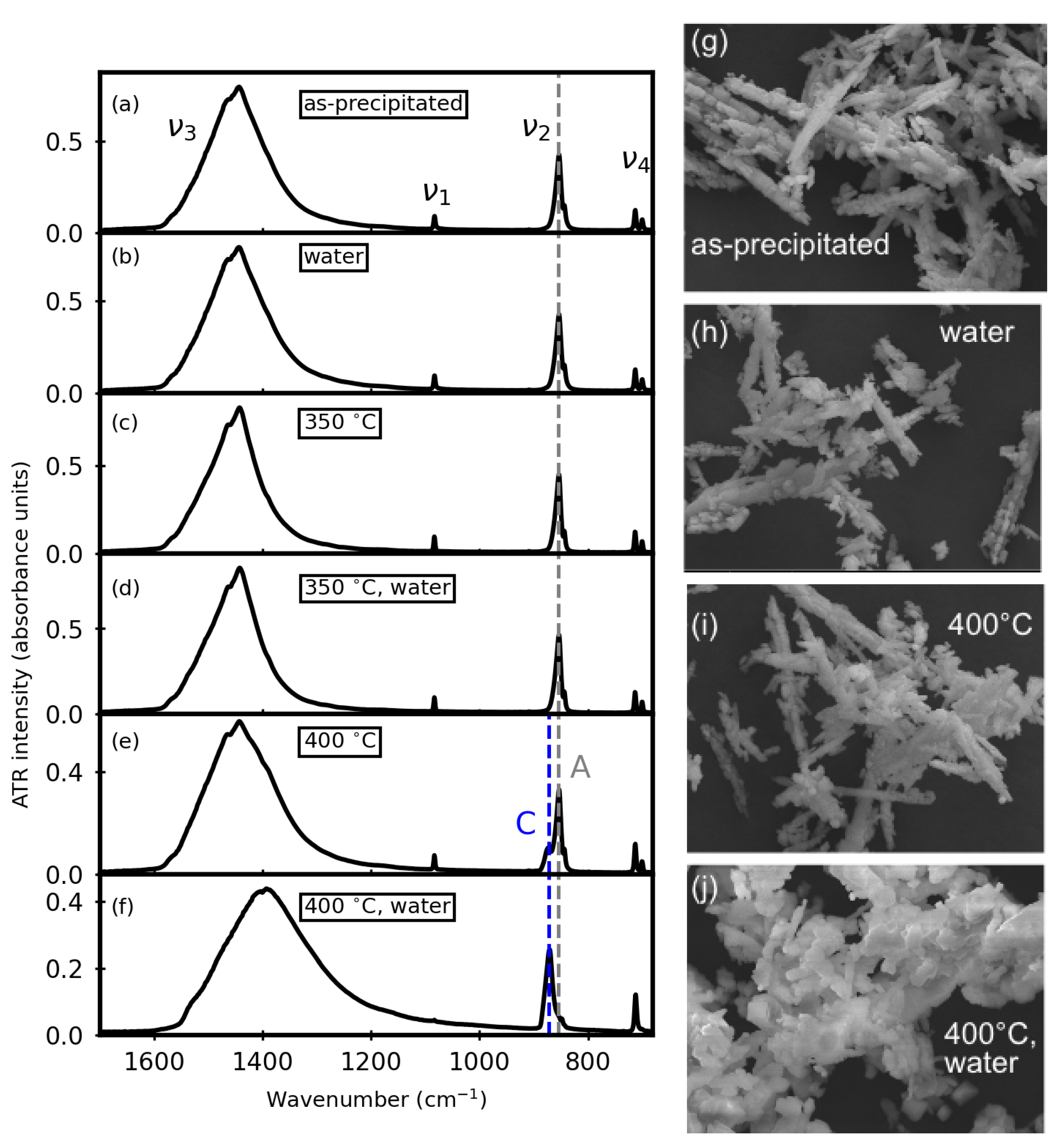
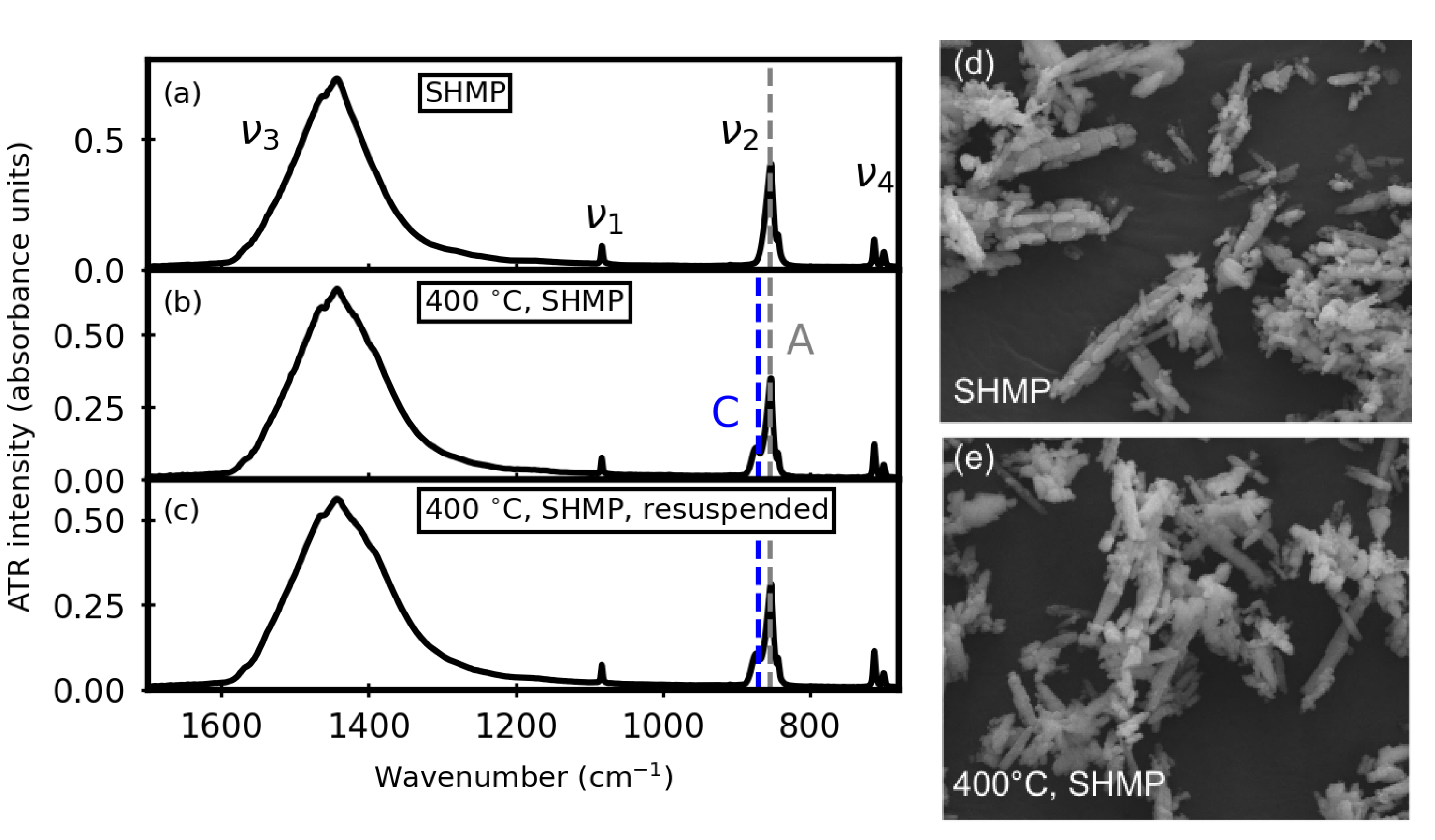
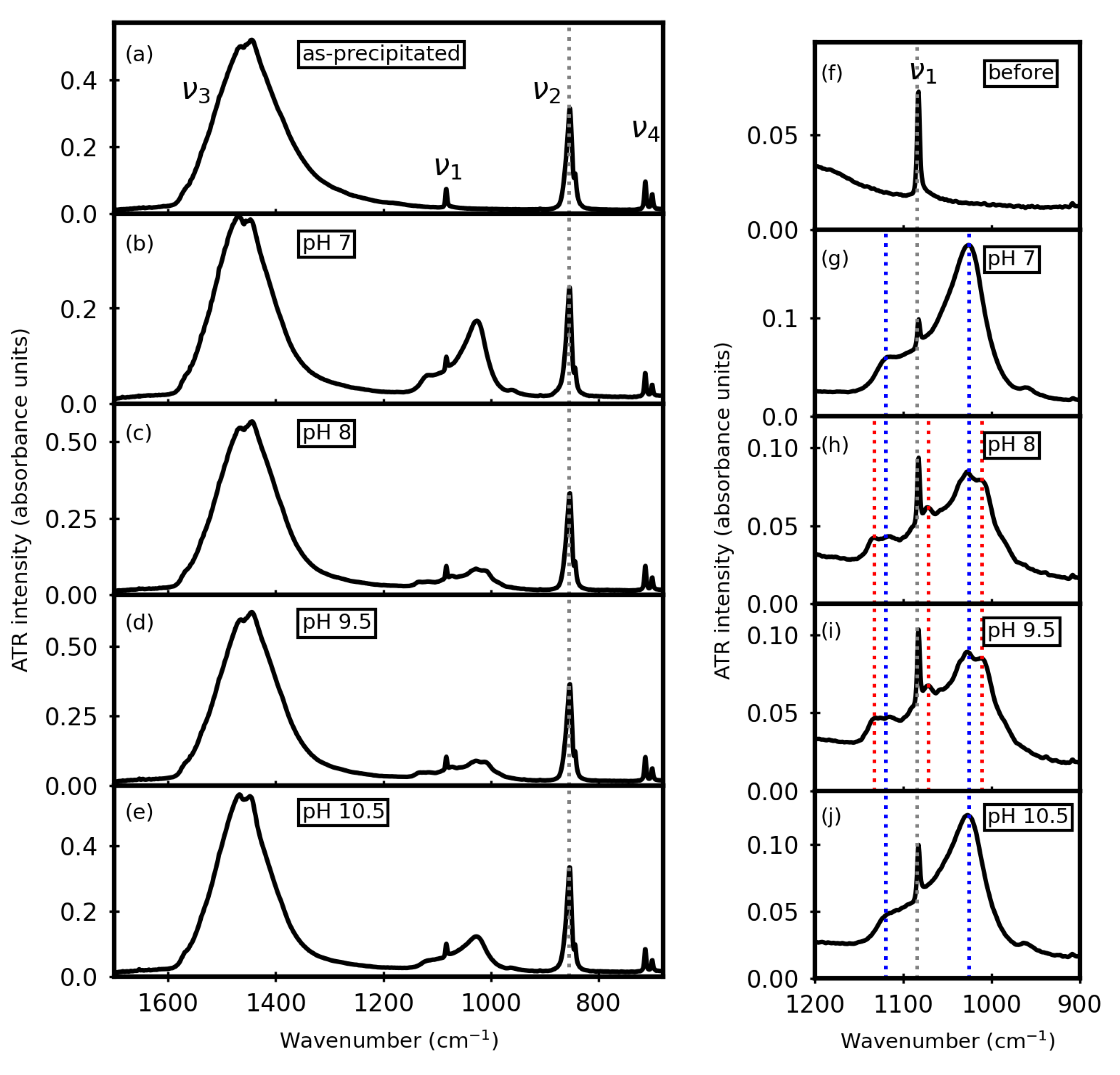
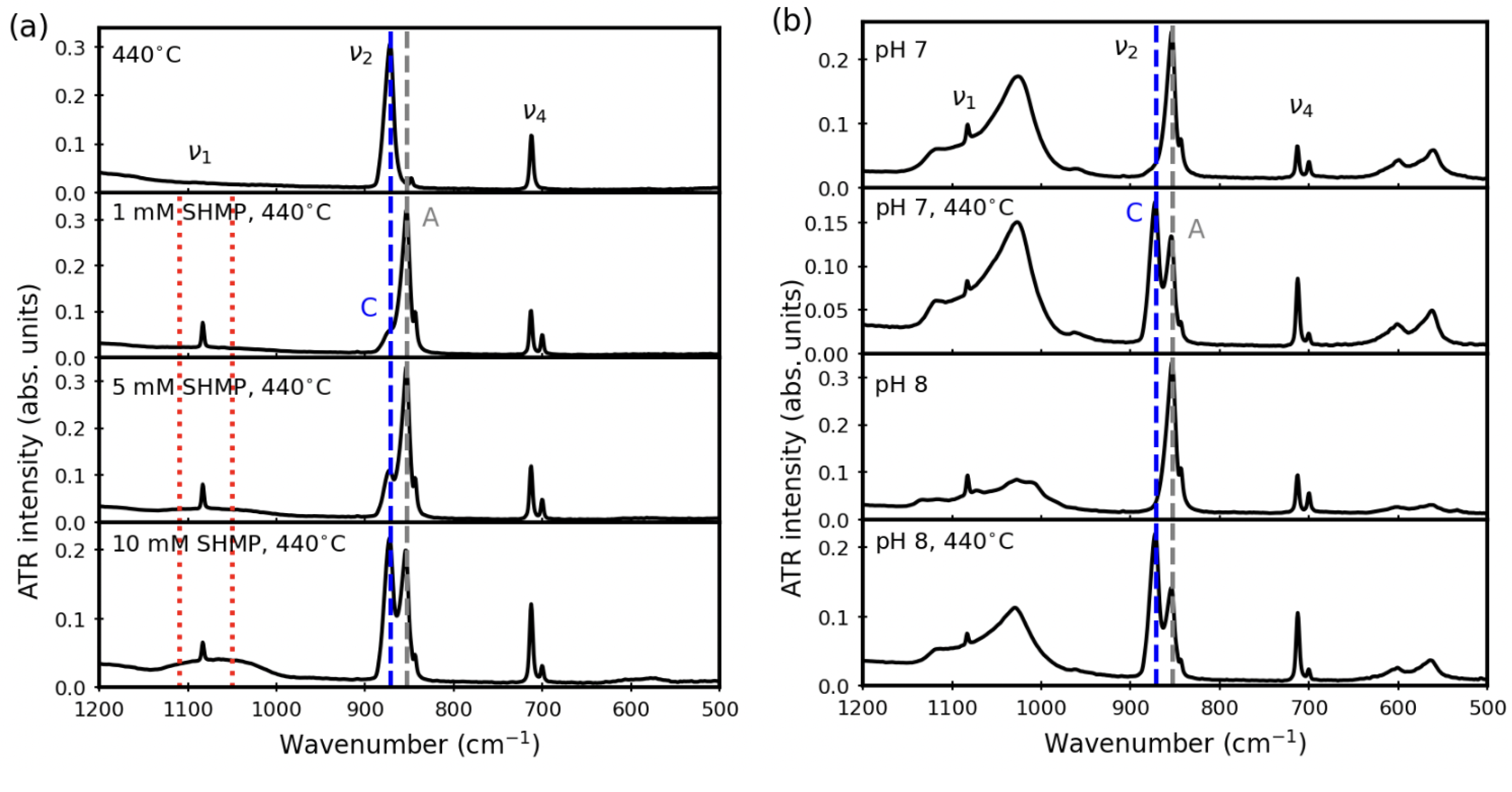
3. Results
3.1. Before Aqueous Treatments
3.2. After Aqueous Treatments
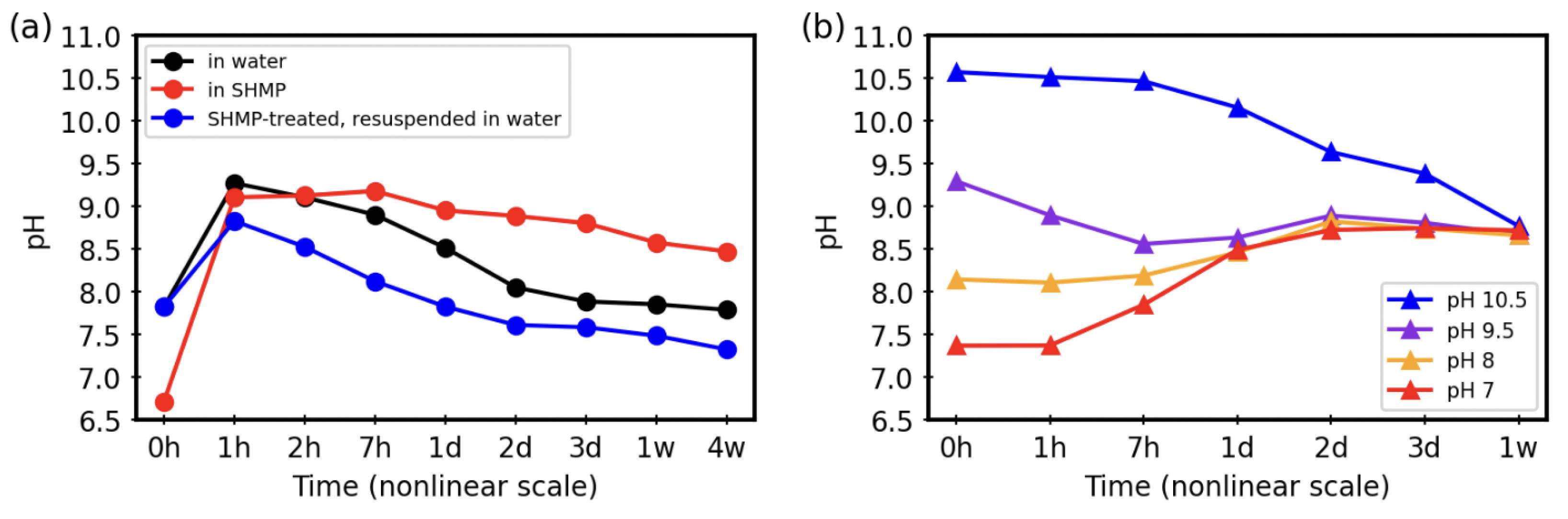
3.3. Time Trends pH during Aqueous Treatments
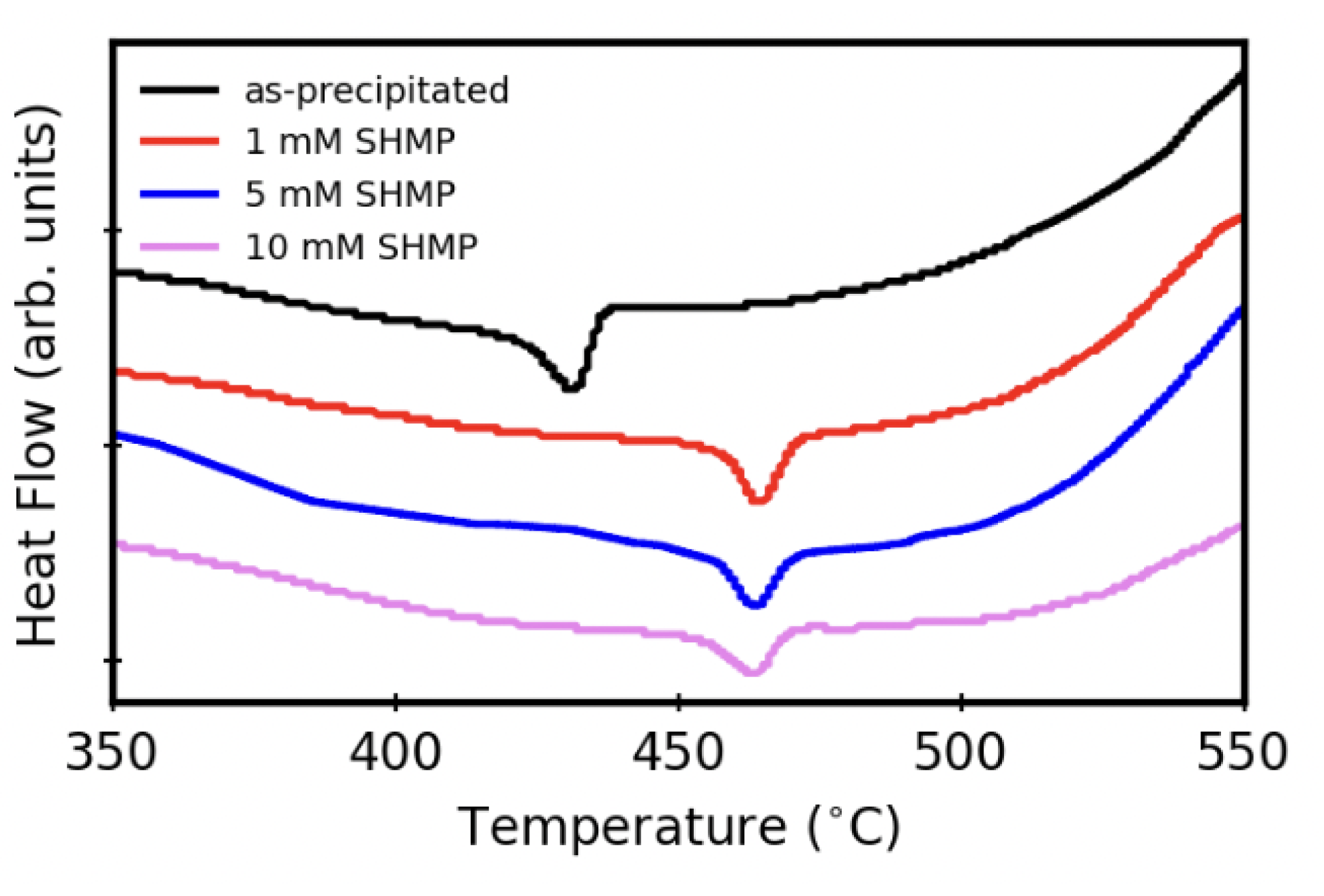
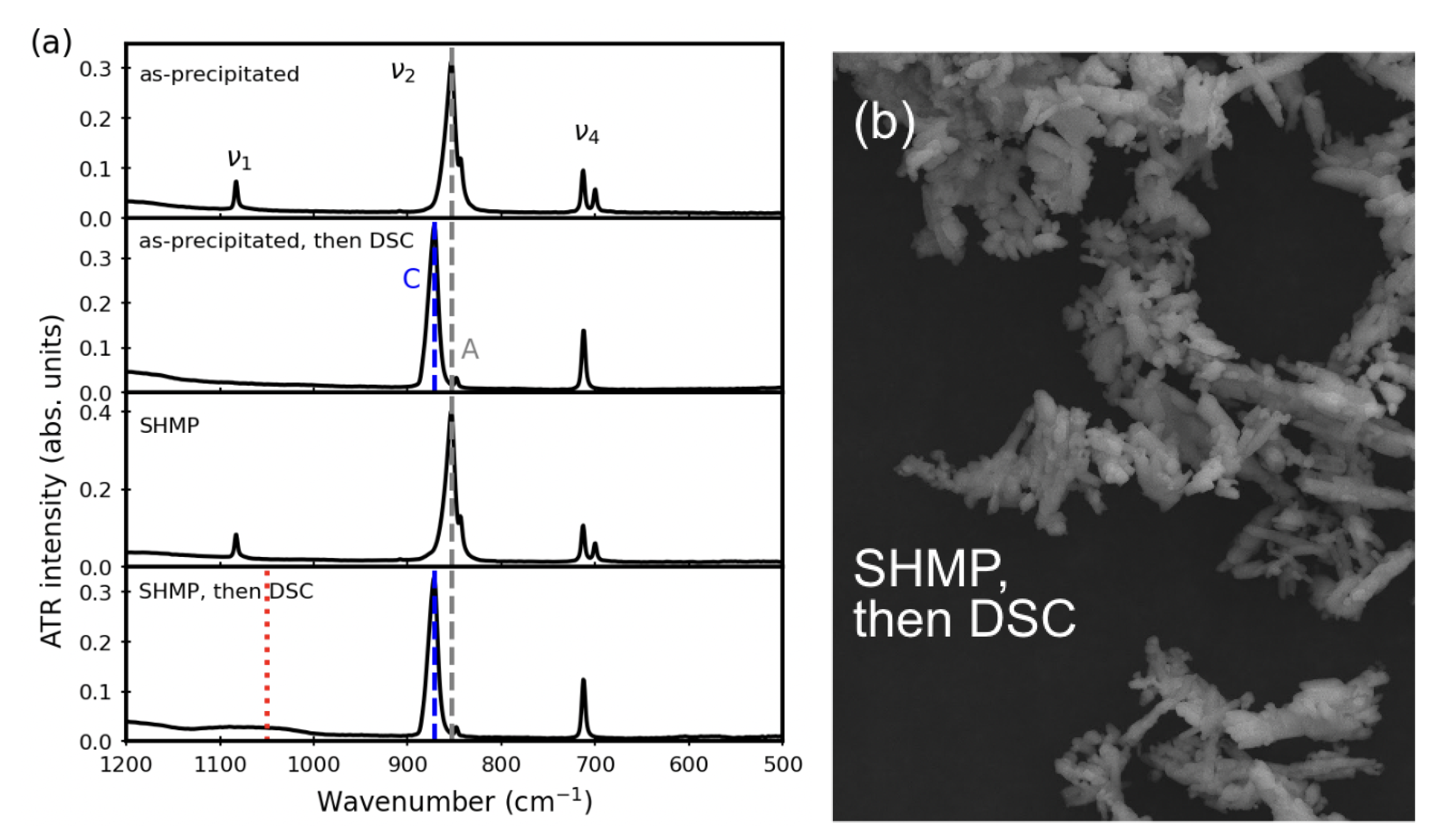
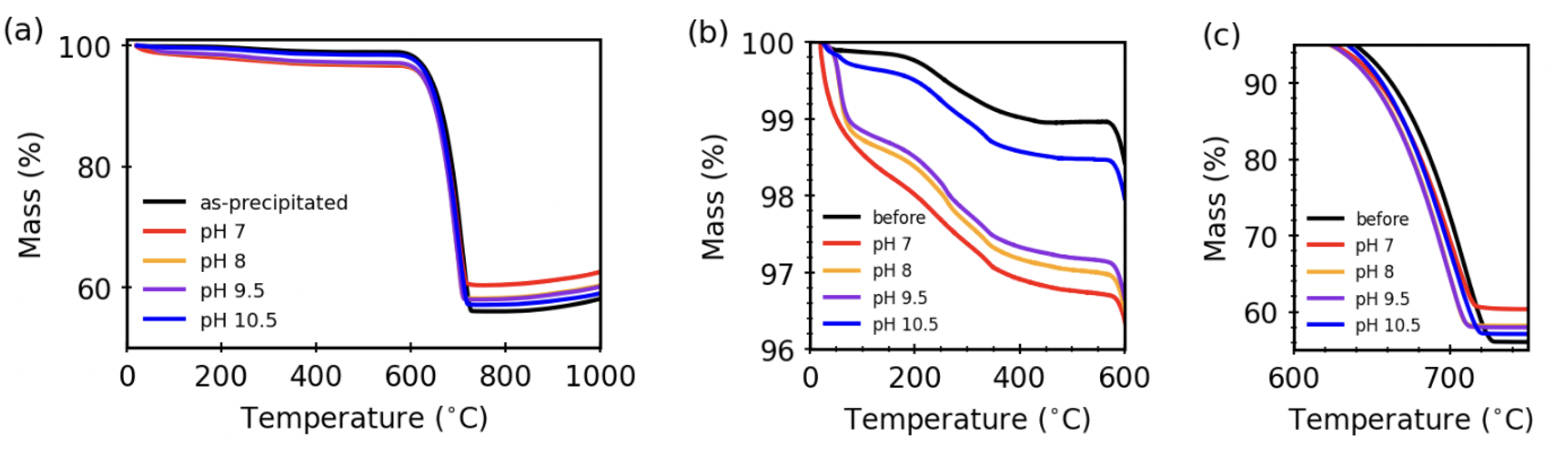
3.4. Thermal Stability after Aqueous Treatments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ramakrishna, C.; Thenepalli, T.; Ahn, J.W. A brief review of aragonite precipitated calcium carbonate (PCC) synthesis methods and its applications. Korean Chem. Eng. Res. 2017, 55, 443–455. [Google Scholar] [CrossRef]
- Nakatsuka, T.; Kawasaki, H.; Itadani, K.; Yamashita, S. Phosphate coupling agents for calcium carbonate filler. J. Appl. Polym. Sci 1982, 27, 259–269. [Google Scholar] [CrossRef]
- Chong, T.; Sheikholeslami, R. Thermodynamics and kinetics for mixed calcium carbonate and calcium sulfate precipitation. Chem. Eng. Sci. 2001, 56, 5391–5400. [Google Scholar] [CrossRef]
- Muryanto, S.; Bayuseno, A.P.; Ma’mun, H.; Usamah, M.J.P.C. Calcium carbonate scale formation in pipes: Effect of flow rates, temperature, and malic acid as additives on the mass and morphology of the scale. Procedia Chem. 2014, 9, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Toffolo, M.B.; Regev, L.; Mintz, E.; Poduska, K.M.; Shahack-Gross, R.; Berthold, C.; Miller, C.E.; Boaretto, E. Accurate radiocarbon dating of archaeological ash using pyrogenic aragonite. Radiocarbon 2017, 59, 231–249. [Google Scholar] [CrossRef]
- Xu, C.; Walsh, C.; Boaretto, E.; Poduska, K.M. Assessing the feasibility of electrophoretic separation of CaCO3 polymorphs for archaeological applications. Anal. Methods 2017, 9, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Zhang, H.; Liu, Q.; Zeng, H. Effects of chemical inhibitors on the scaling behaviors of calcite and the associated surface interaction mechanisms. J. Colloid Interface Sci. 2022, 618, 507–517. [Google Scholar] [CrossRef]
- Hasson, D.; Cornel, A. Effect of residence time on the degree of CaCO3 precipitation in the presence of an anti-scalant. Desalination 2017, 401, 64–67. [Google Scholar] [CrossRef]
- Liu, H.; Huang, H.; Wu, X.; Wu, K.; Hu, J.; Wei, J.; Yu, Q. Interactions between organic chelation agents and ions in seawater for accelerating self-healing of cracks in cement paste. J. Mater. Civ. Eng. 2021, 33, 04021036. [Google Scholar] [CrossRef]
- Combes, C.; Miao, B.; Bareille, R.; Rey, C. Preparation, physical–chemical characterisation and cytocompatibility of calcium carbonate cements. Biomaterials 2006, 27, 1945–1954. [Google Scholar] [CrossRef]
- Combes, C.; Tadier, S.; Galliard, H.; Girod-Fullana, S.; Charvillat, C.; Rey, C.; Auzély-Velty, R.; El Kissi, N. Rheological properties of calcium carbonate self-setting injectable paste. Acta Biomater. 2010, 6, 920–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, N.; Wang, Y.; Xu, X.; Liu, C.; Qian, J.; Feng, G. Mechanisms and applications of the synthesized fusiform aragonite for the removal of high concentration of phosphate. Water Air Soil Pollut. 2016, 227, 1–11. [Google Scholar] [CrossRef]
- Ma, M.; Wang, Y.; Cao, X.; Lu, W.; Guo, Y. Temperature and supersaturation as key parameters controlling the spontaneous precipitation of calcium carbonate with distinct physicochemical properties from pure aqueous solutions. Cryst. Growth Des. 2019, 19, 6972–6988. [Google Scholar] [CrossRef]
- Pismenskaya, N.; Laktionov, E.; Nikonenko, V.; El Attar, A.; Auclair, B.; Pourcelly, G. Dependence of composition of anion-exchange membranes and their electrical conductivity on concentration of sodium salts of carbonic and phosphoric acids. J. Membr. Sci. 2001, 181, 185–197. [Google Scholar] [CrossRef]
- Kimmel Center for Archaeological Science (Weizmann Institute of Science). Available online: https://centers.weizmann.ac.il/kimmel-arch/infrared-spectra-library (accessed on 18 September 2022).
- White, W.B. The carbonate minerals. In The Infrared Spectra of Minerals; Mineralogical Society of Great Britain and Ireland: London, UK, 1974. [Google Scholar] [CrossRef]
- Valenzano, L.; Noel, Y.; Orlando, R.; Zicovich-Wilson, C.; Ferrero, M.; Dovesi, R. Ab initio vibrational spectra and dielectric properties of carbonates: Magnesite, calcite and dolomite. Theor. Chem. Acc. 2007, 117, 991–1000. [Google Scholar] [CrossRef]
- Derkani, M.H.; Fedorov, A.J.F.M.; Abdallah, W.; Sauerer, B.; Anderson, J.; Zhang, Z.J. Mechanisms of Surface Charge Modification of Carbonates in Aqueous Electrolyte Solutions. Colloids Interfaces 2019, 30, 62. [Google Scholar] [CrossRef] [Green Version]
- Perić, J.; Krstulović, R.; Ferić, T.; Vučak, M. The examination of the phase transformation of aragonite into calcite by means of DSC analysis. Thermochim. Acta 1992, 207, 245–254. [Google Scholar] [CrossRef]
- Koga, N.; Kasahara, D.; Kimura, T. Aragonite crystal growth and solid-state aragonite–calcite transformation: A physico–geometrical relationship via thermal dehydration of included water. Cryst. Growth Des. 2013, 13, 2238–2246. [Google Scholar] [CrossRef]
- Kezuka, Y.; Kawai, K.; Eguchi, K.; Tajika, M. Fabrication of Single-Crystalline Calcite Needle-like Particles Using the Aragonite–Calcite Phase Transition. Minerals 2017, 7, 133. [Google Scholar] [CrossRef]
- Hanein, T.; Simoni, M.; Woo, C.L.; Provis, J.L.; Kinoshita, H. Decarbonisation of calcium carbonate at atmospheric temperatures and pressures, with simultaneous CO2 capture, through production of sodium carbonate. Energy Environ. Sci. 2021, 14, 6595–6604. [Google Scholar] [CrossRef]
- Po, H.N.; Senozan, N.M. The Henderson-Hasselbalch Equation: Its History and Limitations. J. Chem. Educ. 2001, 78, 1499. [Google Scholar] [CrossRef]
- Gashti, M.P.; Bourquin, M.; Stir, M.; Hulliger, J. Glutamic acid inducing kidney stone biomimicry by a brushite/gelatin composite. J. Mater. Chem. B 2013, 1, 1501–1508. [Google Scholar] [CrossRef] [PubMed]
- Liao, C.J.; Lin, F.H.; Chen, K.S.; Sun, J.S. Thermal decomposition and reconstitution of hydroxyapatite in air atmosphere. Biomaterials 1999, 20, 1807–1813. [Google Scholar] [CrossRef] [PubMed]
- Dosen, A.; Giese, R.F. Thermal decomposition of brushite, CaHPO4·2H2O to monetite CaHPO4 and the formation of an amorphous phase. Am. Mineral. 2011, 96, 368–373. [Google Scholar] [CrossRef]
- Sø, H.U.; Postma, D.; Jakobsen, R.; Larsen, F. Sorption of phosphate onto calcite; results from batch experiments and surface complexation modeling. Geochim. Cosmochim. Acta 2011, 75, 2911–2923. [Google Scholar] [CrossRef]
- Xu, N.; Yin, H.; Chen, Z.; Liu, S.; Chen, M.; Zhang, J. Mechanisms of phosphate retention by calcite: Effects of magnesium and pH. J. Soils Sediments 2014, 14, 495–503. [Google Scholar] [CrossRef]
- Xu, N.; Chen, M.; Zhou, K.; Wang, Y.; Yin, H.; Chen, Z. Retention of phosphorus on calcite and dolomite: Speciation and modeling. RSC Adv. 2014, 4, 35205–35214. [Google Scholar] [CrossRef]
- Guo, Y.; Zhou, Y.; Jia, D.; Tang, H. Fabrication and characterization of hydroxycarbonate apatite with mesoporous structure. Microporous Mesoporous Mater. 2009, 118, 480–488. [Google Scholar] [CrossRef]
- Wang, Y.; Moo, Y.X.; Chen, C.; Gunawan, P.; Xu, R. Fast precipitation of uniform CaCO3 nanospheres and their transformation to hollow hydroxyapatite nanospheres. J. Colloid Interface Sci. 2010, 352, 393–400. [Google Scholar] [CrossRef]
- de Morais, S.C.; de Lima, D.F.; Ferreira, T.M.; Domingos, J.B.; de Souza, M.A.F.; Castro, B.B.; de Carvalho Balaban, R. Effect of pH on the efficiency of sodium hexametaphosphate as calcium carbonate scale inhibitor at high temperature and high pressure. Desalination 2020, 491, 114548. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample/Mass (%) | Low T Loss | High T Loss | Assumed CaO | Excess Mass |
|---|---|---|---|---|
| (20–575 C) | (575–750 C) | Mass Remaining | Remaining | |
| ideal | 0.0% | 43.0% | 57.0% | 0.0% |
| as-precipitated | 1.1% | 40.9% | 54.2% | 3.8% |
| pH 10.5 | 1.6% | 39.4% | 52.2% | 6.8% |
| pH 9.5 | 2.9% | 37.0% | 49.0% | 11.1 % |
| pH 8 | 3.0% | 36.7% | 48.6% | 11.7 % |
| pH 7 | 3.3% | 34.2% | 45.3% | 17.2 % |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, B.; Poduska, K.M. Comparing Polyphosphate and Orthophosphate Treatments of Solution-Precipitated Aragonite Powders. Solids 2022, 3, 684-696. https://doi.org/10.3390/solids3040042
Gao B, Poduska KM. Comparing Polyphosphate and Orthophosphate Treatments of Solution-Precipitated Aragonite Powders. Solids. 2022; 3(4):684-696. https://doi.org/10.3390/solids3040042
Chicago/Turabian StyleGao, Boyang, and Kristin M. Poduska. 2022. "Comparing Polyphosphate and Orthophosphate Treatments of Solution-Precipitated Aragonite Powders" Solids 3, no. 4: 684-696. https://doi.org/10.3390/solids3040042