

Current Technical Approaches to Study RNA–Protein Interactions in mRNAs and Long Non-Coding RNAs
Institute of Biochemistry, University of Münster, Corrensstr. 36, 48149 Münster, Germany
BioChem 2023, 3(1), 1-14; https://doi.org/10.3390/biochem3010001
Submission received: 25 October 2022
/
Revised: 19 December 2022
/
Accepted: 26 December 2022
/
Published: 30 December 2022
(This article belongs to the Special Issue RNA and Protein Dynamics: Latest Advances and Prospects)
Abstract
:It is commonly understood that RNA-binding proteins crucially determine the fate of their target RNAs. Vice versa, RNAs are becoming increasingly recognized for their functions in protein regulation and the dynamics of RNA-protein complexes. Long non-coding RNAs are emerging as potent regulators of proteins that exert unknown RNA-binding properties and moonlighting functions. A vast array of RNA- and protein-centric techniques have been developed for the identification of protein and RNA targets, respectively, including unbiased protein mass spectrometry and next-generation RNA sequencing as readout. Determining true physiological RNA and protein targets is challenging as RNA–protein interaction is highly dynamic, tissue- and cell-type-specific, and changes with the environment. Here I review current techniques for the analysis of RNA–protein interactions in living cells and in vitro. RNA-centric techniques are presented on the basis of cross-linking or the use of alternative approaches. Protein-centric approaches are discussed in combination with high-throughput sequencing. Finally, the impact of mutations in RNA–protein complexes on human disease is highlighted.
1. Introduction
RNA-binding proteins (RBPs) dictate the life and fate of every RNA, with functions from transcription factors over single-stranded RNA binding proteins to assembly in ribonucleoprotein complexes (RNPs) such as the spliceosome. Vice versa, RNAs are emerging to regulate the functions of many cognate binding proteins. Long non-coding RNAs (lncRNAs) have been demonstrated to act as scaffolds in the organization of RNP assembly, recruit transcription factors or chromatin modifying complexes, and small nuclear RNAs (snRNAs) are functional components of the spliceosome [1,2]. lncRNAs may have indirect functions by weakly associating with RNAs, chromatin or RBPs to contribute to condensate formation to support gene regulation. Thus, RBPs and RNAs mutually influence each other’s fate. Canonical RBPs exhibit common RNA-binding domains such as the hnRNP K homology (KH) domain, RNA recognition motif (RRM), Zinc finger and DEAD box helicase domains [3,4,5], which consist typically of α-helices often complemented by β-sheets, but increasing evidence reveals non-classical RBPs with intrinsically disordered domains that can loosely interact with RNA, proteins, or chromatin [6]. Besides their role in the formation of membrane-less organelles such as stress granules by liquid–liquid phase separation [7], the interactions to RNA through small polar amino acids in conserved RGG or YGG repeats are less specific and are also believed responsible to induce moonlighting actions of metabolic enzymes such as GAPDH [8].
The analysis of RNA–protein interactions is crucial to understand the functions of RNP complexes in cellular metabolism and disease. Especially for lcnRNAs, which are marked by low expression and instability, the identification of RNA-binding proteins requires optimized detection techniques. Here I review current and novel techniques to analyse RNA–protein interactions in cells and in vitro. They are divided into protein- and RNA-centric approaches. For both groups, I will give the most important and established protocols that involve covalent cross-linking of RNA to protein binders or employ alternative ways independent from cross-linking. In the section of RNA-centric approaches, examples of RBP identification are highlighted especially for lncRNA targets. In the protein-centric section, recent developments and state-of-the-art techniques in combination with high-throughput sequencing are highlighted. Finally, a discussion of the importance of RBPs and lncRNAs in human disease is given and is wrapped-up by a presentation of therapeutic tools to intervene in RNA–protein interactions.
2. Techniques to Study RNA-Protein Interactions
Immunoprecipitation (IP) techniques to study RNA–protein interactions can be divided into RNA-centric and protein-centric approaches, in which RNA or protein is used as a bait, respectively (Table 1). The RNA-centric approach is used to discover cognate and novel RBPs bound to a certain RNA of interest, while the protein-centric method identifies RNAs and binding sites that are targeted by a known RBP.
3. RNA-Centric Approaches
3.1. In Vitro RNA-Centric Approaches
RNA-centric approaches involve the isolation of an RNA of interest by IP and identification of the bound proteins by immunoblotting or mass spectrometry. The in vitro approach is best suited for characterising known interactions, for instance by mutating those nucleotides responsible for protein binding, or validating RBP binding to the target RNA under varying cellular conditions such as growth, oxidative stress, protein depletion, or drug treatment.
In vitro pulldown approaches usually focus on an in vitro-transcribed (IVT) RNA of interest that can be immunoprecipitated in several ways, through prior modification of the RNA with affinity tags or 3′ extensions, or by hybridisation of the RNA to target-specific antisense probes (Figure 1a). In addition, poly(dT) oligonucleotide capture has been applied to enrich poly(A) mRNA-associated RBPs [9,10,11]. As affinity handle, an IVT RNA can be 5′-tagged with biotin [12] or an aptamer sequence [13]. For example, a streptavidin-binding aptamer, S1, was inserted into an mRNA 3′ UTR to pull down ARE-binding proteins that bound to the AU-rich element (ARE) of the reporter [14]. An alternative to the four S1 aptamer hairpins are the bacteriophage-derived PP7 and MS2 binding elements. Up to 24 copies of hairpin loop domains are inserted into reporter mRNAs or endogenous loci as a 3′ extension for the recognition and immunoprecipitation by the PP7 [15] and MS2 [16] coat proteins, respectively.
After incubation with cell lysate, the proteins that stay bound to the immobilized RNA are eluted by RNase digestion or boiling in SDS buffer for further analysis by sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) or for mass spectrometry (MS) [12]. For desthiobiotin-modified RNAs, gentle conditions of 12.5 mM biotin are sufficient to elute the RNA-protein complexes [17].
To identify RNA-interacting proteins in MS, proteins are often labelled with a mass tag to derive quantification. Labelling is achieved by metabolic labelling in stable isotope-labelled media (SILAC), chemical labelling of proteolysed peptides, or spike-in of peptide standards. Label-free direct quantification of proteins is used upon comparison to a control condition [18].
3.2. In Vivo RNA-Centric Approaches
3.2.1. Techniques Involving Cross-Linking
In vivo approaches, on the other hand, capture an endogenous RBP usually after cross-linking it to its cognate RNA in the cell. Cells are treated with ultraviolet (UV) light or formaldehyde before purifying the RNA under denaturing conditions to retain the covalent RNA–protein complexes and dispose of weakly associated proteins. In this step, antisense oligonucleotide probes are used to hybridize to and pull down the RNA of interest. A tandem purification approach employed target-specific probes after poly(A)-enrichment of mRNA [19]. Due to a cell lysis-induced RNA fragmentation step, tiled 5′-biotinylated probes were used on the long non-coding RNA Xist to retrieve 81 Xist-interacting proteins from HeLa cells, namely, Spen and hnRNPK [20]. This method, ChIRP-MS or comprehensive identification of RNA-binding proteins by mass spectrometry, also captured novel RBPs of the short snRNAs U1 and U2. RBR-ID (proteomic identification of RNA-binding regions) could map RNA-binding regions of RBPs from embryonic stem cell nuclei by efficiently cross-linking metabolically labelled 4-thiouridine (4sU)-containing transcripts to RBPs [21].
Apart from endogenous transcripts, biotin- or MS2-engineered RNAs with their complexed RBPs can be captured by co-expressing the MS2 coat protein and immobilization to beads (Figure 1b). A streptavidin-biotin pulldown strategy has also been tested from the protein side in MS2-BioTRAP. Here, the MS2 coat protein is fused to an HB tag that contains an in vivo biotinylation site and is enriched together with its MS2-tagged target mRNA. The mRNA, in turn, is complexed with the cross-linked cellular RBPs [22]. In addition to DNA or RNA probes, peptide nucleic acids (PNAs) have been used to hybridize to a target mRNAs localized in dendritic neurons [23]. UV irradiation enables the activated PNA to cross-link with proteins that directly contact the mRNA/PNA complex. mRNA-interacting proteins are then identified by MS after pulldown with an antisense biotinylated probe.
For lncRNAs, the CHART method was used to validate cognate RBPs of the lncRNAs NEAT1 and MALAT1 in human cells and of roX2 in flies by Western blot. In addition, CHART could map genome-wide binding regions of roX2 to chromatin by using desthiobiotin probes for RNA pulldown from cross-linked chromatin extracts and sequencing of the isolated DNA [17]. The lncRNAs Xist and Firre were studied by using 5′-biotinylated 90 nt-long antisense DNA oligos. Initially, this method RAP (RNA antisense purification) was developed to analyse RNA localisation to chromatin, but in combination with MS (RAP-MS) served to identify RBPs that are responsible for X-chromosome inactivation [24,25,26].
The extent, efficiency, and specificity of cross-linking depends on the choice of reagent: for RNA–protein interactions, UV light is usually superior to formaldehyde as it does not induce protein–protein bonding, captures only direct contacts (‘zero distance’), and is irreversible [27]. However, as the recovery of RNA–protein complexes is often low and requires high cell input, formaldehyde can be preferrable due to higher cross-linking efficiency [28]. Cross-linking efficiency for both reagents is also biased towards certain nucleotide sequences.
To circumvent the difficulties of quantitative RNA–protein complex recovery, the in vivo method CARIC (click-chemistry-assisted RNA interactome capture) uses UV cross-linking and click-chemistry to easily pull down and enrich metabolically labelled RNAs with their cognate protein partners (Figure 1b). The researchers dually labelled RNAs with 4sU and 5-ethynyl-uridine (5EU), which enable a photoactivated cross-linking step of bound RBPs and a selective click reaction with biotin-azide, respectively. MS proteomics identified 597 human proteins including 130 previously unknown RBPs, some without known RNA-binding motifs, that targeted lncRNAs, snRNAs, and miRNAs [29]. A similar method, termed RICK (RNA interactome using click chemistry), introduced 5EU-labeling of nascent transcripts and click-chemistry-assisted pulldown for the identification of RBPs from often-neglected non-polyadenylated transcripts [30].
Moreover, there are methods like RNAcompete and RNAMaP that quantitatively measure binding kinetics of a mutational library of a given RNA to an RNA-binding protein on a DNA-sequencing platform. These approaches decompose contributions of RNA primary and secondary structure to binding affinities and determine sequence motif preferences of RBPs [31,32].
3.2.2. Proximity Labelling Techniques
Approaches without cross-linking exploit affinity labelling to mark proteins in the vicinity of the RNA of interest, making RBPs amenable to affinity enrichment and Western blotting or MS analysis. Proximity labelling is widely applied by using engineered biotin ligases (E. coli BirA and homologs BioID2 and RABU) [33,34,35,36] or ascorbate peroxidases (APEX, APEX2) [37,38] in combination with a tagging reagent, which is most commonly biotin. Biotin ligases convert biotin to biotinoyl-5′-AMP that selectively reacts with lysine residues, while the APEX enzymes convert biotin-phenol in the presence of H2O2 to biotin-phenoxyl radicals that preferentially attack tyrosine residues. The labelling enzyme is fused to an RNA-binding protein to target endogenous RNA loci and label the surrounding proteins. APEX2 is also widely used to assess the spatial proteomic landscape of cellular compartments [37,38].
In RaPID (RNA-protein interaction detection), a fusion of the biotin ligase BirA and the RNA-binding λN peptide is co-expressed with an RNA engineered with a BoxB stem-loop (Figure 1c). In biotin-containing medium, macromolecules in a realm of 20 nm around the λN-bound BoxB-RNA will be labelled [33]. RaPID is used to discover novel RNA–protein interactions, but is limited to lysine-specific biotinylation and requires the overexpression of engineered RNAs. A similar approach uses the MS2 coat protein (MCP) system to guide BirA to an mRNA that is tagged with 24 MS2 stem-loops at the 3′ UTR [39]. Although this method, RNA BioID, could identify the dynamic proteome of endogenous β-actin mRNA, the method is equally limited to genetically engineered mRNA loci.
A recently developed method relies equally on biotin proximity labelling, but does so by exploiting the CRISPR-Cas system for a completely different mechanism. A biotin-tagged small peptide, PupE, is covalently attached to proteins surrounding the Cas-targeted RNA. This allows for affinity purification, enrichment, and MS analysis of biotin-PupE-labelled RBPs [40]. Strikingly, an RNA-targeting, nuclease-inactive dCas13a protein acts as the guide to recruit the fused PafA ligase to a defined RNA locus. CRUIS, or CRISPR-based RNA-united interacting system, requires stable cell expression of the dCas13a-PafA fusion protein transfected with single-guide RNA and Biotin-PupE plasmids. CRUIS has been shown to identify novel RBPs of the lncRNA NORAD.
Equally, the CRISPR-based method CBRPP (CRISPR-based RNA proximity proteomics) detected RBPs of NORAD and β-actin mRNA. CBRPP requires a fusion of dCas13b and the smallest biotin ligase, a BirA homolog [35], to be stably expressed in HEK293T cells, and co-expression of β-actin-targeted crRNAs for guidance [41]. Low expression levels under a Tet-On 3 G promoter and incubation with biotin for 18 h guarantees to capture true proximate RBPs, which are repeatedly labelled over time, and reduces background labelled proteins, as these do not accumulate over time.
4. Protein-Centric Approaches
Protein-centric approaches involve targeting of a protein of interest by IP and identifying the RNAs bound to it, usually by downstream RNA-sequencing (Table 1). Depending on the method, RBP binding sites can be mapped from regions spanning up to 100 nucleotides (nt) down to single-nucleotide resolution and determination of preferred binding motifs.
4.1. Techniques Involving Cross-Linking: CLIP and Related Protocols
By far the most widely used method is cross-linking and immunoprecipitation (CLIP), which exists in multiple forms with common basic steps (Figure 2a). The initial freezing of RNA–protein bonds is achieved via an in vivo UV cross-linking step, and after cell lysis and partial RNase digestion the protein of interest is immunoprecipitated with a specific antibody [42]. After separation of the protein–RNA complexes by SDS-PAGE and transfer to a membrane, the RNA is liberated from the excised complexes by proteinase K digestion. In the original protocol, the RNA was 32P-labelled, the cross-linked sites were read-through in RT-PCR, and binding regions were narrowed down by overlaying sequencing clusters [42]. For high-throughput sequencing (HITS-CLIP), preparation of cDNA libraries comprises universal steps, the order of which varies for each protocol [43]. First, for 3′ and 5′ adapter ligation phosphatase and kinase treatment are required, respectively. Reverse transcription (RT) to cDNA, PCR amplification, and massively parallel sequencing reveals RNA targets of the RBP. Of note, quantitative CLIP-seq [44] analysis is challenging as it depends on cross-linking efficiency which is different for each RNA species and sequence. The number of reads per transcript does not directly reflect the number of cross-links formed, and thus is not equivalent to the amount of protein interacting with this transcript [43]. In addition, CLIP suffers from several uncertainties: high false positive rates, limiting antibody efficiency, and high input cell numbers restrict its use to bulk mixtures of millions of cells [45]. Alternative protocols such as iCLIP, irCLIP, eCLIP, GoldCLIP, and CRAC exploit the fact that 80% of RT reads stop at the cross-link site, and use RT stops to map RBP sites at single-nucleotide resolution. The above CLIP protocols show improvements such as 5′ cDNA circularisation, infrared-dye 3′ adapters, on-bead adapter ligation, use of epitope-tagged RBPs, and replacement of gel purification by affinity purification, respectively [46,47,48,49,50].
In the alternative protocol PAR-CLIP (photoactivatable ribonucleoside-enhanced crosslinking and immunoprecipitation), metabolically labelled RNAs containing 4sU or 6-thioguanosine (6sG) are employed as cross-linking enhancers [51]. Treatment of cells with 4sU labels 1 in 40 uridines, and the photoactivatable nucleoside enhances the UV cross-linking efficiency at 365 nm in comparison to conventional CLIP at 265 nm. Since 4sU causes reverse transcriptase to incorporate G in the opposite cDNA strand, sequencing of T-to-C mutations allows to map RBP binding motifs at cross-linking sites. In this way, the RNA recognition element (RRE) of Pumilio 2 was mapped, and thousands of mRNA targets for AGO/miRNA complexes were identified.
RIP-seq is a similar protein-centric approach that leaves out the cross-linking step. Nuclear or cellular lysate containing native RNA–protein complexes is incubated with an antibody against the protein of interest, e.g., the polycomb repressor complex protein PRC2. Immunoprecipitation with beads followed by RT and sequencing of the extracted RNA identified thousands of PRC2-interacting RNAs [52].
Recently, a novel method, GECX-RNA or genetically encoded chemical cross-linking of proteins with target RNA, has applied the incorporation of unnatural amino acids into the Escherichia coli Hfq chaperone to enable cross-linking with nucleic acids directly at the active site. After antibody-assisted pulldown of the protein, reverse transcription and cDNA sequencing can detect the binding sites of the RBP site-specifically and in an accurate way [53].
4.2. Techniques without Cross-Linking: TRIBE and STAMP
An alternative to CLIP is to use the RBP of interest fused to the nucleotide deaminases ADAR or APOBEC1. RNAs bound by the RBP are deaminated close to the binding sites, and sequencing reveals the mutated transcripts as RNA targets [54,55]. One can also fuse a poly(U)-polymerase to the RBP, which adds poly(U)-tails to the 3′ end of targeted RNAs and enables identification via RNA sequencing [56].
TRIBE (targets of RNA-binding protein identified by editing) uses stable expression of the deaminase ADAR fused to an RBP to direct A-to-I editing near RBP binding sites, and after mRNA isolation and cDNA sequencing identifies RBP-specific mRNA targets (Figure 2b). The method was applied to Drosophila S2 cells and to transient expression in fly brain neurons to identify cell-type-specific targets of Hrp48 and two other RBPs [54]. However, drawbacks of TRIBE are false negatives due to its low editing frequency of one edit/transcript and ADAR bias towards bulged As next to double-stranded (ds) RNA regions. To overcome the low editing frequency of TRIBE, the authors optimized the deaminase efficiency of ADAR through a E488Q mutation, which led to hyper-edited sites of >1 edit per read, and increased the numbers of RBP binding sites and of target mRNAs identified [57]. However, it still suffers the structure- and sequencing-bias of ADAR which prefers bulged As in dsRNA regions and nearest neighbour As.
STAMP (surveying targets by APOBEC-mediated profiling) was developed to overcome limitations of low editing by ADAR. Overexpression of an RBP-APOBEC1 fusion leads to binding site-adjacent C-to-U editing by the deaminase. Clusters of 10–100 A mutations in RNA-sequencing reads are enriched relative to the APOBEC-only control (Figure 2c) [55]. STAMP was expanded to single-cell RNA-sequencing using the 10x Genomics bead capture system and to long-read PacBio sequencing to distinguish isoform-specific edits. STAMP was demonstrated to detect ribosome-associated targets correlating to high translation and to extract RBFOX2-binding site motifs de novo from edited regions even from single cell data. However, the edit enrichment in mRNA 3′ UTRs could indicate false positives that are generated by the high levels of overexpression.
The method TRIBE-STAMP combines the ideas of TRIBE and STAMP to simultaneously analyse the binding of two RBPs in a cell and elucidate sequential binding events in time on a single mRNA target [58]. The authors applied the mutational editing by ADAR and APOBEC1 to N6-methyladenosine (m6A)-harbouring transcripts that are bound by the m6A reader proteins YTH-DF1, -DF2, and -DF3. Each combination of tagged DF pairs was co-expressed in HEK293T cells, e.g., DF1-APOBEC and DF2-ADAR. Then, C-to-U- and A-to-I-induced mutations were mapped by cDNA-sequencing, and co-editing events > 1 were assessed for individual reads (Figure 2d). This revealed that 40% of the mRNA targets overlapping for DF1, DF2, and DF3 exhibited co-editing, indicating that the DF paralogs bind and modify the same transcript sequentially in time. Excitingly, not only does it validate the redundancy of DF proteins [59], but it implies that DF binding to m6A sites does not immediately trigger mRNA degradation. Rather, one editing event, i.e., m6A binding of a first DF, e.g., DF1, increased the likelihood of a second editing event on the same molecule and m6A site by a second DF, e.g., DF3 [58].
A different method to identify RBP-targeted transcripts was developed based on the covalent tagging with uridines at the 3′ end of RNAs [56]. A fusion chimera of the yeast PUF3 protein and the polyU-polymerase PUP, obtained via genome engineering in Saccharomyces cerevisiae, successfully tagged PUF3 targets in vivo and allowed identification of >400 PUF3-regulated mRNAs for mitochondrial regulation. Interestingly, the method can also reliably class transcripts by their PUF3-binding affinities, as the number of nucleotides added (1–10 Us) by PUP is directly correlated to the time that the RBP stays bound to the RNA: the longer the U-tail, the more productive the RBP binding event has been in vivo.
Recently, a novel method called RT&Tag has used reverse transcription and transposase-mediated tagmentation of RNA/cDNA hybrids on immunoprecipitated nuclear extracts to map RNA–protein interactions as well as to detect chromatin-associated transcripts and RNA modifications [60]. The authors could capture the non-coding RNA roX2 from flies together with its dosage compensation complex and transcripts associated with chromatin silencing.
Other established protocols like APEX-seq have exploited proximity biotinylation of RNAs in the vicinity of a specific RBP in combination with sequencing [61]. In the presence of biotin and H2O2, the ascorbate peroxidase APEX2 labels any transcript proximal to its fusion partner eIF4A to reveal eIF4A-specific RNA interactions [62]. When combined with metabolic 4sU-labelling of nascent RNAs, the efficiency of biotin-labelling and enrichment of biotinylated RNAs was improved even further when applied to study the subcellular spatial transcriptome [63].
5. Discussion
Databases listing RNA–protein interactions are helpful tools to obtain information on a given target, expression pattern, RNP complex composition, binding sites, affinities, and other parameters. The most recent releases are listed: POSTAR3 is a comprehensive database comprising a large collection of RBPs and binding sites from CLIP-seq data as well as RNA structure-seq data [64]. RNAInter contains RNA interaction networks along with RNA structure and expression [65], RBP2GO lists RBPs of 13 species with interactions and functions [66], RPpocket provides RNA–protein complex interactions and an analysis of RNA-binding pocket topology [67]. Although less actual, ProNIT is useful for experimentally determined thermodynamic parameters between proteins and nucleic acids [68].
Multiple RBP mutations are associated with human disease and underline the necessity to understand RNA–protein interactions. Mutations can lead to changes in splicing, RNA binding, protein interactions, catalysis, or localization [69]. For instance, a mutation in the spinal muscular atrophy (SMA)-associated SMN1 gene leads to silencing of a splicing enhancer, exon skipping, and production of a non-functional protein [70]. Loss-of-function of a specific KH domain is associated with fragile X syndrome of mental retardation [5]. Mutations in the disordered repeat domains of FUS lead to toxic accumulation of protein aggregates in fly models and provide a rational for neurodegenerative disorders like amyotrophic lateral sclerosis (ALS) [71,72]. On the other hand, mutations in the RNA part of the RNP not only directly influence the production of an mRNA-encoded protein, but decreased expression of microRNAs or increased expression of the lncRNA HOTAIR associated with cancer [73]. HOTAIR recruitment of Polycomb repressor complex for histone methylation leads to silencing of target genes and is associated with increased metastatic invasiveness [74].
Several tools to interfere with or rescue RNA–protein interactions have been validated in animal models and some are already being applied as clinical therapeutics. Small molecules can directly bind to an RBP or RNA target. For instance, antitumour drugs such as spliceostatin target two of the U2 snRNP protein components to prevent splice site recognition [69,75]. Of note, an unbiased small molecule screening approach for RNA targets recently guided lead optimization of a novel inhibitor of the lncRNA Xist that is potent to disrupt X chromosome inactivation in a mouse cell model [76]. Antisense oligonucleotides (ASOs) are commonly used to block access of RBPs to their RNA binding sites. The ASO drug nusinersen is injected quarterly to patients suffering from SMA: in mice, the ASO-masking of the splicing silencer site in the SMN1 pre-mRNA rescued splicing and production of the functional SMN1 protein [77]. Other types of disrupting RNA–protein interactions for therapeutic intervention employ RNA interference (RNAi), which is used to trigger decay or inhibit translation of a specific mRNA target by hybridization to its 3′ UTR [73,78], and aptamers, synthetically evolved RNA sequences that specifically bind to a lncRNA or an RBP [79].
Future research will certainly involve the CRISPR/Cas system to exploit the endoribonuclease activity of Cas13 to specifically target RNAs in cells. More excitingly, the RNA-recruiting function of dCas13 variants could be applied to site-specifically revert disease-relevant mutations. Since many methods rely on cross-linking but suffer from low cross-linking efficiency, alternative methods involving editing enzymes or tagging enzymes will be of high interest. These methods can be further improved and applied to other targets. Still, alternative cross-linking reagents or the 4sU-labelling approach will also be of high demand to capture RNA-protein complexes in vivo. Regardless of the methodology, every newly identified target should be cross-validated with orthogonal methods. In CBRPP, for instance, dCAS13-identified RBPs of the lncRNA NORAD were validated by the protein-centric methods RIP and CLIP [41]. Overall, it will be exciting to see applications of other RNA-manipulating enzymes for novel method developments in the study of RNA–protein interactions.
Funding
This research was funded by Deutsche Forschungsgemeinschaft DFG by grants MA 8356/1–1 and MA 8356/2–1 to Johanna Mattay.
Data Availability Statement
Not applicable.
Conflicts of Interest
The author declares no conflict of interest.
References
- Cech, T.R.; Steitz, J.A. The noncoding RNA revolution-trashing old rules to forge new ones. Cell 2014, 157, 77–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geisler, S.; Coller, J. RNA in unexpected places: Long non-coding RNA functions in diverse cellular contexts. Nat. Rev. Mol. Cell Biol. 2013, 14, 699–712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linder, P.; Jankowsky, E. From unwinding to clamping—the DEAD box RNA helicase family. Nat. Rev. Mol. Cell Biol. 2011, 12, 505–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cléry, A.; Blatter, M.; Allain, F.H.-T. RNA recognition motifs: Boring? Not quite. Curr. Opin. Struct. Biol. 2008, 18, 290–298. [Google Scholar] [CrossRef] [PubMed]
- Valverde, R.; Edwards, L.; Regan, L. Structure and function of KH domains. FEBS J. 2008, 275, 2712–2726. [Google Scholar] [CrossRef]
- Hentze, M.W.; Castello, A.; Schwarzl, T.; Preiss, T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018, 19, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Protter, D.S.W.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.-H.; Curtis, J.D.; Maggi, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.-C.; van der Windt, G.J.W.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239–1251. [Google Scholar] [CrossRef] [Green Version]
- Castello, A.; Fischer, B.; Eichelbaum, K.; Horos, R.; Beckmann, B.M.; Strein, C.; Davey, N.E.; Humphreys, D.T.; Preiss, T.; Steinmetz, L.M.; et al. Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 2012, 149, 1393–1406. [Google Scholar] [CrossRef] [Green Version]
- Baltz, A.G.; Munschauer, M.; Schwanhäusser, B.; Vasile, A.; Murakawa, Y.; Schueler, M.; Youngs, N.; Penfold-Brown, D.; Drew, K.; Milek, M.; et al. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell 2012, 46, 674–690. [Google Scholar] [CrossRef]
- Mitchell, S.F.; Jain, S.; She, M.; Parker, R. Global analysis of yeast mRNPs. Nat. Struct. Mol. Biol. 2013, 20, 127–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Cho, S.; Moon, H.; Loh, T.J.; Jang, H.N.; Shen, H. Detecting RNA-protein interaction using end-labeled biotinylated RNA oligonucleotides and immunoblotting. Methods Mol. Biol. 2016, 1421, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.Y.; Haurwitz, R.E.; Apffel, A.; Zhou, K.; Smart, B.; Wenger, C.D.; Laderman, S.; Bruhn, L.; Doudna, J.A. RNA-protein analysis using a conditional CRISPR nuclease. Proc. Natl. Acad. Sci. USA 2013, 110, 5416–5421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leppek, K.; Stoecklin, G. An optimized streptavidin-binding RNA aptamer for purification of ribonucleoprotein complexes identifies novel ARE-binding proteins. Nucleic Acids Res. 2014, 42, e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, F.; Downey, T.P.; Peabody, D.S. Translational repression and specific RNA binding by the coat protein of the Pseudomonas phage PP7. J. Biol. Chem. 2001, 276, 22507–22513. [Google Scholar] [CrossRef] [Green Version]
- Bardwell, V.J.; Wickens, M. Purification of RNA and RNA-protein complexes by an R17 coat protein affinity method. Nucleic Acids Res. 1990, 18, 6587–6594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, M.D.; Wang, C.I.; Kharchenko, P.V.; West, J.A.; Chapman, B.A.; Alekseyenko, A.A.; Borowsky, M.L.; Kuroda, M.I.; Kingston, R.E. The genomic binding sites of a noncoding RNA. Proc. Natl. Acad. Sci. USA 2011, 108, 20497–20502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bantscheff, M.; Schirle, M.; Sweetman, G.; Rick, J.; Kuster, B. Quantitative mass spectrometry in proteomics: A critical review. Anal. Bioanal. Chem. 2007, 389, 1017–1031. [Google Scholar] [CrossRef] [Green Version]
- Matia-González, A.M.; Iadevaia, V.; Gerber, A.P. A versatile tandem RNA isolation procedure to capture in vivo formed mRNA-protein complexes. Methods 2017, 118–119, 93–100. [Google Scholar] [CrossRef] [Green Version]
- Chu, C.; Zhang, Q.C.; da Rocha, S.T.; Flynn, R.A.; Bharadwaj, M.; Calabrese, J.M.; Magnuson, T.; Heard, E.; Chang, H.Y. Systematic discovery of Xist RNA binding proteins. Cell 2015, 161, 404–416. [Google Scholar] [CrossRef]
- He, C.; Sidoli, S.; Warneford-Thomson, R.; Tatomer, D.C.; Wilusz, J.E.; Garcia, B.A.; Bonasio, R. High-resolution mapping of RNA-binding regions in the nuclear proteome of embryonic stem cells. Mol. Cell 2016, 64, 416–430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsai, B.P.; Wang, X.; Huang, L.; Waterman, M.L. Quantitative profiling of in vivo-assembled RNA-protein complexes using a novel integrated proteomic approach. Mol. Cell. Proteom. 2011, 10, M110.007385. [Google Scholar] [CrossRef] [Green Version]
- Zielinski, J.; Kilk, K.; Peritz, T.; Kannanayakal, T.J.; Miyashiro, K.Y.; Eiríksdóttir, E.; Jochems, J.; Langel, U.; Eberwine, J. In vivo identification of ribonucleoprotein-RNA interactions. Proc. Natl. Acad. Sci. USA 2006, 103, 1557–1562. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McHugh, C.A.; Chen, C.-K.; Chow, A.; Surka, C.F.; Tran, C.; McDonel, P.; Pandya-Jones, A.; Blanco, M.; Burghard, C.; Moradian, A.; et al. The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature 2015, 521, 232–236. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Pandya-Jones, A.; McDonel, P.; Shishkin, A.A.; Sirokman, K.; Surka, C.F.; Kadri, S.; Xing, J.; Goren, A.; Lander, E.S.; et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science 2013, 341, 1237973. [Google Scholar] [CrossRef] [Green Version]
- Hacisuleyman, E.; Goff, L.A.; Trapnell, C.; Williams, A.; Henao-Mejia, J.; Sun, L.; McClanahan, P.; Hendrickson, D.G.; Sauvageau, M.; Kelley, D.R.; et al. Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 2014, 21, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Song, J.; Yi, C. Genome-wide mapping of cellular protein-RNA interactions enabled by chemical crosslinking. Genom. Proteom. Bioinform. 2014, 12, 72–78. [Google Scholar] [CrossRef] [Green Version]
- Sutherland, B.W.; Toews, J.; Kast, J. Utility of formaldehyde cross-linking and mass spectrometry in the study of protein-protein interactions. J. Mass. Spectrom. 2008, 43, 699–715. [Google Scholar] [CrossRef]
- Huang, R.; Han, M.; Meng, L.; Chen, X. Transcriptome-wide discovery of coding and noncoding RNA-binding proteins. Proc. Natl. Acad. Sci. USA 2018, 115, E3879–E3887. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.; Guo, X.; Yin, M.; Tariq, M.; Lai, Y.; Kanwal, S.; Zhou, J.; Li, N.; Lv, Y.; Pulido-Quetglas, C.; et al. Capturing the interactome of newly transcribed RNA. Nat. Methods 2018, 15, 213–220. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Araya, C.L.; Chircus, L.M.; Layton, C.J.; Chang, H.Y.; Snyder, M.P.; Greenleaf, W.J. Quantitative analysis of RNA-protein interactions on a massively parallel array reveals biophysical and evolutionary landscapes. Nat. Biotechnol. 2014, 32, 562–568. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.; Kazan, H.; Chan, E.T.; Peña Castillo, L.; Chaudhry, S.; Talukder, S.; Blencowe, B.J.; Morris, Q.; Hughes, T.R. Rapid and systematic analysis of the RNA recognition specificities of RNA-binding proteins. Nat. Biotechnol. 2009, 27, 667–670. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, M.; Majzoub, K.; Rao, D.S.; Neela, P.H.; Zarnegar, B.J.; Mondal, S.; Roth, J.G.; Gai, H.; Kovalski, J.R.; Siprashvili, Z.; et al. RNA-protein interaction detection in living cells. Nat. Methods 2018, 15, 207–212. [Google Scholar] [CrossRef] [Green Version]
- Roux, K.J.; Kim, D.I.; Raida, M.; Burke, B. A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J. Cell Biol. 2012, 196, 801–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, D.I.; Jensen, S.C.; Noble, K.A.; Kc, B.; Roux, K.H.; Motamedchaboki, K.; Roux, K.J. An improved smaller biotin ligase for BioID proximity labeling. Mol. Biol. Cell 2016, 27, 1188–1196. [Google Scholar] [CrossRef]
- Branon, T.C.; Bosch, J.A.; Sanchez, A.D.; Udeshi, N.D.; Svinkina, T.; Carr, S.A.; Feldman, J.L.; Perrimon, N.; Ting, A.Y. Efficient proximity labeling in living cells and organisms with TurboID. Nat. Biotechnol. 2018, 36, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Rhee, H.-W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef] [Green Version]
- Lam, S.S.; Martell, J.D.; Kamer, K.J.; Deerinck, T.J.; Ellisman, M.H.; Mootha, V.K.; Ting, A.Y. Directed evolution of APEX2 for electron microscopy and proximity labeling. Nat. Methods 2015, 12, 51–54. [Google Scholar] [CrossRef]
- Mukherjee, J.; Hermesh, O.; Eliscovich, C.; Nalpas, N.; Franz-Wachtel, M.; Maček, B.; Jansen, R.-P. β-Actin mRNA interactome mapping by proximity biotinylation. Proc. Natl. Acad. Sci. USA 2019, 116, 12863–12872. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Sun, W.; Shi, T.; Lu, P.; Zhuang, M.; Liu, J.-L. Capturing RNA-protein interaction via CRUIS. Nucleic Acids Res. 2020, 48, e52. [Google Scholar] [CrossRef]
- Li, Y.; Liu, S.; Cao, L.; Luo, Y.; Du, H.; Li, S.; Zhang, Z.; Guo, X.; Tian, W.; Wong, C.C.; et al. CBRPP: A new RNA-centric method to study RNA-protein interactions. RNA Biol. 2021, 18, 1608–1621. [Google Scholar] [CrossRef] [PubMed]
- Ule, J.; Jensen, K.B.; Ruggiu, M.; Mele, A.; Ule, A.; Darnell, R.B. CLIP identifies Nova-regulated RNA networks in the brain. Science 2003, 302, 1212–1215. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, M.; Porter, D.F.; Khavari, P.A. Methods to study RNA-protein interactions. Nat. Methods 2019, 16, 225–234. [Google Scholar] [CrossRef]
- Licatalosi, D.D.; Mele, A.; Fak, J.J.; Ule, J.; Kayikci, M.; Chi, S.W.; Clark, T.A.; Schweitzer, A.C.; Blume, J.E.; Wang, X.; et al. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature 2008, 456, 464–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darnell, R.B. HITS-CLIP: Panoramic views of protein-RNA regulation in living cells. Wiley Interdiscip. Rev. RNA 2010, 1, 266–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- König, J.; Zarnack, K.; Rot, G.; Curk, T.; Kayikci, M.; Zupan, B.; Turner, D.J.; Luscombe, N.M.; Ule, J. iCLIP reveals the function of hnRNP particles in splicing at individual nucleotide resolution. Nat. Struct. Mol. Biol. 2010, 17, 909–915. [Google Scholar] [CrossRef] [Green Version]
- Zarnegar, B.J.; Flynn, R.A.; Shen, Y.; Do, B.T.; Chang, H.Y.; Khavari, P.A. irCLIP platform for efficient characterization of protein-RNA interactions. Nat. Methods 2016, 13, 489–492. [Google Scholar] [CrossRef]
- van Nostrand, E.L.; Pratt, G.A.; Shishkin, A.A.; Gelboin-Burkhart, C.; Fang, M.Y.; Sundararaman, B.; Blue, S.M.; Nguyen, T.B.; Surka, C.F.; Elkins, K.; et al. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat. Methods 2016, 13, 508–514. [Google Scholar] [CrossRef]
- Gu, J.; Wang, M.; Yang, Y.; Qiu, D.; Zhang, Y.; Ma, J.; Zhou, Y.; Hannon, G.J.; Yu, Y. GoldCLIP: Gel-omitted ligation-dependent CLIP. Genom. Proteom. Bioinform. 2018, 16, 136–143. [Google Scholar] [CrossRef]
- Granneman, S.; Kudla, G.; Petfalski, E.; Tollervey, D. Identification of protein binding sites on U3 snoRNA and pre-rRNA by UV cross-linking and high-throughput analysis of cDNAs. Proc. Natl. Acad. Sci. USA 2009, 106, 9613–9618. [Google Scholar] [CrossRef]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.; Jungkamp, A.-C.; Munschauer, M.; et al. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Wang, N.; Liu, H.; Yu, B.; Jin, L.; Ren, X.; Shen, Y.; Wang, L. Genetically encoded chemical crosslinking of RNA in vivo. Nat. Chem. 2022, 1–12. [Google Scholar] [CrossRef] [PubMed]
- McMahon, A.C.; Rahman, R.; Jin, H.; Shen, J.L.; Fieldsend, A.; Luo, W.; Rosbash, M. TRIBE: Hijacking an RNA-editing enzyme to identify cell-specific targets of RNA-binding proteins. Cell 2016, 165, 742–753. [Google Scholar] [CrossRef] [Green Version]
- Brannan, K.W.; Chaim, I.A.; Marina, R.J.; Yee, B.A.; Kofman, E.R.; Lorenz, D.A.; Jagannatha, P.; Dong, K.D.; Madrigal, A.A.; Underwood, J.G.; et al. Robust single-cell discovery of RNA targets of RNA-binding proteins and ribosomes. Nat. Methods 2021, 18, 507–519. [Google Scholar] [CrossRef]
- Lapointe, C.P.; Wilinski, D.; Saunders, H.A.J.; Wickens, M. Protein-RNA networks revealed through covalent RNA marks. Nat. Methods 2015, 12, 1163–1170. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Rahman, R.; Rosbash, M. Mechanistic implications of enhanced editing by a HyperTRIBE RNA-binding protein. RNA 2018, 24, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Flamand, M.N.; Ke, K.; Tamming, R.; Meyer, K.D. Single-molecule identification of the target RNAs of different RNA binding proteins simultaneously in cells. Genes Dev. 2022, 36, 1002–1015. [Google Scholar] [CrossRef]
- Zaccara, S.; Jaffrey, S.R. A unified model for the function of YTHDF proteins in regulating m6A-modified mRNA. Cell 2020, 181, 1582–1595.e18. [Google Scholar] [CrossRef]
- Khyzha, N.; Henikoff, S.; Ahmad, K. Profiling RNA at chromatin targets in situ by antibody-targeted tagmentation. Nat. Methods 2022, 19, 1383–1392. [Google Scholar] [CrossRef]
- Fazal, F.M.; Han, S.; Parker, K.R.; Kaewsapsak, P.; Xu, J.; Boettiger, A.N.; Chang, H.Y.; Ting, A.Y. Atlas of subcellular RNA localization revealed by APEX-seq. Cell 2019, 178, 473–490.e26. [Google Scholar] [CrossRef]
- Padrón, A.; Iwasaki, S.; Ingolia, N.T. Proximity RNA labeling by APEX-seq reveals the organization of translation initiation complexes and repressive RNA granules. Mol. Cell 2019, 75, 875–887.e5. [Google Scholar] [CrossRef]
- Huang, J.; Zhao, R.; Qin, S.; Yang, S.; Li, W.; Mo, J.; Wang, F.; Du, Y.; Weng, X.; Zhou, X. 4-Thiouridine-enhanced peroxidase-generated biotinylation of RNA. ChemBioChem 2021, 22, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Zhang, S.; Zhu, Y.; Xi, X.; Bao, P.; Ma, Z.; Kapral, T.H.; Chen, S.; Zagrovic, B.; Yang, Y.T.; et al. POSTAR3: An updated platform for exploring post-transcriptional regulation coordinated by RNA-binding proteins. Nucleic Acids Res. 2022, 50, D287–D294. [Google Scholar] [CrossRef]
- Kang, J.; Tang, Q.; He, J.; Le, L.; Yang, N.; Yu, S.; Wang, M.; Zhang, Y.; Lin, J.; Cui, T.; et al. RNAInter v4.0: RNA interactome repository with redefined confidence scoring system and improved accessibility. Nucleic Acids Res. 2022, 50, D326–D332. [Google Scholar] [CrossRef] [PubMed]
- Caudron-Herger, M.; Jansen, R.E.; Wassmer, E.; Diederichs, S. RBP2GO: A comprehensive pan-species database on RNA-binding proteins, their interactions and functions. Nucleic Acids Res. 2021, 49, D425–D436. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Liu, H.; Yang, L.; Zhou, T.; Li, X.; Zhao, Y. RPpocket: An RNA-protein intuitive database with RNA pocket topology resources. Int. J. Mol. Sci. 2022, 23, 6903. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.D.S.; Bava, K.A.; Gromiha, M.M.; Prabakaran, P.; Kitajima, K.; Uedaira, H.; Sarai, A. ProTherm and ProNIT: Thermodynamic databases for proteins and protein-nucleic acid interactions. Nucleic Acids Res. 2006, 34, D204–D206. [Google Scholar] [CrossRef] [Green Version]
- Gebauer, F.; Schwarzl, T.; Valcárcel, J.; Hentze, M.W. RNA-binding proteins in human genetic disease. Nat. Rev. Genet. 2021, 22, 185–198. [Google Scholar] [CrossRef]
- Hua, Y.; Sahashi, K.; Hung, G.; Rigo, F.; Passini, M.A.; Bennett, C.F.; Krainer, A.R. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010, 24, 1634–1644. [Google Scholar] [CrossRef]
- Bogaert, E.; Boeynaems, S.; Kato, M.; Guo, L.; Caulfield, T.R.; Steyaert, J.; Scheveneels, W.; Wilmans, N.; Haeck, W.; Hersmus, N.; et al. Molecular dissection of FUS points at synergistic effect of low-complexity domains in toxicity. Cell Rep. 2018, 24, 529–537.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.A.; Shah, N.; Wang, K.C.; Kim, J.; Horlings, H.M.; Wong, D.J.; Tsai, M.-C.; Hung, T.; Argani, P.; Rinn, J.L.; et al. Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature 2010, 464, 1071–1076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cretu, C.; Agrawal, A.A.; Cook, A.; Will, C.L.; Fekkes, P.; Smith, P.G.; Lührmann, R.; Larsen, N.; Buonamici, S.; Pena, V. Structural basis of splicing modulation by antitumor macrolide compounds. Mol. Cell 2018, 70, 265–273.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aguilar, R.; Spencer, K.B.; Kesner, B.; Rizvi, N.F.; Badmalia, M.D.; Mrozowich, T.; Mortison, J.D.; Rivera, C.; Smith, G.F.; Burchard, J.; et al. Targeting Xist with compounds that disrupt RNA structure and X inactivation. Nature 2022, 604, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Mercuri, E.; Darras, B.T.; Connolly, A.M.; Kuntz, N.L.; Kirschner, J.; Chiriboga, C.A.; Saito, K.; Servais, L.; Tizzano, E.; et al. Nusinersen versus sham control in infantile-onset spinal muscular atrophy. N. Engl. J. Med. 2017, 377, 1723–1732. [Google Scholar] [CrossRef] [Green Version]
- Cooper, T.A.; Wan, L.; Dreyfuss, G. RNA and disease. Cell 2009, 136, 777–793. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.-C.; Spitale, R.C.; Chang, H.Y. Long intergenic noncoding RNAs: New links in cancer progression. Cancer Res. 2011, 71, 3–7. [Google Scholar] [CrossRef]
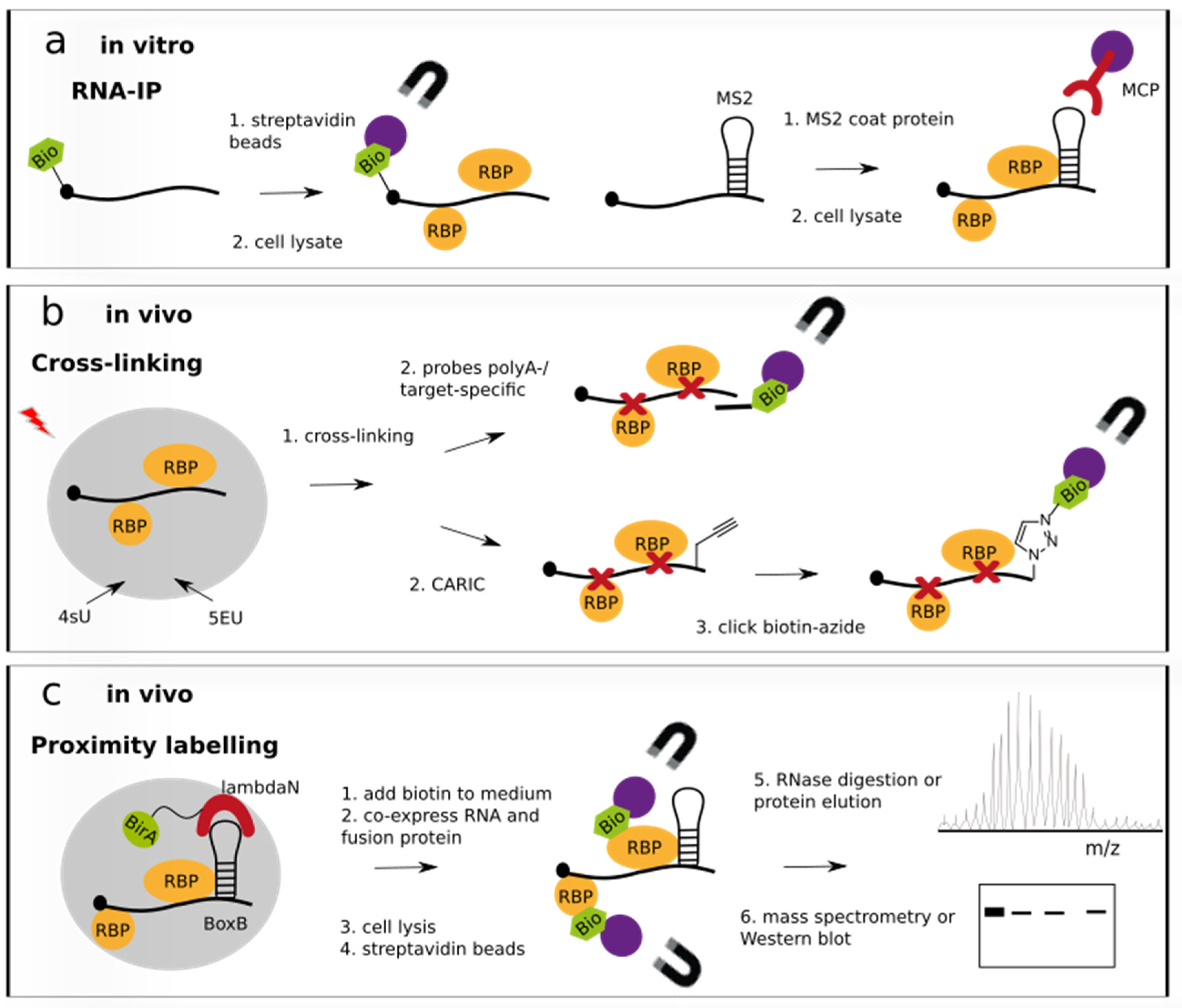
Figure 1.
RNA-centric approaches to study RNA–protein interactions. (a) In vitro-transcribed RNA tagged with biotin, aptamer or stem-loop MS2 is incubated with cell lysate or purified protein and pulled down with streptavidin beads or MS2-coat protein immobilized on beads for analysis of bound proteins. Protein analysis is performed by Western blot or mass spectrometry similar to steps 5. and 6. shown in (c). (b) UV-cross-linked RNA-protein complexes are recovered by IP with target-specific probes or poly(dT) oligonucleotides coupled to beads (upper case). In the click-chemistry-based method CARIC, metabolically labelled 5-ethinyluridine (5EU)-transcripts are clicked to biotin-azide for purification (lower case). Metabolic labelling with 4-thiouridine (4sU) serves to enhance cross-linking efficiency. Although not shown, recovered RNA-protein complexes are subjected to similar analysis steps 5. and 6. as shown in (c). (c) Proximity labelling is performed by recruiting a biotin ligase BirA in close vicinity to an RNA targeted by the fusion protein. A BoxB or MS2 stem-loop is recognised and bound by the λN peptide or MS2 coat protein, respectively. Subsequent IP of the biotinylated RBPs in complex with the RNA and elution of the RBPs allows for protein analysis by mass spectrometry or Western blotting.
Figure 1.
RNA-centric approaches to study RNA–protein interactions. (a) In vitro-transcribed RNA tagged with biotin, aptamer or stem-loop MS2 is incubated with cell lysate or purified protein and pulled down with streptavidin beads or MS2-coat protein immobilized on beads for analysis of bound proteins. Protein analysis is performed by Western blot or mass spectrometry similar to steps 5. and 6. shown in (c). (b) UV-cross-linked RNA-protein complexes are recovered by IP with target-specific probes or poly(dT) oligonucleotides coupled to beads (upper case). In the click-chemistry-based method CARIC, metabolically labelled 5-ethinyluridine (5EU)-transcripts are clicked to biotin-azide for purification (lower case). Metabolic labelling with 4-thiouridine (4sU) serves to enhance cross-linking efficiency. Although not shown, recovered RNA-protein complexes are subjected to similar analysis steps 5. and 6. as shown in (c). (c) Proximity labelling is performed by recruiting a biotin ligase BirA in close vicinity to an RNA targeted by the fusion protein. A BoxB or MS2 stem-loop is recognised and bound by the λN peptide or MS2 coat protein, respectively. Subsequent IP of the biotinylated RBPs in complex with the RNA and elution of the RBPs allows for protein analysis by mass spectrometry or Western blotting.

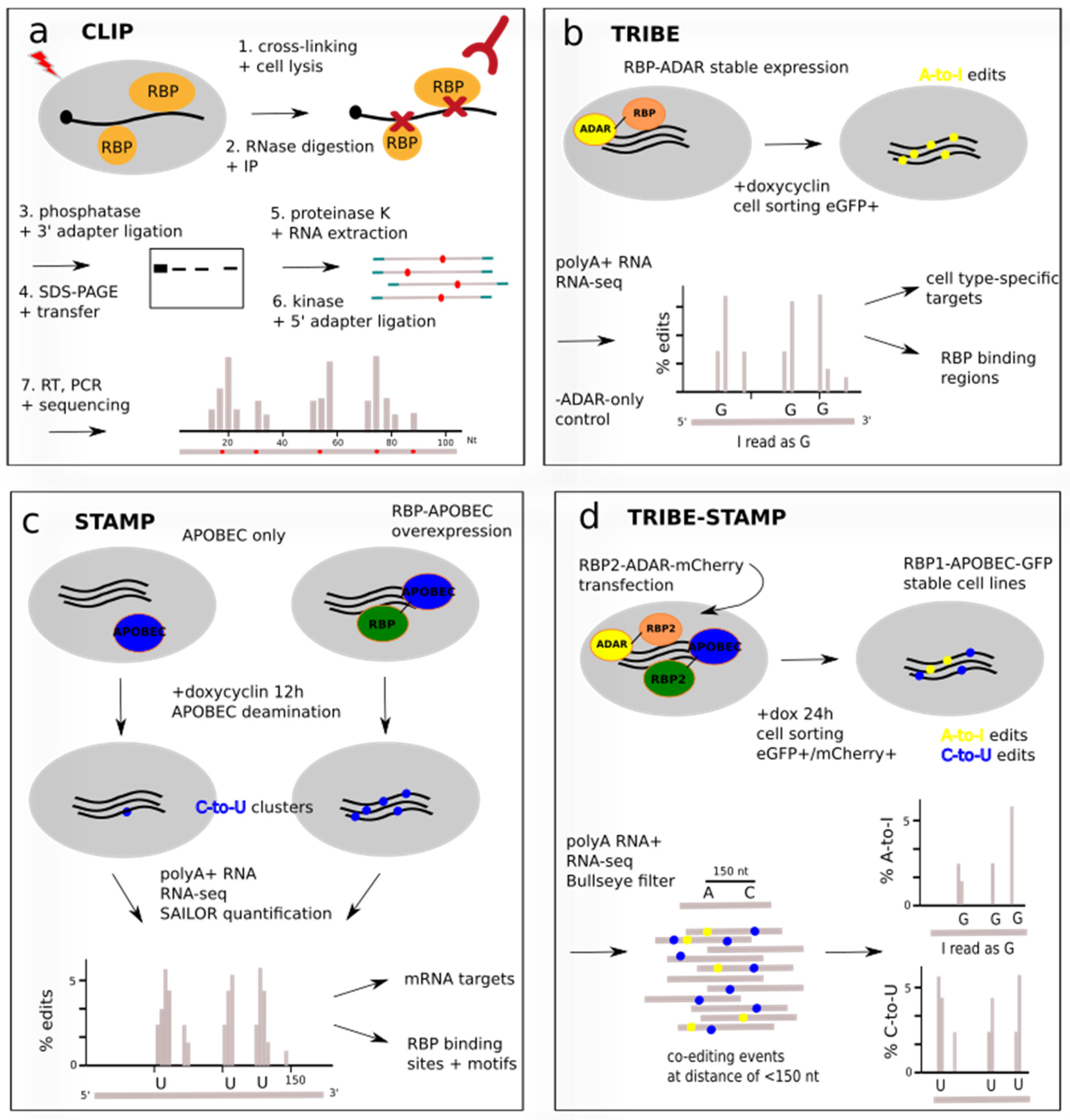
Figure 2.
Protein-centric approaches to study RNA–protein interactions. (a) Schematic CLIP protocol. UV cross-linked RNA-protein complexes are immunoprecipitated with an RBP-specific antibody, separated by size via gel electrophoresis and transferred to a membrane. Target complexes are excised and RNA is extracted by proteinase K digestion of protein components. A cDNA library protocol is followed and transcripts and RBP binding sites are identified by next-generation sequencing. Note that the order of steps 3. to 7. varies depending on the CLIP protocol. (b) TRIBE uses a fusion of the deaminase ADAR and the RBP to induce in-cell A-to-I editing near RBP binding sites. Inducible expression in distinct cell types allows identification of cell-type-specific targets and of binding regions such as 3′ UTR or coding sequence. (c) STAMP detects RBP binding sites by induced mutations in RNA-sequencing reads. A fusion of deaminase APOBEC to the RBP of interest is overexpressed as stable cell line, and C-to-U conversions are inserted in the RNA in the vicinity of the RBP binding site. Subtraction of background edits yields the editing clusters on target mRNAs. (d) Combination of STAMP and TRIBE using a first RBP (RBP1) fused to APOBEC and a transfected second RBP (RBP2) fused to ADAR. Fluorescent cell sorting guarantees parallel expression. cDNA-sequencing maps co-edited sites on the same read and identifies shared RNA targets of RBP1 and RBP2. Editing percentage is defined as the number of A-to-I or C-to-U edits at a given site divided by the number of reads at that site.
Figure 2.
Protein-centric approaches to study RNA–protein interactions. (a) Schematic CLIP protocol. UV cross-linked RNA-protein complexes are immunoprecipitated with an RBP-specific antibody, separated by size via gel electrophoresis and transferred to a membrane. Target complexes are excised and RNA is extracted by proteinase K digestion of protein components. A cDNA library protocol is followed and transcripts and RBP binding sites are identified by next-generation sequencing. Note that the order of steps 3. to 7. varies depending on the CLIP protocol. (b) TRIBE uses a fusion of the deaminase ADAR and the RBP to induce in-cell A-to-I editing near RBP binding sites. Inducible expression in distinct cell types allows identification of cell-type-specific targets and of binding regions such as 3′ UTR or coding sequence. (c) STAMP detects RBP binding sites by induced mutations in RNA-sequencing reads. A fusion of deaminase APOBEC to the RBP of interest is overexpressed as stable cell line, and C-to-U conversions are inserted in the RNA in the vicinity of the RBP binding site. Subtraction of background edits yields the editing clusters on target mRNAs. (d) Combination of STAMP and TRIBE using a first RBP (RBP1) fused to APOBEC and a transfected second RBP (RBP2) fused to ADAR. Fluorescent cell sorting guarantees parallel expression. cDNA-sequencing maps co-edited sites on the same read and identifies shared RNA targets of RBP1 and RBP2. Editing percentage is defined as the number of A-to-I or C-to-U edits at a given site divided by the number of reads at that site.


{kind=link}
{kind=link}
Table 1.
Summary of techniques to study RNA–protein interactions covered in this review. Year of development, target studied, advantages and limitations are listed.
Table 1.
Summary of techniques to study RNA–protein interactions covered in this review. Year of development, target studied, advantages and limitations are listed.
| Method | Year | Single Target or Transcriptome? | Target Studied | Advantages | Limitations |
|---|---|---|---|---|---|
| in vitro RNA-centric approaches | |||||
| IVT RNA IP | single RNA | multiple | choice of target RNA, mutagenesis possible | no endogenous loci, large tags | |
| Engineered RNA IP | single RNA | multiple | in cells | large tags, genetic engineering or overexpression | |
| in vivo RNA-centric approaches | |||||
| Techniques involving cross-linking | |||||
| CL and IP | single RNA | multiple | direct RNA/protein contacts | UV also captures RNA/RNA and protein/protein interactions | |
| ChIRP-MS | 2015 | single RNA | lncRNA Xist, snRNAs U1, U2 | several tiled probes for long RNAs | - |
| RBR-ID | 2016 | transcriptome-wide | nuclear RNAs | 10–100x less input (106 cells) than for polyA+ | false negatives due to low CL efficiency to some 4sU-transcripts |
| MS2-BioTRAP | 2011 | single RNA | IRES-containing reporter mRNA | in vivo biotinylation site of HB-tagged MCP | engineering of MS2-RNA and overexpression of MCP |
| PAIR | 2006 | single RNA | ankylosis mRNA | PNA activation by UV-photocleavable group | transport of PNA into cells via cell-penetrating peptide |
| CHART | 2011 | single RNA | lncRNAs NEAT1, MALAT1, roX2 | map chromatin binding sites of target RNA | no evidence of direct RNA/DNA binding |
| RAP-MS | 2015/2018 | single RNA | lncRNAs Xist, Firre | efficient pulldown of lncRNAs | multiple controls to exclude false positives |
| CARIC | 2018 | transcriptome-wide | mRNAs, lncRNAs, snRNAs, miRNAs, rRNAs | quantitative pulldown via click reaction to 5EU | no control over stoichiometry of 4sU/5EU labelling |
| RICK | 2018 | transcriptome-wide | non-poly(A) RNAs | quantitative pulldown via click reaction to 5EU | no control over stoichiometry of 5EU labelling |
| Proximity labelling techniques | |||||
| RAPID | 2018 | single RNA | EDEN15 RNA | applicable to organs/tissues, low cell input | endogenously biotinylated proteins, engineered RNAs |
| RNA BioID | 2019 | single RNA | β-actin mRNA | captures transient and loose interactions | unspecific biotin labelling, engineered RNAs |
| CRUIS | 2020 | single RNA | lncRNA NORAD | dCas13-based targeting of endogenous RNA locus | large peptide for labelling, transfection of 2 plasmids into stable cell line |
| CBRPP | 2021 | single RNA | lncRNA NORAD, β-actin mRNA | reduced background labelling | transfection of 2 plasmids into stable cell line |
| in vivo protein-centric approaches | |||||
| Techniques involving cross-linking | |||||
| CLIP and variants | 2003/2008–today | single protein | multiple | many protocols with improved steps | false positives, low antibody efficiency, high cell input |
| GECX-RNA | 2022 | single protein | Hfq chaperone, YTH domain | active site-specific mapping of RNA binding | engineered proteins with unnatural amino acids |
| Techniques without cross-linking | |||||
| RIP-seq | 2010 | single protein | multiple | stable interactions | no transient interactions |
| TRIBE | 2016/2018 | single protein | Hrp48 | identification of cell-type-specific RNA targets | editing bias of ADAR deaminase |
| STAMP | 2021 | single protein | RBFOX2, RPS2, RPS3 | applicable to single cell scale and direct long-read RNA sequencing | false positives |
| TRIBE-STAMP | 2022 | single protein | YTHDF1, YTHDF2, YTHDF3 | sequential binding events via monitoring co-editing sites | low editing efficiency of ADAR |
| RNA-tagging | 2015 | single protein | PUF3 | endogenous proteins, RBP binding affinity correlated to polyU length | - |
| RT&Tag | 2022 | single protein | histone modifications | chromatin-associated RNA targets | laborious protocol |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Mattay, J. Current Technical Approaches to Study RNA–Protein Interactions in mRNAs and Long Non-Coding RNAs. BioChem 2023, 3, 1-14. https://doi.org/10.3390/biochem3010001
AMA Style
Mattay J. Current Technical Approaches to Study RNA–Protein Interactions in mRNAs and Long Non-Coding RNAs. BioChem. 2023; 3(1):1-14. https://doi.org/10.3390/biochem3010001
Chicago/Turabian StyleMattay, Johanna. 2023. "Current Technical Approaches to Study RNA–Protein Interactions in mRNAs and Long Non-Coding RNAs" BioChem 3, no. 1: 1-14. https://doi.org/10.3390/biochem3010001