1. Introduction
Bilayers are typical structure formations of mostly organic biological and non-biological systems in which an anisometric molecular shape and amphiphilic character predetermine their behaviour in an aqueous environment. It is a basic formation in living and non-living organisms. In addition to the amphiphilic compounds, such as the salts of fatty acids, phospholipids, etc., the biological structures, including fibrous proteins showing relatively long, well-defined hydrophobic–hydrophilic formations of amino acids, can also show organized lyotropic (hydrotropic) liquid structural behaviour. Polyelectrolyte multilayer (PEM) assemblies (e.g., planar films or walls of hollow capsules) are interesting as well, consisting of bilayers formed of polyionoactive macromolecules pairs (bilayers) of opposite ionogenic character.
The ability of those formations to create organized structures is well observed in anisotropic liquid crystal (LC) systems [
1,
2]. Liquid crystals are categorized into two forms: thermotropic liquid crystalline LCs, which are formed by a change in temperature, and lyotropic LCs, which are formed when amphiphilic molecules are mixed with an aqueous phase (hydromesophase); the formation of these two forms is dependent on both temperature and concentration [
3,
4,
5,
6]. Self-assembly molecules in LCs are highly ordered, but in a liquid, they are free to diffuse in a random way [
4]. Hexagonal and cubic mesophases are of particularly high interest in the drug delivery field due to their exceptional potential as drug vehicles [
7,
8]. However, as yet, there is not an adequate understanding of the theoretical explanation for the targeted release of these mesophases into selected locations of illness in the body, usually due to varying temperatures between the ill and the healthy location.
With an increase in the concentration of amphiphilic components, e.g., lipids, different well-defined structures are created, such as the normal micelle (oil in water), the normal cubic phase, the normal hexagonal phase, the lamellar phase, the reversed cubic phase (water in oil), and the reversed hexagonal phase. Additionally, the reversed cubic phases can be divided into two categories: the reversed bicontinuous cubic phase and the reversed micellar cubic phase [
9].
The lamellar phase appears extensively in all organisms, e.g., it forms the basic building block of cell membranes [
9]. The lamellar phase is a planar structure, consisting of surfactant bilayers separated by water, where polar head groups protrude in the intervening layers of the aqueous interface. The surfactant bilayers are separated by water. The polar head groups of the amphiphilic molecules and are associated and in direct contact with water, while the hydrophobic tails are located away from water [
4,
10,
11,
12,
13].
The cubic phase structure contains spherical packing and consists of continuous curved layers in which the polar part of the molecule appears on the surface of the sphere, and the non-polar part (water-insoluble lipomesophase) is directed toward the centre of the sphere.
The hexagonal phase (the long-range order is two-dimensional) can be seen as an array of hexagonally closely packed water curved layers sheltered by a surfactant monolayer, similar to the cubic phase structure.
It is possible to conceive of both the cubic and the hexagonal phases as formations of continuous curved bilayers where the polar part of the molecule is located on the surface, as well as in middle of the sphere, with the rod-like particles in contact with water [
4].
The phase behaviour of the LC system is sensitive to temperature and pressure [
14,
15,
16,
17,
18,
19], as well as to light, magnetic field, and other factors, such as the presence of amphiphilic molecules, water content, and third-party additives [
9]. The internal structure of the LC materials often includes the lamellar phase, bicontinuous cubic phase, or inverse hexagonal phase, which thrive with an excess of aqueous solution (such as an excess of bodily fluids) [
8]. For example, it has been found that in a glyceryl monooleate (GMO)—water system, at temperatures between 25–80 °C, the lamellar phase, and different types of bicontinuous cubic phases are formed, but if the temperature rises to about 80 °C, the cubic-to-reversed hexagonal phase transition occurs [
9,
15]. It has also been suggested [
18,
19] that only glyceryl oleate (OG) and phytanyl glycerate (PG) are able to form the reversed hexagonal phase in excess water at the standard physiological temperature. Concurrently, GMO, one of the monoglycerides with the same molecular weight as OG could only form the cubic phase under the same conditions [
9,
18,
19].
The structural transitions affected by the factors mentioned above can be explained by use of the so-called critical packing parameter (CPP), which is based on the spatial stacking of amphiphilic molecules [
9,
16,
17]. The CPP is defined as CPP = V/al, where V represents the hydrophobic chain volume, a represents the cross-sectional area of the hydrophilic head group, and l represents the hydrophobic chain length in the molten state. According to the theory of CPP, different phases correspond to different CPP values. When CPP < 1, normal phases are formed, which means that the cross-sectional area of the hydrophilic head group is larger than that of the hydrophobic tails, such as the normal micelle (L
1), the normal discontinuous cubic (I
1) phase, the normal hexagonal (H
1) phase, and the normal bicontinuous cubic (Q
1) phase. Inversely, reversed phases such as the reversed bicontinuous cubic (Q
2) phase, the reversed hexagonal (H
2) phase, the reversed discontinuous cubic (I
2) phase, and the reversed micelle (L
2) form with CPP > 1. When the polar head group area and the tail are nearly equal, the lamellar phase forms. According to this simple theory, the increasing temperature leads to a decrease in the hydration of the lipid’s polar head and, consequently, a decrease in the cross-sectional area. Increasing temperature also has a profound effect on hydrophobic tails, resulting in a shorter tail length but a larger volume. In general, an increase in temperature leads to an increase in CPP. However, the transition of a normal lamellar or micelle phase into normal cubic or hexagonal phases, etc., remains to be explained.
In the literature [
4,
7], three properties of surfactant(s) are mentioned as affecting the formation of the lyotropic liquid crystalline phase. These are [
7]: the magnitude of the repulsive forces between adjacent head groups at the interface of the surfactant and water, the degree of contact between water and the alkyl chain, and the conformational disorders in the alkyl chains. Nevertheless, the phase behaviour of different amphiphilic molecules is not the same, which can be attributed to the difference in the hydrophilic hydration intensity, the number and length of alkyl chains, and the degree of unsaturation. However, the ways in which water influences the epimolecular behaviour of these structures are yet to be determined.
2. Water Role in Formation and Behaviour of Bilayers
An idea of a hydration bonding system embodies an important and key role of water molecules in the behaviour of hydrotropic systems. Hydrophilic systems are characterized by their hydration layers on a hydrophilic phase interface denoted in literature, under various names such as immobilized water, vicinal water, non-solute water, gel water, unfreezable hydration water, etc. [
20,
21,
22]. As is known, water in these hydration layers evokes a weak hydration interaction between interacting opposite hydrophilic interfaces due to an interplay of long-range forces characterized by both the attractive and repulsive hydration forces, i.e., hydration bonding and de-bonding activities between those interfaces, respectively [
23] (pp. 222–241). The quality of these interactions is controlled by basic orientation of water molecules during interactions between the opposite sides of the interface’s domains, i.e., the attractive or the repulsive hydration forces are determined as inverse or identical based on the orientation of water molecules, respectively. Quantification of such a hypothesis, according to SCHL (Structural Changes in Hydration Layers) theory, enables us to better understand the behaviour of the hydrotropic systems [
24]. For instance, we can imagine (see
Figure 1) as the interacting hydrophilic interfaces regularly distributed into approx. 0.15 nm × 0.15 nm domains with different basic orientation of water molecules. For example, hydration water surrounding a protein molecule plays a crucial role in protein dynamics and functioning in coherence with a temperature-hysteresis phenomenon. Yamamoto and co-workers [
25] experiments suggest that hydration by unfreezable and freezable water is a necessary and sufficient condition for the activation of protein dynamics. By use of neutron scattering, it has been revealed that only unfreezable hydration water contributes to the activation of protein dynamics via the coupling of their dynamics.
Domain’s distribution of the interacting interfaces determines the character of hydration forces, i.e., their mutually hydration bonding or de-bonding activities. If the domain’s distribution has simple regular mosaic character [
26,
27] this action is already complicated because the character of this interaction depends upon the distance between interacting interfaces. At the nearest distance, attraction prevails, turning into repulsion at higher distances and changing further with the temperature. This difference appears more clearly in the interactions of “heterogeneous” nano-surfaces, in which both the repulsive and the attractive hydration forces are affected simultaneously.
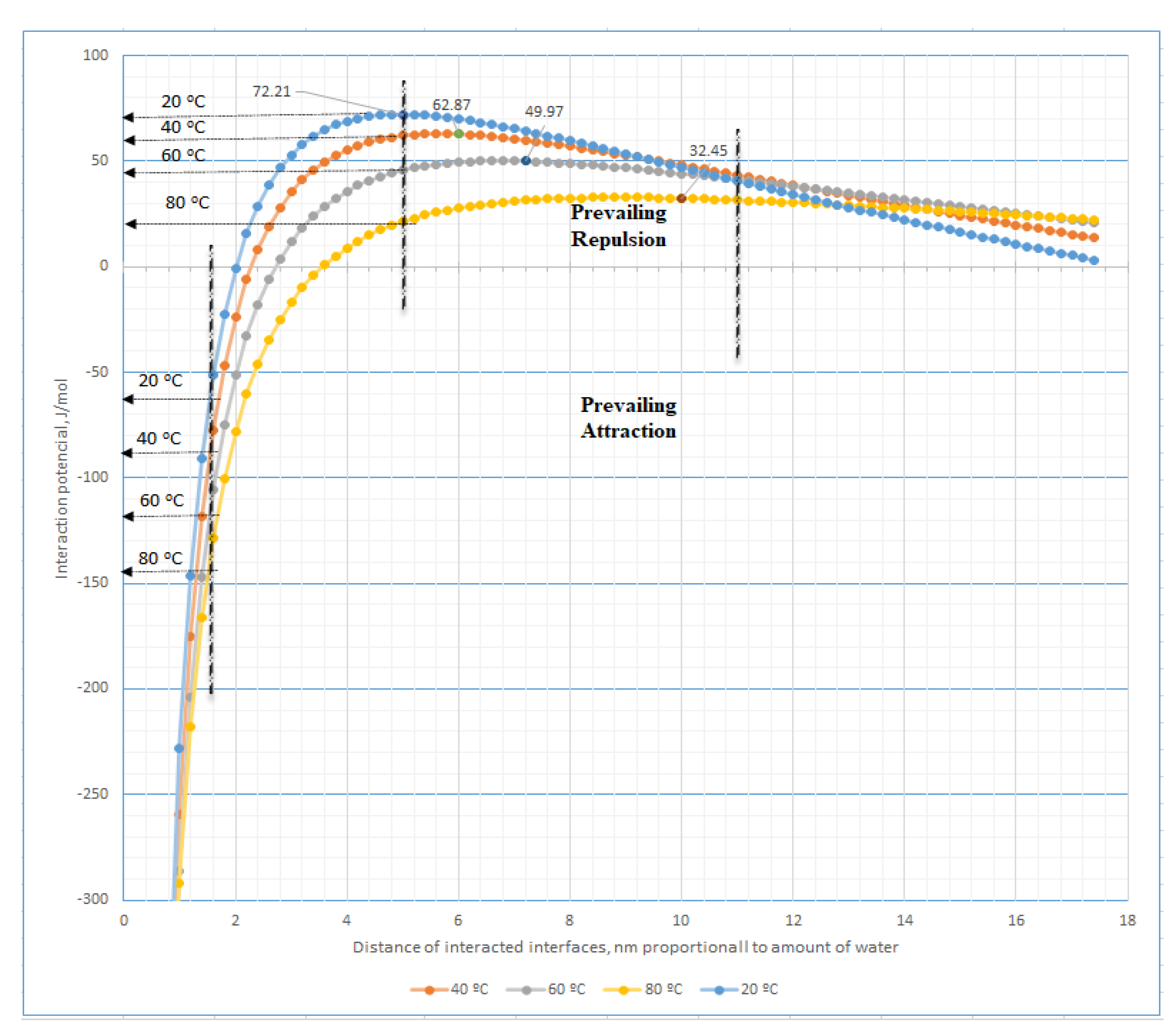
Figure 1 introduces the simplest model of those interactions between two plane formations of mosaic-type arrangement of the acting hydration forces, i.e., those covered with regularly alternating quantitatively coincident sites of attractive and repulsive hydration forces. The theoretical relationships between the interaction potential and the distance of these nano-spots appointed by the amount of water content in the hydrated system at different temperatures are presented in
Figure A1,
Appendix A. The relationships have been derived (see
Appendix A) according to a mosaic model (see
Figure 1) by adding the theoretical repulsive and attractive potential in each point of distance for the corresponding temperature [
28].
In oriented hydrotropic liquid, i.e., liquid crystals, this role connects to a specific molecular structure of the hydrophilic end groups of amphiphilic molecules, evoking basic orientation of water molecules at the hydrophilic interface. The mutual distance between these end groups predetermines the quantity of the acting hydration forces decreasing non-linearly with the increase in this one. In comparable conditions, (see
Figure A1), i.e., at constancy of water content in the hydrotropic system appointing a constant distance between interacting interfaces, with ascending temperature, both the attractive and the repulsive hydration forces in hydromesophase increase or decrease, respectively.
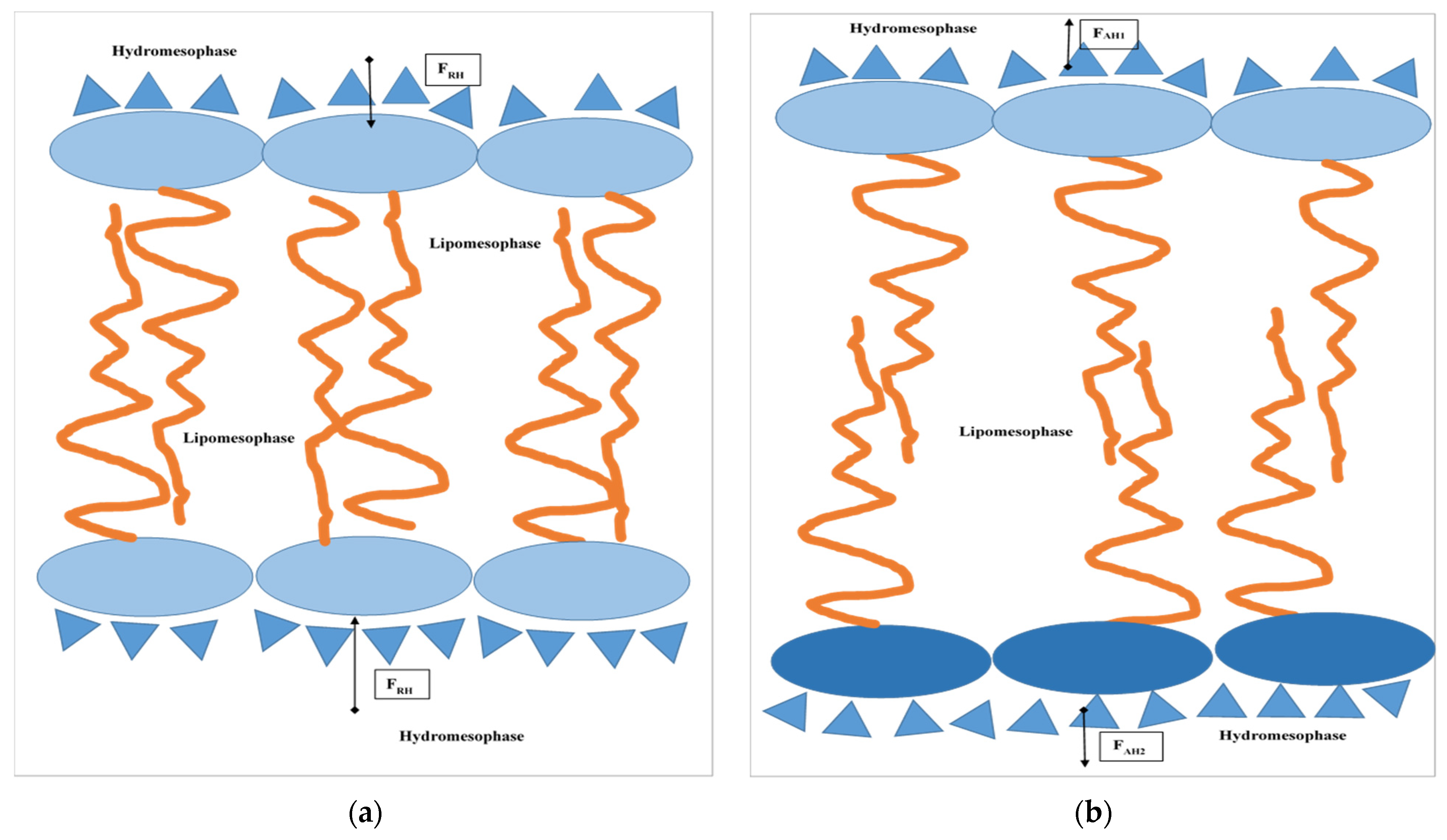
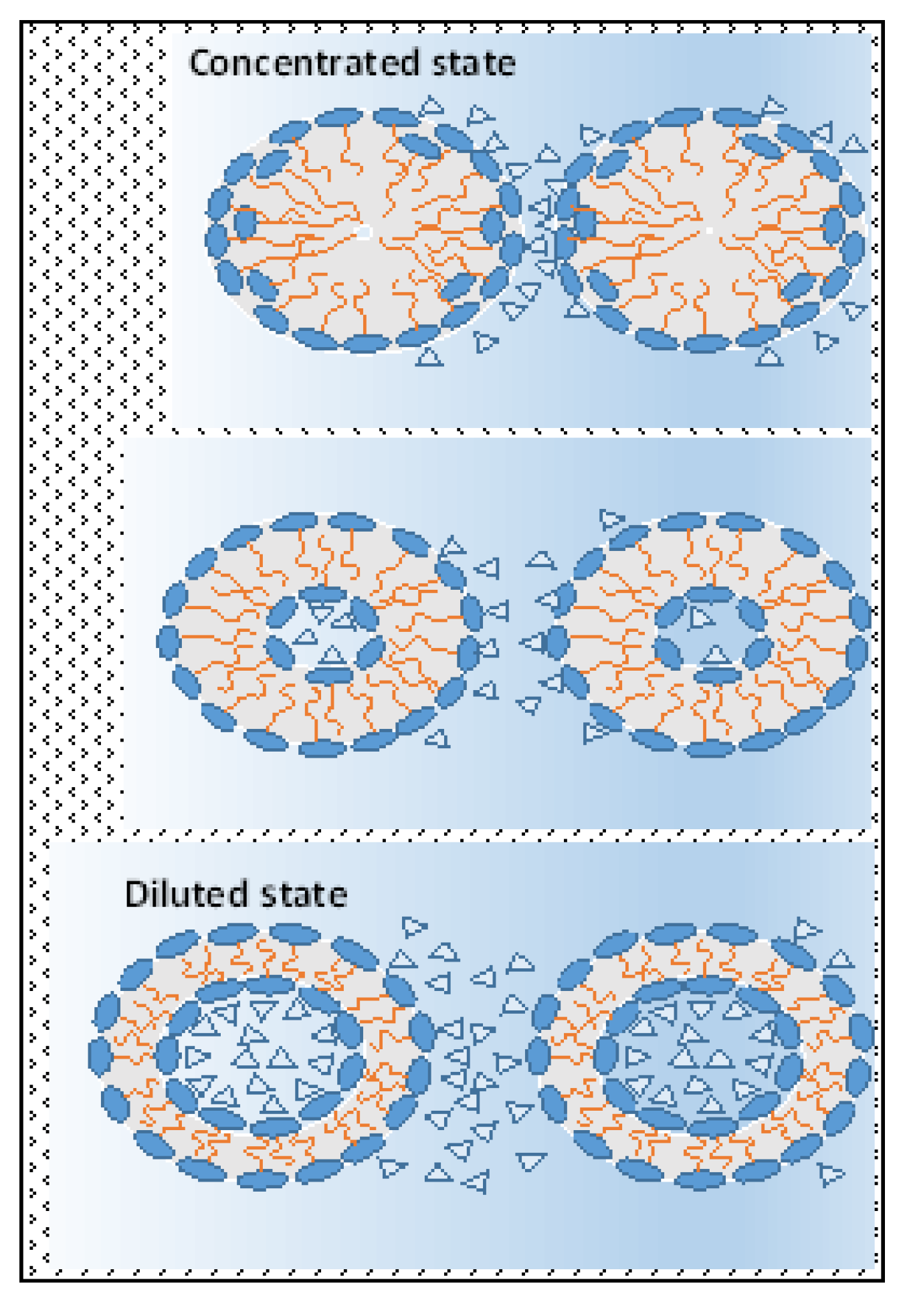
A similar real model represents bilayers created by amphiphilic molecules in a water environment in hydrotropic (lyotropic) liquid crystal systems (see
Figure 2). For example, at the distance between interacting hydrophilic interfaces formed by molecules of an amphiphilic character, 2.8–3.2 nm, repulsion prevails among these interfaces at a temperature extent of 20–60 °C due to prevailing repulsive hydration forces in contrast with the adduction prevailing among those at 80 °C, followed by outstretching of those bilayers (compare
Figure A1 and
Figure 2b). Obviously, in contrast with a state in which the attractive hydration forces are prevailing, the prevailing repulsive hydration forces among interacting polar parts squeeze their bilayers (
Figure 2a) during those interactions in hydrotropic liquid crystal systems. It is also important to mention that with increasing temperature, in comparable conditions, the repulsive hydration forces slightly decrease in contrast with the more intensive increase in the attractive hydration forces. Furthermore, with the decreasing distance among interacting lamellar interfaces, e.g., with decreasing water content in the system, the attraction hydration forces continuously increase in contradiction with the repulsive ones. At a certain distance, they reach a maximum which decreases with the increase in the temperature (see
Figure A1).
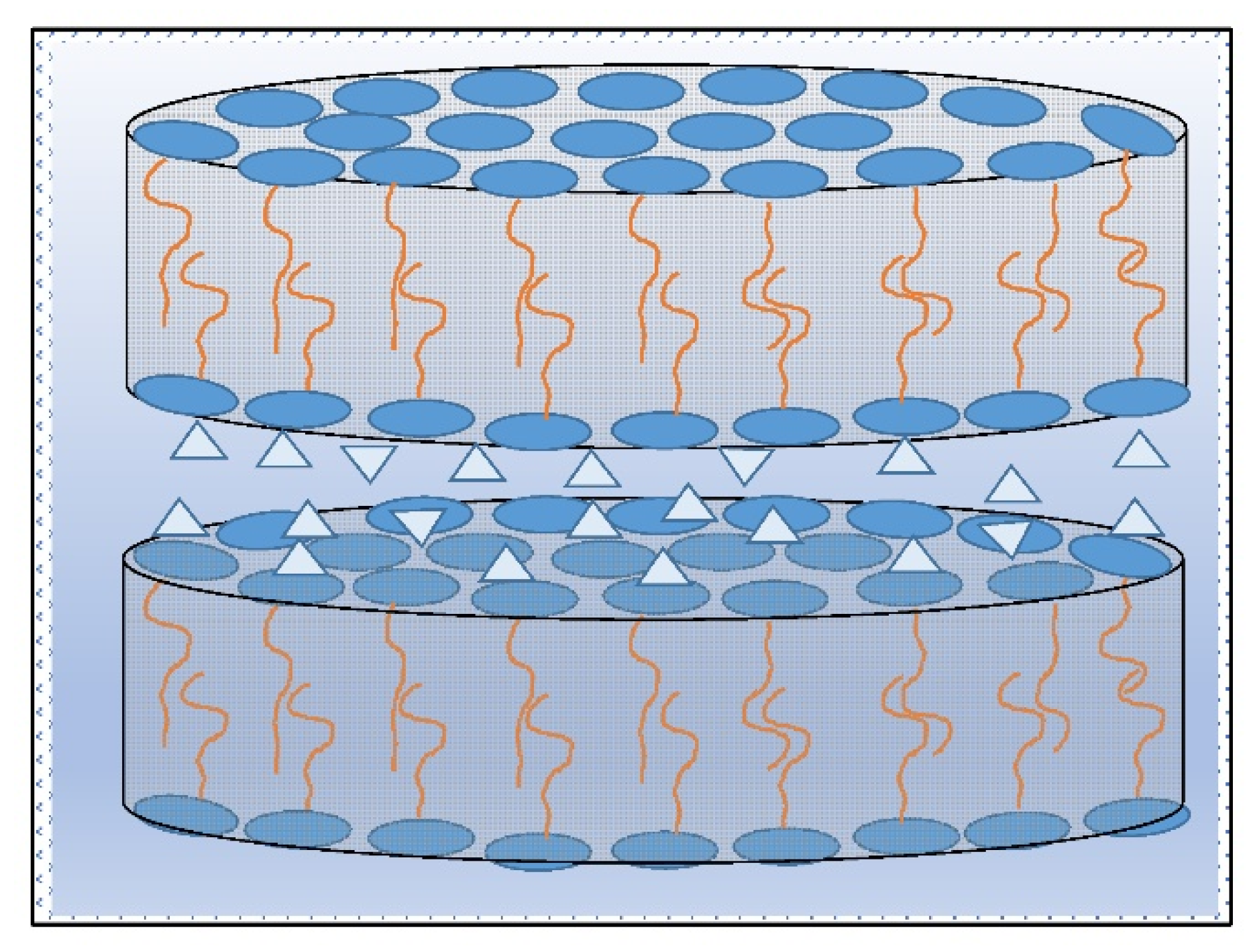
However, if the actual intermolecular forces (van der Waal’s respective London’s, Debye’s, or Keesom’s forces) among lipophilic parts of amphiphilic molecules are relatively high and protect the lipophilic chains of molecules to move mutually, a spatial reorientation of these ones is possible within a well-defined interlayer spacing of the hydromesophases. Another important aspect of this process is intermolecular distance fluctuation. Usually, due to intermolecular energy fluctuation, the distance between interacting forming bilayers is not constant. The interfaces then curl up due to differences in interactions between the hydration forces. The curling up of lamellar interfaces depends upon the difference of both the outer hydration forces relative to the surface area of a lamellar phase, ∆F
H/A = ∆p
H. Due to these forces, the lamellar originating structures form hexagonal, and planar–lamellar formations (
Figure 3) as micro-rod space-oriented micro- and nanostructures of microspheres (i.e., micelles—see
Figure 4a), and micro-disc structures (oval rod-like—see
Figure 5a or oval disc-like—see
Figure 5b) with different space architectures. These include discontinuous cubic, bicontinuous cubic, and helical chiral formations.
2.1. Explanation Formation of Hydrotropic Liquid Crystals’ (LCs) Systems and Their Thermotropic Behaviour
It is supposed that the organization and structural configuration of LCs systems [
3,
4,
5,
6] is controlled by prevailing hydration forces between inner interfaces in hollow parts of micelles and hexagonal structures, acting among outer interfaces of those structures. Thus, they create architecture of anisotropy character of liquid crystals. For instance:
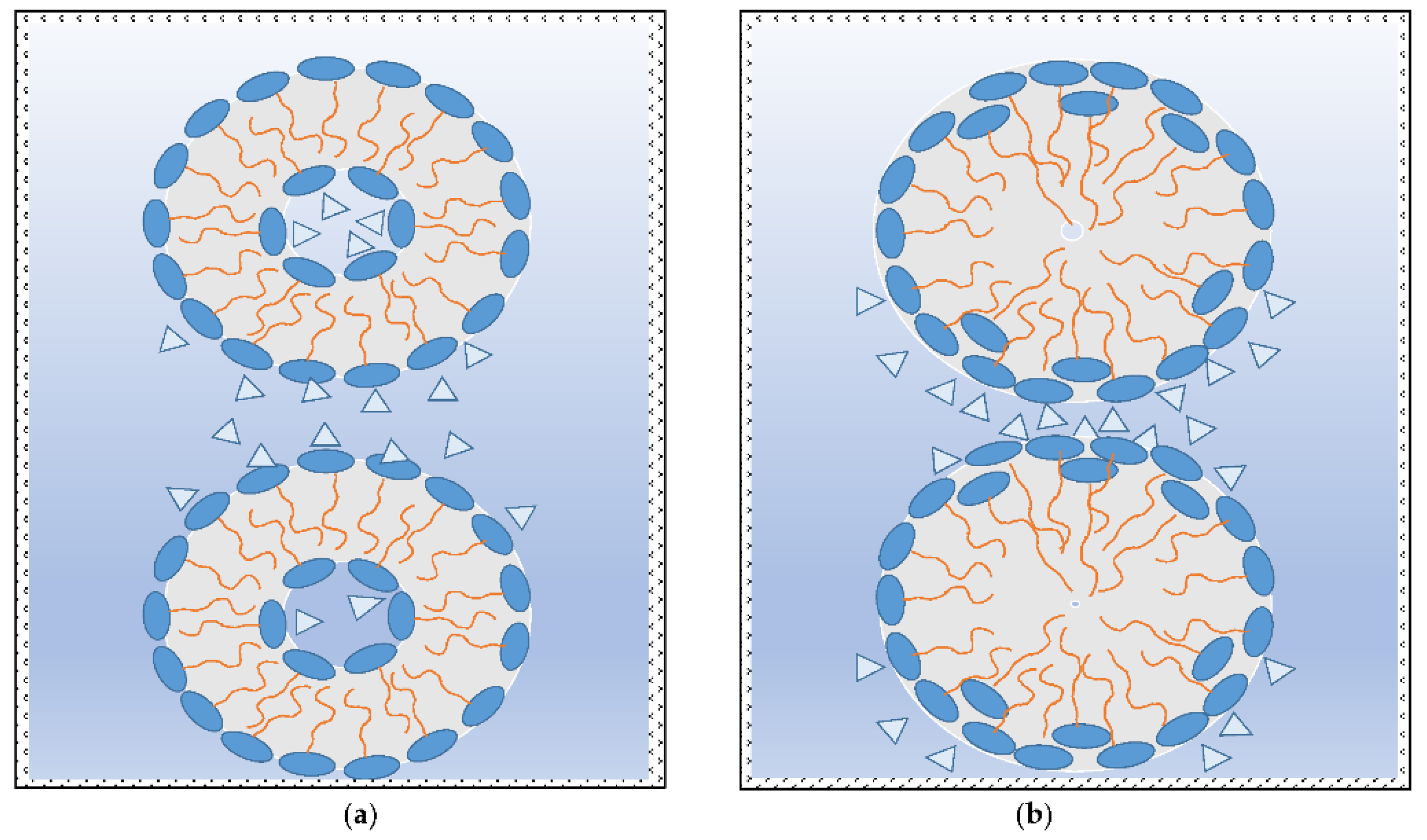
If, in LCs systems, the attractive hydration forces (∆p
H = (F
AH1 − F
AH2)/A > 0) prevail, then the outer interfaces have positive mean curvature (convex), i.e., normal micelle and normal discontinuous cubic, normal hexagonal, and normal bicontinuous cubic structures (critical packing parameter, CPP < 1) are forming. The inner interfaces of these objects have negative mean curvature (concave) with zero or a small amount of water in middle of the closed micro and submicro objects because, with a decrease in distance between interacting interfaces, the attraction between them is continuously increased (see
Figure 4a). Obviously, this is caused by possible and favourable reorientation of the hydrophilic parts of amphiphilic molecules to the outer water medium (
Figure 4b). Helical structures of cholesteric chiral smectic (soap-like) and nematic (thread-like) LCs are probably also formed by this intermolecular mechanism.
If the repulsive hydration forces among polar parts of amphiphilic molecules (∆p
RH = (F
RH1 − F
RH2)/A < 0) prevail, then these ones tend to curl into opposite structures with higher water content being confined in the middle hollow part of those formations (see
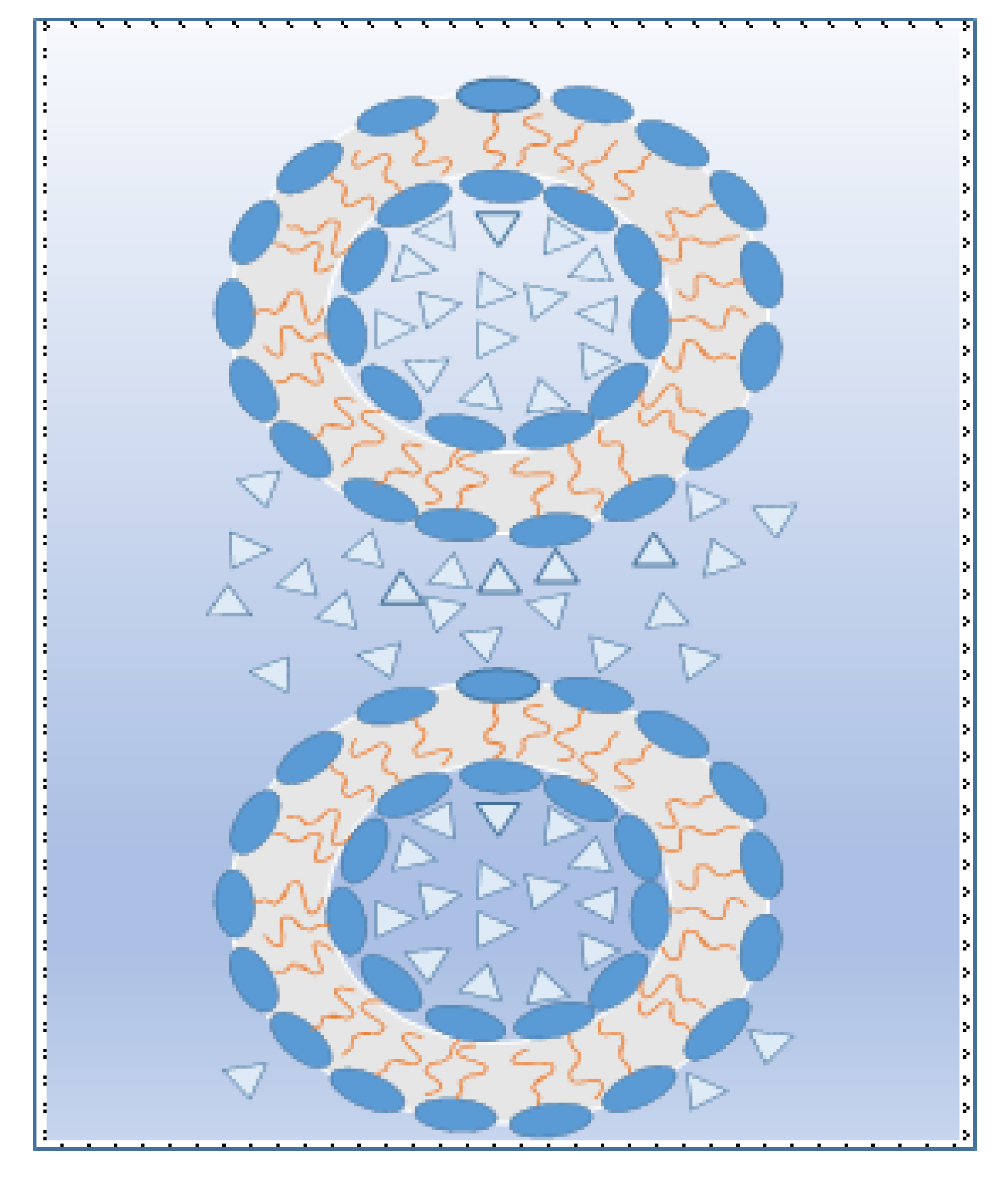
Figure 6). Reverse bicontinuous cubic, reverse hexagonal, and reverse discontinuous cubic objects or reverse micelles (CPP > 1) then form. Regardless, at constant temperature, the existence of distances between interacting lamellar interfaces in which a maximum repulsive hydration forces acts gives cause for occurrence of a relatively high amount of water in the middle part of those forming objects.
If prevailing repulsive or attractive hydration forces are equilibrated, i.e., ∆FH/A = ∆pH = zero, the interfaces have planar lamellar smectic structure.
Nevertheless, as follows from
Figure 7, the size, shape, and structure of the arising oriented micro and submicro curled objects divided among hydromesophases depend upon the distance between interacting interfaces of lipomesophases, i.e., upon the amount of water in the system. So, the system with higher water content where repulsive hydration forces dominate have less “packed” micelle, discontinuous cubic, hexagonal, and bicontinuous cubic structure. On other side, more “packed” and smaller, curled up micro- and submicro-objects are formed in areas of prevailing attractive hydration forces activity, i.e., at small distances between interacting interfaces in the systems with low water content.
However, in contrast with convex surfaces, the concave surfaces of middle hollow parts have approximately the same distances in the space of hydromesophases. This reality subsequently predicts different behaviours of those micro and submicro-objects. Thus, it is also rational to note that convex surfaces at comparable conditions are logically more active than concave surfaces because these ones are more accessible. In comparison with situation of the concave hydromesophases (see
Figure 4b), the interactions between the convex of the interfaces are complicated due to the inconstant spatial distances between those interacting objects. Thus, in appropriate situations, the qualitatively different hydration forces among interacting convex surfaces, i.e., the attractive and repulsive ones, might be influenced at the same time, whereas among interacting concave surfaces, only one sort of hydration forces, e.g., the attractive ones, are active.
Notably, the hydration forces are also acting among hydrophilic head groups of amphiphilic molecules of bilayers. Due to the small distances between these, the attractive hydration forces are arising, i.e., a hydration bonding system is forming. For instance, if the distance between two head groups is 1.6 nm, approximately 10 water molecules are located (see
Figure 8a or
Figure A1) and a strong bonding system is created. This is strengthened further with increasing temperature, followed by formation of a partly irreversible H-bond or fully irreversible chemical bond. Instead of reversible bilayers, an envelope creates an irreversible bilayer skin around the micro-objects.
2.2. Temperature Influence upon Behaviour of Hydrotropic LC Systems
Lyotropic LCs, in contrast with thermotropic LCs, are systems that change their character based on temperature, as well as the concentration of amphiphilic molecules. Formation of different types of LCs structures and even bilayered and multiwalled aggregates is typical in solution. Due to the formation of these structures, the dependence of temperature upon the concentration of amphiphilic molecules has a typical character of extreme function with maximum [
9,
15,
29,
30], i.e., the existence of every structure is limited at isothermal conditions by the concentration zone, which decreases with the increase in the temperature.
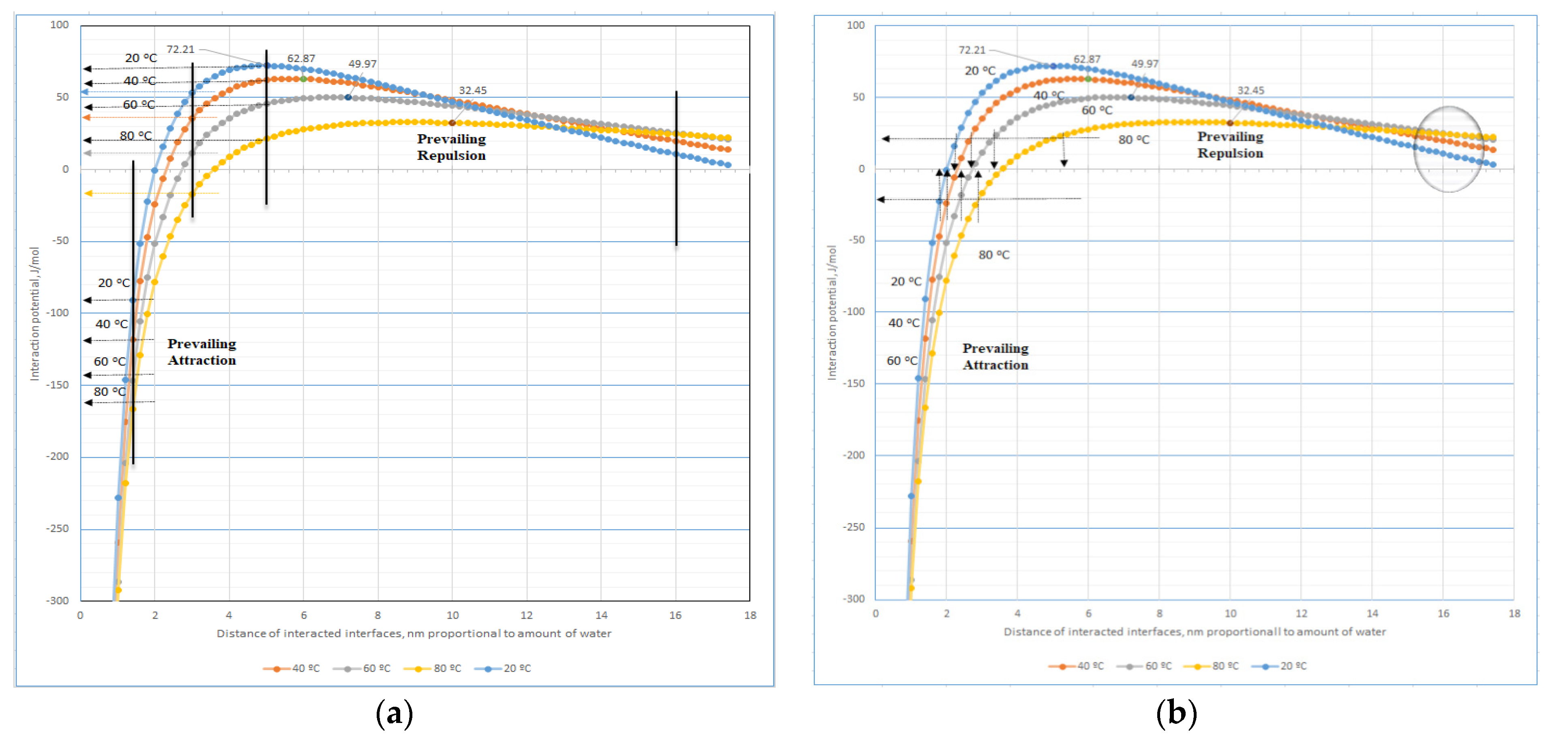
As usual, with increases in temperature, the intermolecular bonding systems are weakened, but the behaviours of the hydration bonding system are more complicated, especially in range of interfaces distances 2–4 nm (see
Figure A1 and
Figure 8a). The theoretical calculations represented in these Figures document increases in the repulsive hydration and attraction forces with temperature among interacting regular mosaic distributed interfaces in hydromesophase at distances > 14 nm and <2 nm, respectively. However, in higher concentrated systems dominating the interface’s distances in the range 2–4 nm, a special effect can be observed. With increased temperature, an antibonding hydrated system and the bonding hydrated system is weakened and strengthened, respectively, as in the highest concentration of bilayers systems, i.e., at interfaces distances < 2 nm. For instance, it is a situation similar to eggs’ protein behaviours during heating. The reversible hydration bonding system is functioning as a predecessor to formation of H-bonds, i.e., reversible H-bonds of the oxygen-H type or partly reversible bonds of the nitrogen-H type are usually finished by a fully irreversible chemical bond, e.g., the peptide bond.
As follows from
Figure 8a, behaviour of the high concentrated LCs systems should change more deeply. At the highest concentration of LCs, i.e., at interfaces distances < 2 nm, the consistency of those systems is increased with the temperature. In the range of the transitional concentrations, i.e., predominantly at distances of interacting interfaces in the range 2–4 nm, with increased temperature, the viscosity of LCs systems should be increased. Additionally, in contrast with lower concentrated LCs systems, the viscosity should decrease with increasing temperature (the prevailing repulsion hydration forces increase regularly with temperature only at interface distances > 16 nm). Furthermore,
Figure 8b indicates more precisely at comparable conditions, i.e., at constant potential of the interactions, that prevailing repulsive hydration forces are less sensitive to consistency than the attractive ones (this one controls the distance between interacting outer and inner hydro-surfaces of LCs) of a hydro-system of mutually interacting micro-planes with mosaic organization of hydrated surfaces.
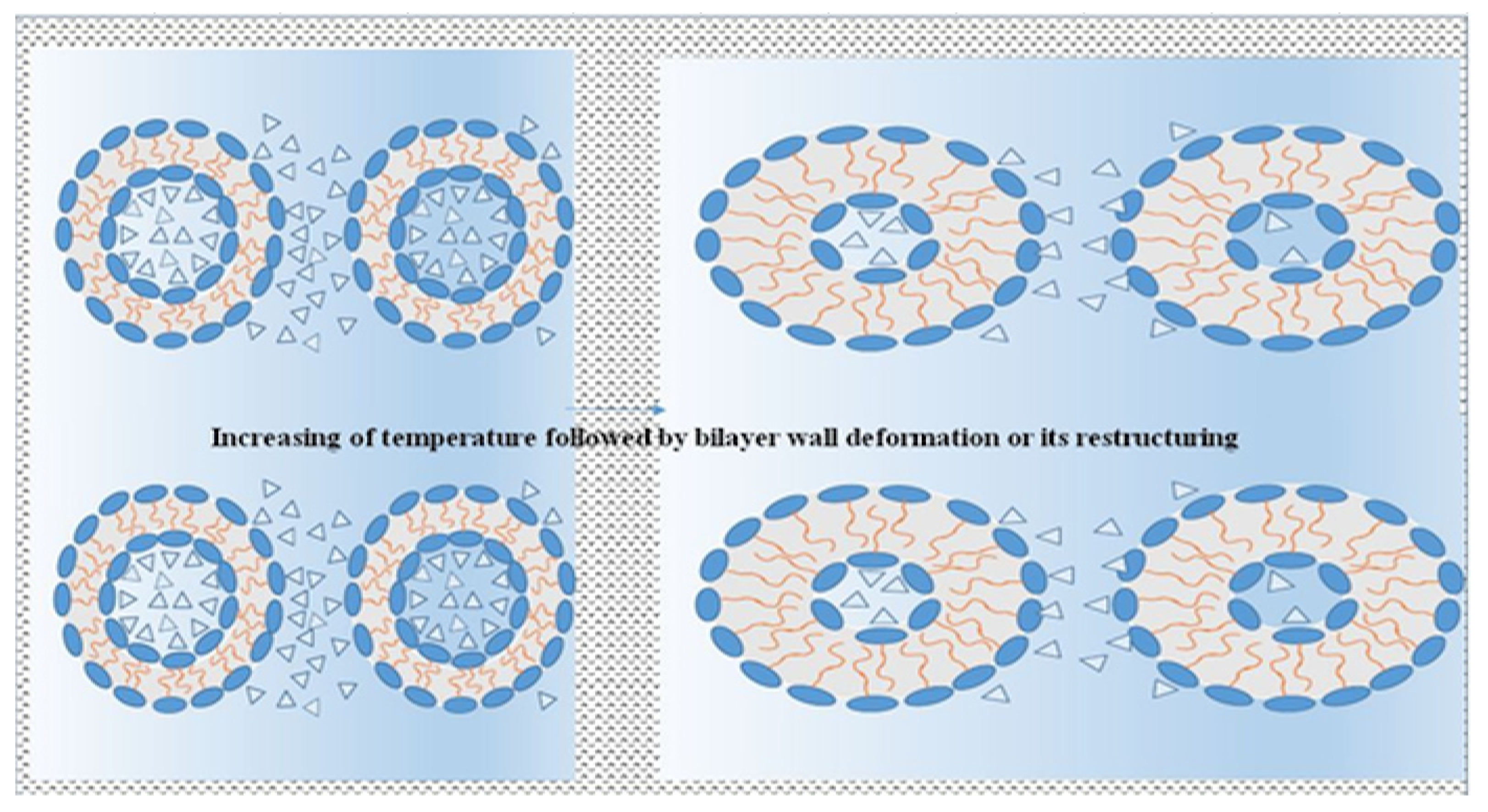
The difference between hydration forces of the outer and the inner interfaces of interacting micro-objects formed by the bilayers evokes subsequent shape deformations of the cross-section from circular to elliptic form (see
Figure 9). However, if the pressure of those hydration forces exceeds the acceptable level given by compactness of a bilayer wall, the micro-objects are squeezed or restructured into a reversed form of phases, i.e., the outer convex into the inner concave part and vice versa. It is important that the restructuring is accompanied by complete disintegration of the bilayer wall. Additionally, it is important that in both cases, the water solution closed in the inner parts of micro-objects is released into outer continuous hydromesophase. If squeezing has taken place only in contrast with mere deformation, the volume of closed water in the middle hollow part of the micro-object is decreased, e.g., from 13 to 10 zeptoL in sphere-shaped micro-objects with diameter, d
o = 10 nm, and the free water solution is released into the outer continuous hydromesophase. Contrariwise, a restructuring of the bilayer wall accompanies an increase in the temperature and an increase in the volume of closed water in the middle hollow part of the micro-object. In more detail, it is possible to study this restructuring process in
Figure 10.
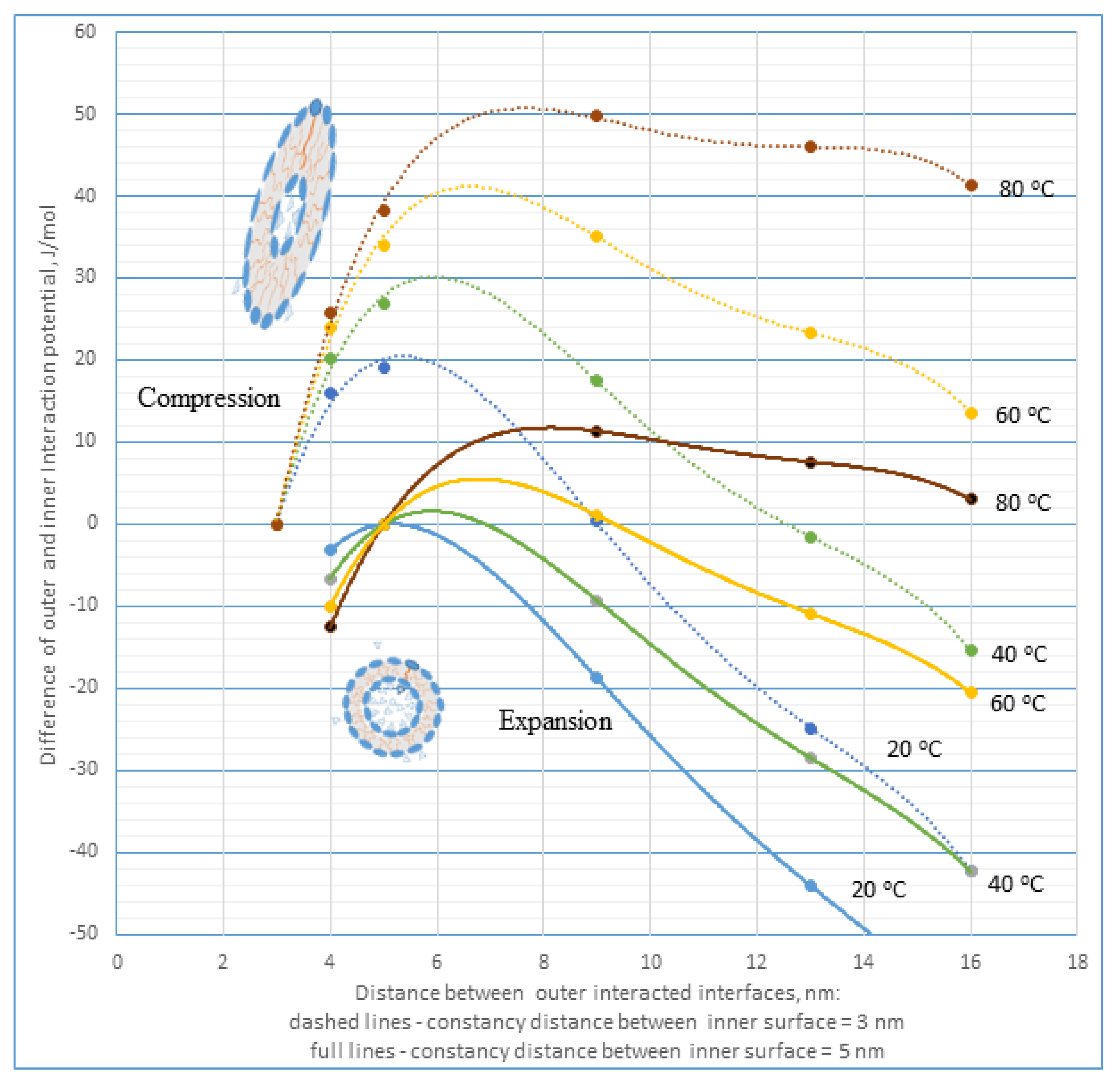
Figure 10 depicts theoretical dependences of the pressure of hydration forces upon the bilayer walls of interacting micro-objects vs. distance at the constant temperature of outer interacting interfaces relative to the constancy distance of inner interface d
o = 3 or 5 nm (see
Appendix B). This pressure is possible to interpret as a difference between the outer and inner interaction potential of hydration forces during mutual interaction of two micro-planes with mosaic-type organization of the hydrated sites, influencing deformation of closed hydromesophase in micro-objects with bilayer walls. The theoretical calculations were derived according to data presented in
Figure 8a. As follows, the pressure of hydration forces impacting closed inner hydromesophase in micro-objects with bilayer walls is dependent upon the distance between interacting interfaces in the outer hydromesophase, i.e., upon a concentration of those systems, and is strongly influenced by the temperature. An increased temperature intensifies this influence. This dependence is non-linear, with maximum at a given distance difference between outer and inner interacting interfaces shifting with increases in the temperature to greater values. For example, the spherical bilayer’s micro-objects with inner diameter d
o = 3 nm and concentration that enables us to achieve distances of their outer interfaces of d = 6 nm are restructured after reaching the temperature ≥ 40 °C. Thus, they reach a new equilibrium reverse state comprised of spherical micro-objects with diameter d
o = 5 nm, i.e., the inner part of the micro-object has increased its volume from 0.35 to 1.64 zeptoL.
3. Polyelectrolyte Bilayers
Systems such as polyelectrolyte multilayer (PEM) assemblies (e.g., planar films or walls of hollow capsules) are interesting, as well being well known to be sensitive to external parameters such as ionic strength and pH [
31,
32,
33,
34], temperature [
35] or humidity [
36]. The PEMs consist of bilayers formed of polyionoactive macromolecule pairs with opposite charge character. Suitable films can be prepared by the layer-by-layer (LbL) method [
37], where polyanions and polycations are alternately adsorbed from aqueous solutions, i.e., a coated wafer or hollow polymeric capsules can be prepared by adsorbing polyelectrolyte multilayers [
38,
39]. In analogy with biological membranes, the uniform lipid bilayers were also deposited on polyelectrolyte multilayers [
40,
41,
42,
43,
44,
45]. Firstly, the PEMs from PSS/PAH poly (styrene sulfonate)/poly (allylamine hydrochloride) and PSS/PDADMAC (PDADMAC: poly (diallyl dimethylammonium chloride) bilayer pairs were studied intensively [
46,
47]. The studies of the multilayers containing strong polyelectrolytes PSS and PDADMAC against weak polyelectrolytes such as PAH show similar or different effects of temperature and the so-called “odd–even effect” [
48] in their behaviour, respectively.
PEMs can be seen as electrostatically cross-linked hydrogel systems, which are highly sensitive to changes in the pH or the ionic strength of the aqueous environment [
49,
50]. Apart from the electrostatic interactions between polyions, non-electrostatic interactions are typical for interactions of weak polyelectrolytes, for which the group charge is strongly dependent on pH. Just those interactions taking place in the aqueous environment and being evoked by water molecules are explainable by the mutual impact of hydration forces [
24]. Microcalorimetric measurements have confirmed that amine groups interactions, predominantly secondary amine groups with cellulosic fibrous slurries, exhibit measurable exothermal or endothermal effects, i.e., the firmness of the mutual bonds was corroborated, and the mechanism of their formation was identified. This type of interactions is due to the mechanism of releasing water molecules from active centres on the cellulose substrate and the formation of stronger H-bonds of the “nitrogen–hydrogen” type with amine groups of amine substances, instead of the weaker “oxygen–hydrogen” type with water. Those interactions are accompanied by formation of an irreversible H-bond between the –OH group of cellulosic materials and the –NH
2 group of the polyamine [
23,
51]. In this context, the hydration bond–de-bonding concept serves as an intermediator of those activities.
Temperature-sensitive microcapsules with walls consisting of PAH/PSS or PDADMAC/PSS bilayers irreversibly shrink upon heating from 4.5 to 1.3 μm with a concomitant increase in the shell thickness [
52,
53]. The results show a partial dehydration of the multilayers during the temperature-induced rearrangement. Additionally, the water content of the capsule walls is much higher than that of the same material prepared on planar interfaces (42–56% water). This indicates a more loosely packed structure in the capsule wall than at the planar substrate, and a strong effect of the solid substrate on the PEM structure [
46]. In the case of strong polyelectrolytes, an influence of increased temperature is explained by the weakening of extrinsic charge compensation of polyionic polymers with counterions coming from added salt, followed by increasing intrinsic charge compensation due to electrostatic forces among interacting strong polyionoactive macromolecule pairs [
50]. Similar behaviour of weak polyelectrolytes’ hydrophilic macromolecules, inclusive in the PEMs, is explainable only in terms of the increase in the hydration attraction among interacting nano-domains of macromolecules with an increase in temperature according to the abovementioned mechanism—see chapter “2.2. Temperature influence …” and also
Figure A1 or
Figure 8a (for the distance of interacting domains < 3 nm).
The alternating deposition of polycations and polyanions commonly varies the surface potential of the outermost layer, terminating the assembly between positive and negative values. A few multilayer properties, reversible variations with the sign of charge of the terminating layer, were observed, which became evident as ‘odd–even’ effects [
48] in dependence on the number of single layers; n. Very pronounced odd–even effects were found in the mobility of the hydration water in multilayers [
38]. It was thus concluded that the odd–even effect is not controlled by the hydration properties of the terminating layer itself, but it is the sign of the surface potential that induces changes in the water mobility in the internal layers. The adsorption of PAH to multilayers was accompanied by a large decrease in the net water mobility [
54]. Upon adsorption of a PAH layer, the water mobility decreases, whereas upon adsorption of a PSS layer, the water mobility increases again. Interestingly, this odd–even effect was observed for PAH/PSS, but not for PSS/PDADMAC internal layers [
38,
54]. This means that the water content of a PEM depends on the composition of the outermost layer. At high RH (98%), PSS/PAH multilayers with PSS as outermost layer are thicker than the ones with PAH in the terminated layer [
36]. In cases in which PSS is the outermost layer, a potential with an exponential decay towards the inner part of the multilayer has been monitored, but not for PAH-terminated PEM. It has not been clarified why the potential in case of PAH terminated multilayers remains constant within the multilayer [
55]. The thickness of multilayers in the water swollen state depends on the outermost layer and increases in a zigzag shape. After the deposition of PSS, the film swells and PAH adsorption leads to shrinking. The decrease in thickness upon PAH adsorption indicated that water is pressed out of the multilayer when PAH is adsorbed, while again, more water diffuses into the multilayer upon adsorption of the next PSS layer [
33]. Using a noticeably different interaction mechanism among polyionic polymers (typical electrostatic a charge neutralization mechanism) and among entirely hydrophilic macromolecules (typical interaction due to hydration bonding system according to the SCHL theory of the hydration forces emergence [
28]), it is possible to clarify why the potential remains constant within the multilayer in case of PAH [
55]. Logically, in comparable conditions, osmotic pressure accountable for swelling of PEMs is comparatively higher in the ionic than in the non-ionic systems.
Similar to bilayers are the more isotropic structures of ionic liquids (ILs), predominantly comprised of water containing clusters so-called biobased ILs serving as attractive candidates for effective dissolution and fractionation of lignin during the pretreatment of biomass. Bio-based solvents, such as choline amino acid ionic liquids, e.g., choline lysinate, are typical [
56,
57]. New experiments [
56] using neutron diffraction confirm that water forms oriented own finite domains inside clusters, which are contiguous and connect by way of H-bonds to the lysinate [Lys] anion and to choline [Ch] hydroxyl groups, i.e., the hydration intermolecular bonding system among water molecules creates. Overall, in the [Ch][Lys]–H
2O system, water molecules (up to 26.5% w/w) are accommodated into the IL H-bonding network without significantly altering its local structure, allowing the amphiphilic nanostructure to be preserved [
56].
4. Confinement Bilayers Processes
What happens in the bilayer if water disappears in any hydromesophase, e.g., via evaporation during drying, expelling or superseding, etc., or if foreign nano-objects, e.g., molecules or colloidal particles, etc., intrude and disrupt the bilayer?
Different activities stepwise eliminating water molecules from outer and inner hydromesophase disturbs the reversibility of bilayers system behaviour, which is thus irreversible. Often, these ones are destroyed. As a rule, the water molecules are more easily eliminated from outer than inner hydromesophase. A porous system is originating because of the prevailing strong bonding hydration system followed by formation of H-bonding or an irreversible chemical bonding system between interacting hydrophilic and polar groups of outer interfaces of bilayers. The porous system of bigger or smaller pores forms if, in inner hydromesophase, there is prevailing action of repulsive or attractive hydration forces. Practically, these processes are realized, for instance, by evaporation of water from droplets containing a structuralized bilayers matter, e.g., bilayers of lipid walls of bacteria or viruses, etc. A further possibility is expelling outer hydromesophase water by action of capillary forces of highly hydrophilic and hygroscopic porose material, e.g., by use of a cellulosic product such as tissue paper. An inspiring challenge suggests focusing attention upon superseding of water molecules from inner hydromesophase vacancies of bilayers structures by use of some molecules of essentials oils with bactericidal, fungicidal, and even sporicidal activities [
58,
59].
Certainly, molecules, particles, etc., of foreign substances contained in hydromesophases significantly influence the behaviour of bilayers, inclusive of their disruption. While soluble ionic or non-ionic molecules or colloidal particles deteriorate or contrarily increase their hydration activities, the amphiphilic substances with adhesion comparable to or better than cohesion of the bilayer’s molecules incorporate among those and with different selectivity potentially disrupt these ones. Therefore, a great deal of attention has focused on the types of self-assembled aggregates that may be responsible for destroying the integrity of bacterial and fungal cell membranes. Recently, two mechanistic hypotheses have been presented for the disruption of microbial cell membranes [
60,
61,
62]:
A “barrel-stave” model where amphiphilic peptides in the bilayer form a single pore or two aligned water-filled pores.
A “carpet” model in which a series of disruptive molecules is thought to aggregate on the surface of target lipid bilayers (e.g., cholesterol-rich liposomes made from 1-palmitoyl, 2-oleoyl-sn-glycero-3-phosphocholine/cholesterol), insert into the bilayer, and create defects or pores along with mixed-micelle-like structures.
It should be noted, in this regard, that the membrane-disrupting molecules in their monomeric state can have very different selectivity features compared with aggregated forms, i.e., monomers were found to promote leakage processes, while aggregates favoured the catastrophic rupture of the membrane [
63,
64]. For instance, it was shown that for a series of simple quaternary ammonium compounds derived from L-phenylalanine, significant antibacterial activity was observed in the monomer state, while aggregation resulted in both haemolytic as well as the antibacterial activity. It should also be mentioned that membrane potential is another important factor for the selective destruction of bacteria, where the potential across the inner membrane is significantly higher than that across the plasma membrane of mammalian cells [
65,
66].
Recently, SARS, MERS, and most recently COVID-19 have emerged from novel Coves, and all three have resulted in varying degrees of mortality in humans. To date, several viral proteins have been identified as potential targets for SARS-CoV-2. However, little attention has been paid to the virus’ lipid envelope as a possible bilayer target. One possible mechanism for antiviral action in such a case would involve changes in the lateral pressure in the lipid envelope and changes in conformation and/or the lateral organization of membrane proteins that are necessary for fusion with mammalian cells [
67]. In this context, it was presented that certain surfactants can destroy the membrane integrity of liposomes with relatively low cholesterol concentrations [
68]. Results indicated the COVID-19 virus’ size is about 100 nm, but after achieving of a spherical water droplet, it may be larger. The total average size distribution of the droplet nuclei was 0.58–5.42 μm, and 82% of the droplet nuclei was centred in 0.74–2.12 μm. The entire average size distribution of the coughed droplets was 0.62–15.9 μm, and the average mode size was 8.35 μm. When a contagious person is breathing normally, they exhale about 5 L of air per min, with 60% of water droplets under 1 μm and a concentration of more than 10,000 droplets/cm
3, at a total water mass concentration of water of 30 μg/cm
3, the equivalent of 100% humidity at 310 K. This mass increases when one is sneezing and coughing, and the water-spit droplet gets the peak at about 8 μm, which is easy to stop with a filter [
69]. In this context, a water evaporation/condensation process in droplets is important.
As we know, the evaporation/condensation processes being controlled by the RH of the air environment and by a vapor’s tension around water droplets relative to this one around water’s flat interface are given by the size and shape of water in the droplet’s interface. At constant the temperature, if the vapor’s tension is not equilibrated with the air RH, water is evaporated, leading to higher tension or, if condensed, a lower tension of the water vapor is achieved. Obviously (see
Figure A2 and
Appendix C), in comparable conditions, water of the outer convex shell will be given preference to evaporate in contrast with concave inner water of a corona particle, but after the full elimination of the outer water shell only. In this situation, the vapor molecules penetrate the bare virus particle, condensing inside. In connection with the increase in its volume, destruction follows. As demonstrated in
Figure 11, all these phenomena are strongly dependent upon the size of water droplets, i.e., with increases in the size of droplets, the relative pressure, p
rel, forcing water molecules to evaporate (convex interface) or to condensate (concave interface) non-linearly diminishes. An increase in temperature depresses those influences as well, but to a lesser extent. Additionally, one theory (see
Appendix C) predicts that with a decrease in air RH, the relative pressure, p
rel, further increases or diminishes in cases of convex or concave surfaces, respectively.
We should also note certain behaviour of the trilayer, i.e., lipophilic tail–hydrophilic head group of the first hydrophilic head group–lipophilic tail of the second, the lipophilic tail–hydrophilic head group of the third amphiphilic molecule, etc., created on lipophilic and hydrophilic interface of liquid or liquid–gaseous phases. By evaporating water from droplets containing bilayer structures, a favourable trilayer shell is likely to form around bilayer structures containing aqueous domains, e.g., spores.
5. Conclusions
The miscellaneous complexity of a bilayer’s aqueous systems is studied in more detail. An attempt has been made to describe the bilayers’ behaviour in a water environment with the help of a simple theoretical model of interaction of planes with regular mosaic arrangement of repulsive and attractive hydration forces around hydrophilic nano-sites composing interfaces of the bilayers.
It was shown that the behaviour of structuralized concentrated liquid aqueous systems created by micro-objects with bilayer walls, e.g., LC systems, has sophisticated character due to the action of hydration forces taking place in bottom-up mode. The theoretical model of bilayers enables us to better understand the behaviour of the amphiphilic molecules comprised of hydrotropic (lyotropic) systems in that complicated state.
It was shown that temperature strongly influences the orientation of polar water molecules among interacting interfaces of bilayers, being controlled right here by composition and structure of end groups of amphiphilic molecules. Due to this complication, a temperature influence upon bilayers systems is more complex, but explainable with the abovementioned theoretical tool. An attempt was made to better understand the behaviour of LCs systems with increasing volume of the hydromesophase.
The PEMs comprise a further interesting sort of bilayers consisting of bilayers formed by pairs of polyionoactive macromolecules with opposite charge characteristics. Nevertheless, the behaviour of the multilayers containing strong polyelectrolytes is possible to explain using the classical theory of electrostatic charge neutralization, but the behaviour of bilayers in PEMs consisting of weak polyelectrolytes cannot be explained by this theory. Just those interactions taking place in the aqueous environment that have been evoked by water molecules are explainable in terms of the mutual impact of hydration forces.
The confinement processes have also been acknowledged as being connected with disruption such as destruction of bilayers, i.e., the processes of greatest importance in combat with bacteria, fungi, and viruses. As documented above, a breathed-out puff of micro-droplets containing virus nanoparticles (e.g., COVID-19 virus) should be cleaned in three steps. Firstly, at higher flow, the filter fibres break the droplets, acting as an atomizer, and make them smaller than 1 μm. Secondly, water from outer convex interface shells of smaller droplets is more easily evaporated, followed by uncovering of virus’ nanoparticles (about 100 nm). Thirdly, due to the high RH of the atmosphere, water vapor condensates in the inner part of the bare nanoparticles and destroys them. Obviously, a safety mask, respirator, or filter should be designed which contains an exchangeable coarse hydrophilic filter of soft weak-elastic character with high adhesion to water drops, e.g., tissue paper, followed by a small free chamber. Finally, it should contain an outer high-efficiency supporting filter shell composed of nanofibers.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












