
Native Protein Template Assisted Synthesis of Non-Native Metal-Sulfur Clusters


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Covalently vs. Non-Covalently Coordinated Metal-Cofactors
3. Iron–Sulfur Proteins
3.1. Overview of Rubredoxin
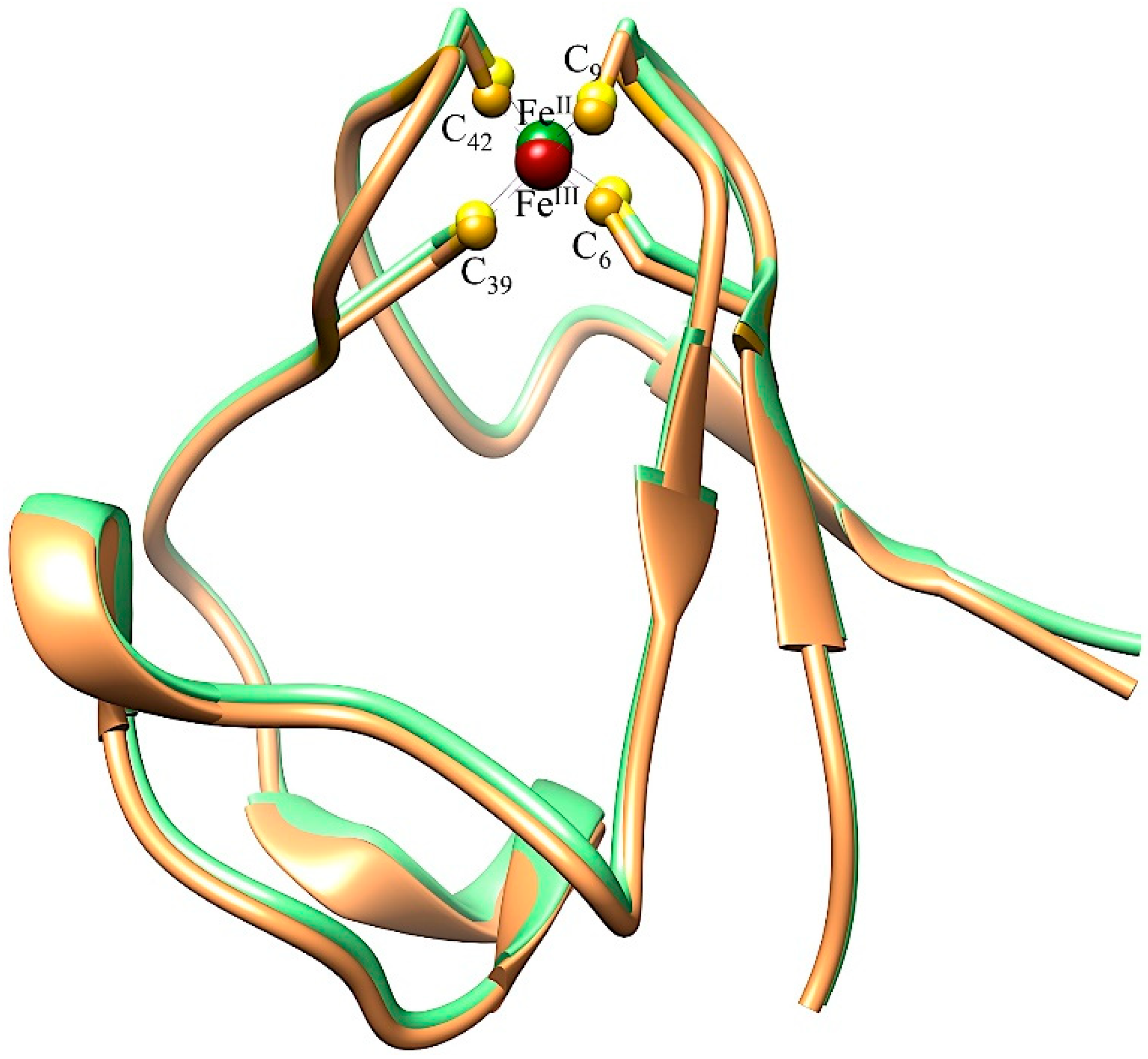
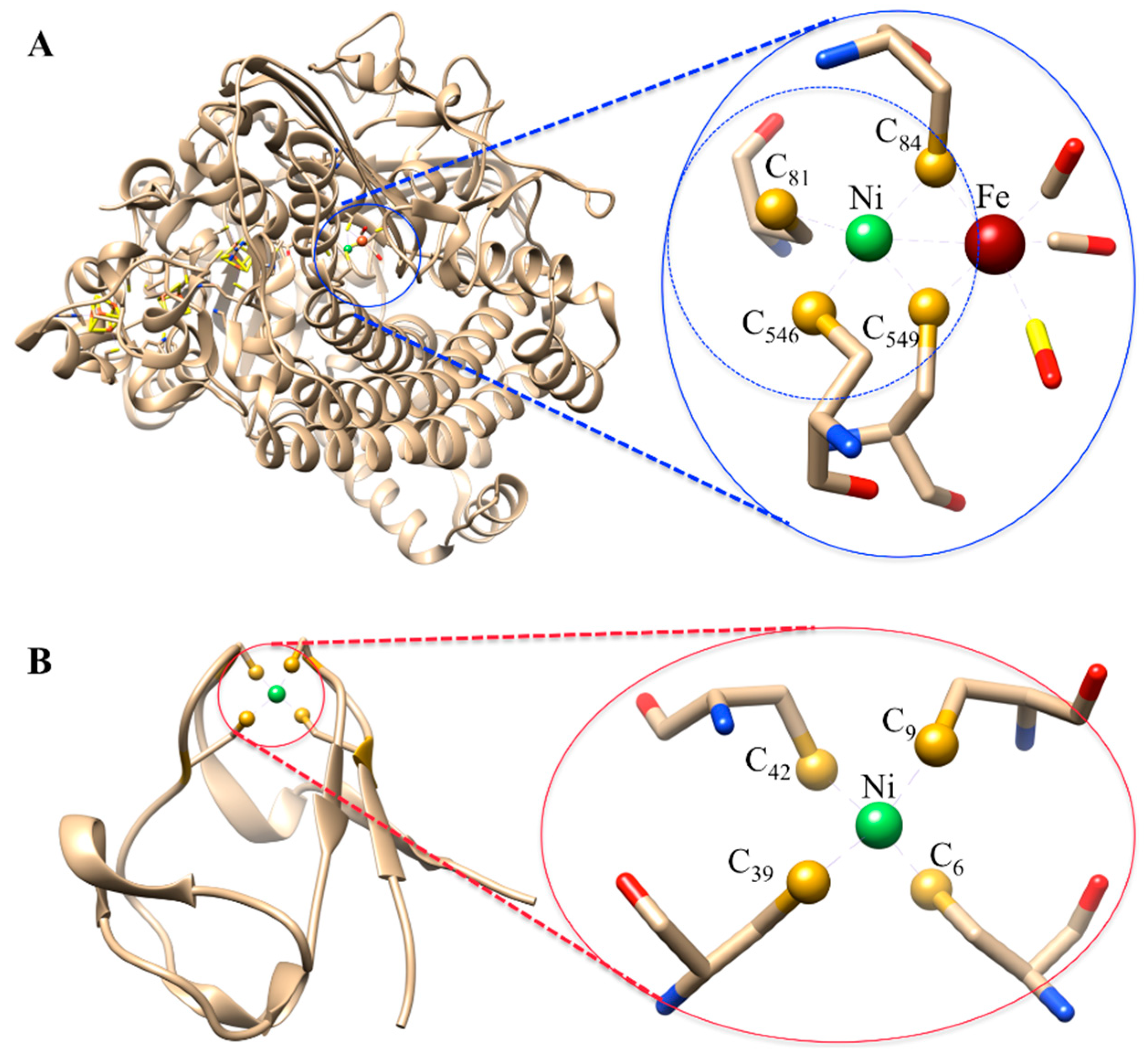
3.1.1. Ni-Substituted Rd: Model of [Ni-Fe]-Hydrogenase
3.1.2. Spectroscopic Probes-M-Rd
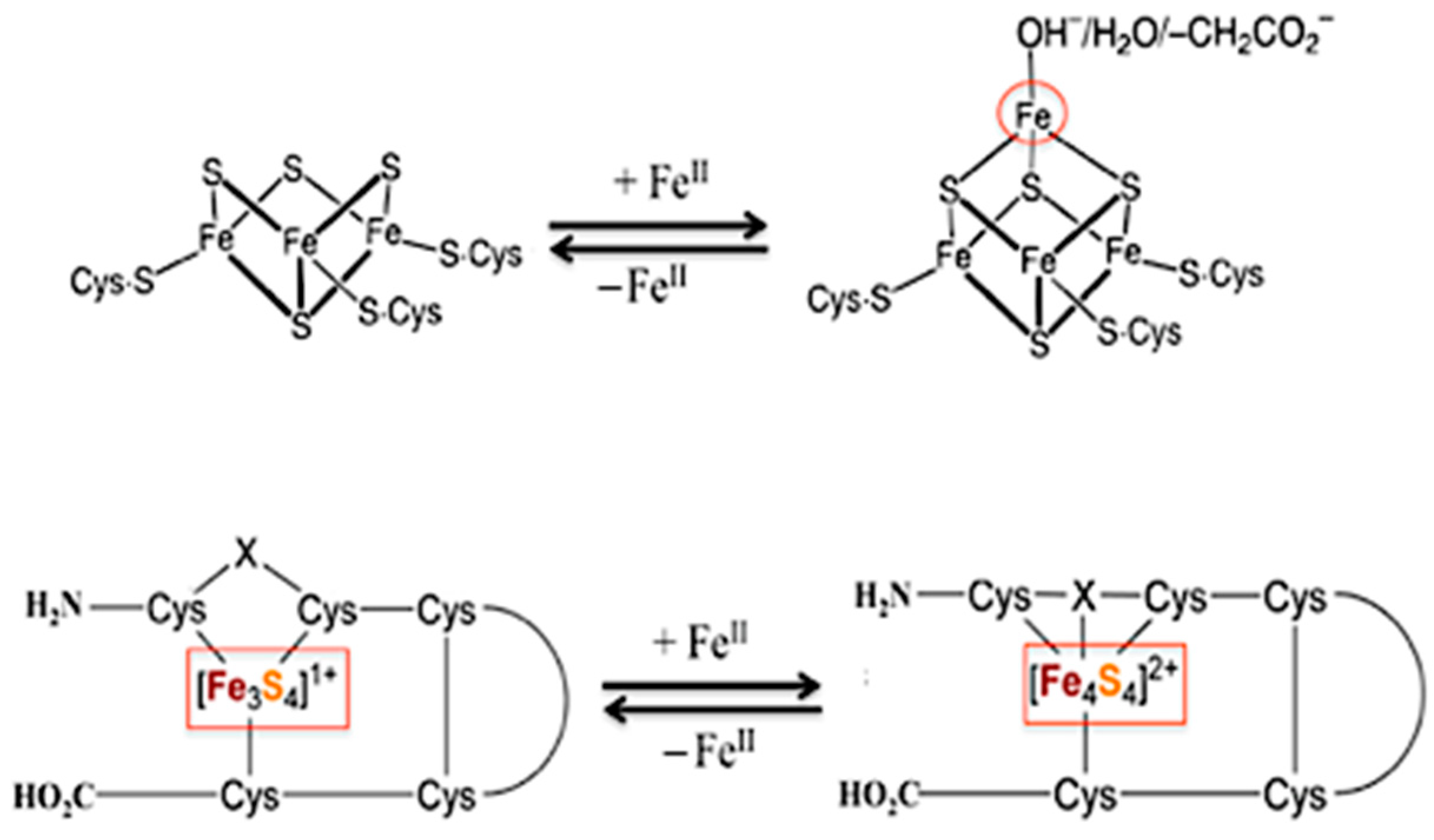
3.2. Overview of [3Fe-4S] Ferredoxin
3.2.1. Ni-Incorporated in [3Fe-4S]-Fd: Model of Ni-Containing CODH
3.2.2. Spectroscopic Probes-[M,3Fe-4S] Ferredoxin
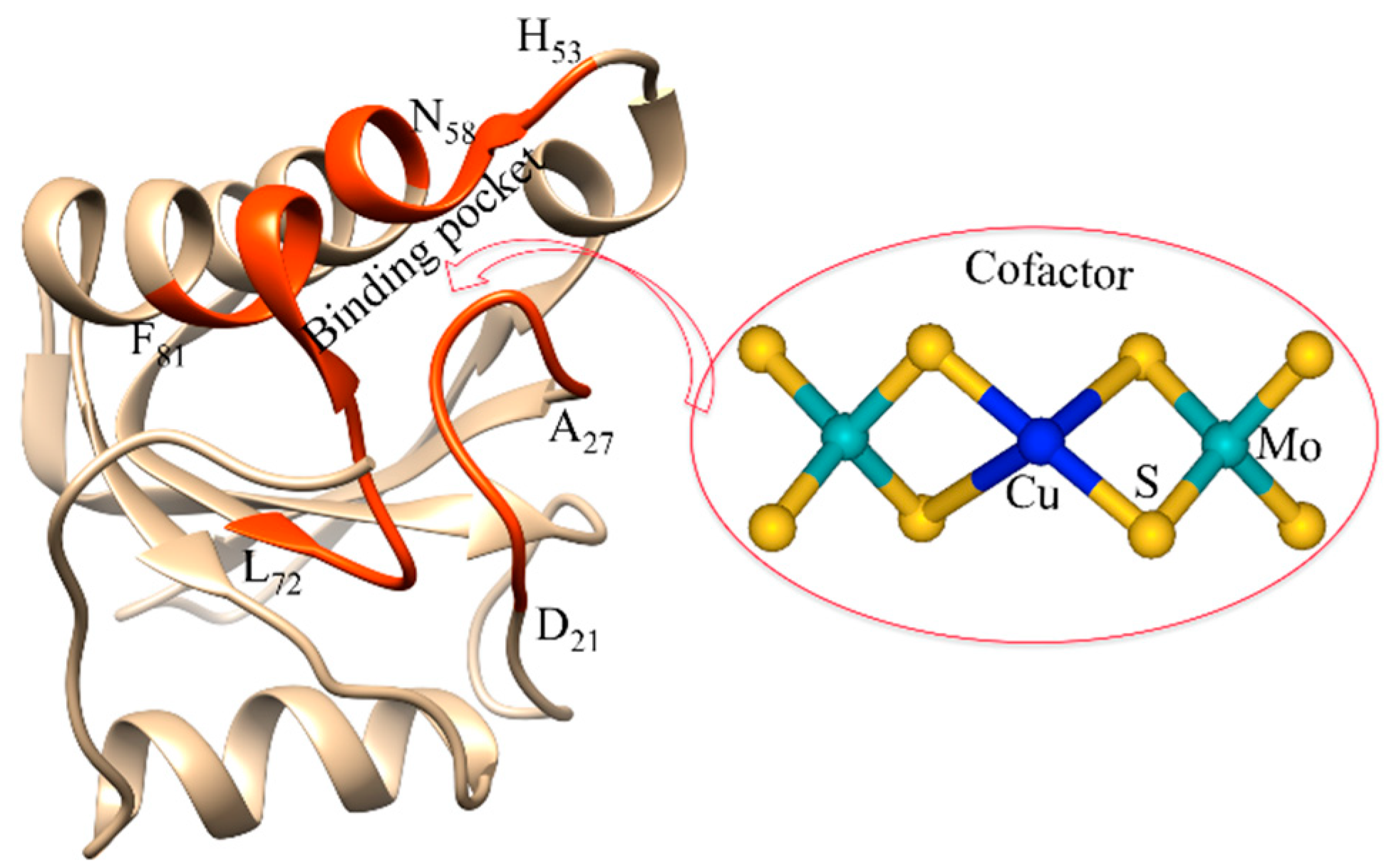
4. Overview of Orange Proteins
Spectroscopic Probes ORP
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Rd | Rubredoxin |
| ORP | Orange Protein |
| Fd | Ferredoxin |
| CODH | Carbon monoxide dehydrogenase |
| cyt-c3 | cytochrome c3 |
| Pf | Pyrococcus furiosus |
| Dg | Desulfovibrio gigas |
| Dd | Desulfovibrio desulfuricans |
| Ch | Carboxydothermus hydrogenoformans |
| Rr | Rhodospirillum rubrum |
| Mt | Moorella thermoacetica |
| TTM | Tetrathiomolybdate |
| EXAFS | Extended X-ray absorption fine structure |
| PDB | Protein data bank |
| ET | Electron transfer |
| NHE | Normal hydrogen electrode |
| NMR | Nuclear magnetic resonance |
References
- Ragsdale, S.W. Metals and Their Scaffolds to Promote Difficult Enzymatic Reactions. Chem. Rev. 2006, 106, 3317–3337. [Google Scholar] [CrossRef] [PubMed]
- Nastri, F.; D’Alonzo, D.; Leone, L.; Zambrano, G.; Pavone, V.; Lombardi, A. Engineering Metalloprotein Functions in Designed and Native Scaffolds. Trends Biochem. Sci. 2019, 44, 1022–1040. [Google Scholar] [CrossRef] [PubMed]
- Bell, E.L.; Finnigan, W.; France, S.P.; Green, A.P.; Hayes, M.A.; Hepworth, L.J.; Lovelock, S.L.; Niikura, H.; Osuna, S.; Romero, E.; et al. Biocatalysis. Nat. Rev. Methods Primers 2021, 1, 46. [Google Scholar] [CrossRef]
- Liu, J.; Chakraborty, S.; Hosseinzadeh, P.; Yu, Y.; Tian, S.; Petrik, I.; Ambika Bhagi, A.; Lu, Y. Metalloproteins Containing Cytochrome, Iron–Sulfur, or Copper Redox Centers. Chem. Rev. 2014, 114, 4366–4469. [Google Scholar] [CrossRef]
- Valdez, C.E.; Smith, Q.A.; Nechay, M.R.; Alexandrova, A.N. Mysteries of Metals in Metalloenzymes. Acc. Chem. Res. 2014, 47, 3110–3117. [Google Scholar] [CrossRef] [PubMed]
- Mirts, E.N.; Bhagi-Damodaran, A.; Lu, Y. Understanding and Modulating Metalloenzymes with Unnatural Amino Acids, Non-Native Metal Ions, and Non-Native Metallocofactors. Acc. Chem. Res. 2019, 52, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Maiti, B.K.; Maia, L.B.; Moura, J.J.G. Sulfide and transition metals-A partnership for life. J. Inorg. Biochem. 2022, 227, 111687. [Google Scholar] [CrossRef]
- Solomon, E.I.; Heppner, D.E.; Johnston, E.M.; Ginsbach, J.W.; Cirera, J.; Qayyum, M.; Kieber-Emmons, M.T.; Kjaergaard, C.H.; Hadt, R.G.; Tian, L. Copper Active Sites in Biology. Chem. Rev. 2014, 114, 3659–3853. [Google Scholar] [CrossRef] [Green Version]
- Maiti, B.K.; Almeida, R.M.; Moura, I.; Moura, J.J.G. Rubredoxins derivatives: Simple sulphur-rich coordination metal sites and its relevance for biology and chemistry. Coord. Chem. Rev. 2017, 352, 379–397. [Google Scholar] [CrossRef]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and Chemistry of Cytochrome P450. Chem. Rev. 2005, 105, 2253–2278. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Yuan, G.; Yuan, T.; Zeng, H.; Zou, Z.-R.; Tu, Z.-C.; Gao, J.; Zou, Y. Set of Cytochrome P450s Cooperatively Catalyzes the Synthesis of a Highly Oxidized and Rearranged Diterpene-Class Sordarinane Architecture. J. Am. Chem. Soc. 2022, 144, 3580–3589. [Google Scholar] [CrossRef]
- Hille, R.; Hall, J.; Basu, P. The Mononuclear Molybdenum Enzymes. Chem. Rev. 2014, 114, 3963–4038. [Google Scholar] [CrossRef] [Green Version]
- Einsle, O.; Rees, D.C. Structural Enzymology of Nitrogenase Enzymes. Chem. Rev. 2020, 120, 4969–5004. [Google Scholar] [CrossRef] [PubMed]
- Seefeldt, L.C.; Yang, Z.-Y.; Lukoyanov, D.A.; Harris, D.F.; Dean, D.R.; Raugei, S.; Hoffman, B.M. Reduction of Substrates by Nitrogenases. Chem. Rev. 2020, 120, 5082–5106. [Google Scholar] [CrossRef]
- McEvoy, J.P.; Brudvig, G.W. Water-splitting chemistry of photosystem II. Chem. Rev. 2006, 106, 4455–4483. [Google Scholar] [CrossRef] [PubMed]
- Marchiori, D.A.; Debus, R.J.; Britt, R.D. Pulse EPR Spectroscopic Characterization of the S 3 State of the Oxygen-Evolving Complex of Photosystem II Isolated from Synechocystis. Biochemistry 2020, 59, 4864–4872. [Google Scholar] [CrossRef] [PubMed]
- Lubitz, W.; Ogata, H.; Rüdiger, O.; Reijerse, E. Hydrogenases. Chem. Rev. 2014, 114, 4081–4148. [Google Scholar] [CrossRef]
- Tai, H.; Hirota, S.; Stripp, S.T. Proton Transfer Mechanisms in Bimetallic Hydrogenases. Acc. Chem. Res. 2021, 54, 232–241. [Google Scholar] [CrossRef]
- Cohen, S.E.; Can, M.; Wittenborn, E.C.; Hendrickson, R.A.; Ragsdale, S.W.; Drennan, C.L. Crystallographic Characterization of the Carbonylated A-Cluster in Carbon Monoxide Dehydrogenase/Acetyl-CoA Synthase. ACS Catal. 2020, 10, 9741–9746. [Google Scholar] [CrossRef]
- Reginald, S.S.; Etzerodt, M.; Fapyane, D.; Chang, I.S. Functional Expression of a Mo–Cu-Dependent Carbon Monoxide Dehydrogenase (CODH) and Its Use as a Dissolved CO Bio-microsensor. ACS Sens. 2021, 6, 2772–2782. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskas, R.J. Enhancing catalytic promiscuity for biocatalysis. Curr. Opin. Chem. Biol. 2005, 9, 195–201. [Google Scholar] [CrossRef] [PubMed]
- Khersonsky, O.; Roodveldt, C.; Tawfik, D.S. Enzyme promiscuity: Evolutionary and mechanistic aspects. Curr. Opin. Chem. Biol. 2006, 10, 498–508. [Google Scholar] [CrossRef] [PubMed]
- Toscano, M.D.; Woycechowsky, K.J.; Hilvert, D. Minimalist active-site redesign: Teaching old enzymes new tricks. Angew. Chem. Int. Ed. 2007, 46, 3212–3236. [Google Scholar] [CrossRef]
- Fernandez-Gacio, A.; Codina, A.; Fastrez, J.; Riant, O.; Soumillion, P. Transforming Carbonic Anhydrase into Epoxide Synthase by Metal Exchange. ChemBioChem 2006, 7, 1013–1016. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Berry, S.M.; Pfister, T.D. Engineering Novel Metalloproteins: Design of Metal-Binding Sites into Native Protein Scaffolds. Chem. Rev. 2001, 101, 3047–3080. [Google Scholar] [CrossRef]
- Yu, F.; Cangelosi, V.M.; Zastrow, M.L.; Tegoni, M.; Plegaria, J.S.; Tebo, A.G.; Mocny, C.S.; Ruckthong, L.; Qayyum, H.; Pecoraro, V.L. Protein Design: Toward Functional Metalloenzymes. Chem. Rev. 2014, 114, 3495–3578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beinert, H.; Kennedy, M.C.; Stout, C.D. Aconitase as Ironminus signSulfur Protein, Enzyme, and Iron-Regulatory Protein. Chem. Rev. 1996, 96, 2335–2374. [Google Scholar] [CrossRef] [PubMed]
- Holm, R.H.; Lo, W. Structural Conversions of Synthetic and Protein-Bound Iron–Sulfur Clusters. Chem. Rev. 2016, 116, 13685–13713. [Google Scholar] [CrossRef] [PubMed]
- George, G.N.; Pickering, I.J.; Yu, E.Y.; Prince, R.C.; Bursakov, S.A.; Gavel, O.Y.; Moura, I.; Moura, J.J.G. A Novel Protein-Bound Copper–Molybdenum Cluster. J. Am. Chem. Soc. 2000, 122, 8321–8322. [Google Scholar] [CrossRef]
- Maiti, B.K.; Almedia, R.; Maia, L.B.; Moura, I.; Moura, J.J.G. Insights into the Molybdenum/Copper Heterometallic Cluster Assembly in the Orange Protein: Probing Intermolecular Interactions with an Artificial Metal-Binding ATCUN Tag. Inorg Chem. 2017, 56, 8900–8911. [Google Scholar] [CrossRef]
- Coleman, J.E. Cadmium-113 nuclear magnetic resonance applied to metalloproteins. Methods Enzymol. 1993, 227, 16–43. [Google Scholar] [PubMed]
- Maret, W.; Vallee, B.L. Cobalt as probe and label of proteins. Methods Enzymol. 1993, 226, 52–71. [Google Scholar]
- Münck, E.; Ksurerus, K.; Hendrich, M.P. Combining Mossbauer spectroscopy with integer spin electron paramagnetic resonance. Methods Enzymol. 1993, 227, 463–479. [Google Scholar] [PubMed]
- Moura, J.J.; Macedo, A.L.; Nuno Palma, P. Ferredoxins. Methods Enzymol. 1994, 243, 165–188. [Google Scholar] [PubMed]
- Butt, J.N.; Fawcett, S.E.J.; Breton, J.; Thomson, J.A.; Armstrong, F.A. Electrochemical Potential and pH Dependences of [3Fe-4S] ↔ [M3Fe-4S] Cluster Transformations (M = Fe, Zn, Co, and Cd) in Ferredoxin III from Desulfovibrio africanus and Detection of a Cluster with M = Pb. J. Am. Chem. Soc. 1997, 119, 9729–9737. [Google Scholar] [CrossRef]
- Conover, R.C.; Park, J.B.; Adams, M.W.W.; Johnson, M.K. Formation and properties of an iron-nickel sulfide (NiFe3S4) cluster in Pyrococcus furiosus ferredoxin. J. Am. Chem. Soc. 1990, 112, 4562–4564. [Google Scholar] [CrossRef]
- Slater, J.W.; Marguet, S.C.; Monaco, H.A.; Shafaat, H.S. Going beyond Structure: Nickel-Substituted Rubredoxin as a Mechanistic Model for the [NiFe] Hydrogenases. J. Am. Chem. Soc. 2018, 140, 10250–10262. [Google Scholar] [CrossRef]
- Saint-Martin, P.; Lespinat, P.A.; Fauque, G.; Berlier, Y.; Legall, J.; Moura, I.; Teixeira, M.; Xavier, A.V.; Moura, J.J. Hydrogen production and deuterium-proton exchange reactions catalyzed by Desulfovibrio nickel(II)-substituted rubredoxins. Proc. Natl. Acad. Sci. USA 1988, 85, 9378–9380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, L.C.; Shafaat, H.S. Reversible Electron Transfer and Substrate Binding Support [NiFe3S4] Ferredoxin as a Protein-Based Model for [NiFe] Carbon Monoxide Dehydrogenase. Inorg. Chem. 2021, 60, 13869–13875. [Google Scholar] [CrossRef]
- Maiti, B.K.; Maia, L.B.; Pauleta, S.R.; Moura, I.; Moura, J.J.G. Protein-Assisted Formation of Molybdenum Heterometallic Clusters: Evidence for the Formation of S2MoS2–M–S2MoS2 Clusters with M = Fe, Co, Ni, Cu, or Cd within the Orange Protein. Inorg. Chem. 2017, 56, 2210–2220. [Google Scholar] [CrossRef]
- Rivas, M.G.; Carepo, M.S.P.; Mota, C.S.; Korbas, M.; Durand, M.-C.; Lopes, A.T.; Brondino, C.D.; Pereira, A.S.; George, G.N.; Dolla, A.; et al. Molybdenum induces the expression of a protein containing a new heterometallic Mo-Fe cluster in Desulfovibrio alaskensis. Biochemistry 2009, 48, 873–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiti, B.K.; Pal, K.; Sarkar, S. Plasticity in [(R4–xR1x)4N]4[Cu4{S2C2(CN)2}4] (x = 0–4) is Molded by a Guest Cation on an Elastic Anionic Host. Eur. J. Inorg. Chem. 2008, 2008, 2407–2420. [Google Scholar] [CrossRef]
- Lehn, J.-M. Supramolecular Chemistry: Concepts and Perspectives; VCH: Weinheim, Germany, 1995. [Google Scholar]
- Spiro, T.G. Iron-Sulfur Proteins: Metal Ions in Biology; Thomas, G., Spiro, Eds.; Wiley-Interscience: New York, NY, USA, 1982; Volume IV. [Google Scholar]
- Sofia, R.; Pauleta, S.R.; Grazina, R.; Carepo, M.S.P.; Moura, J.J.M.; Moura, I. Iron-Sulfur Clusters–Functions of an Ancient Metal Site, Comprehensive Inorganic Chemistry III from Biology to Nanotechnology. In Bioinorganic Chemistry and Homogeneous Biomimetic Inorganic Catalysis; Pecoraro, V., Guo, Z., Eds.; Elsevier: Amsterdam, The Netherlands, 2022; Volume 2, ISBN 9780081026885. in press. [Google Scholar]
- Sykes, A.G.; Cammack, R. Iron-Sulfur Proteins. In Advances in Inorganic Chemistry; Academic Press: San Diego, CA, USA, 1999; Volume 47. [Google Scholar]
- Maiti, B.K.; Moura, I.; Moura, J.J.G.; Pauleta, S.R. The small iron-sulfur protein from the ORP operon binds a [2Fe-2S] cluster. Biochim. Biophys. Acta (BBA)–Bioenerg. 2016, 1857, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Tavares, P.; Pereira, A.S.; Krebs, C.; Ravi, N.; Moura, J.J.; Moura, I.; Huynh, B.H. Spectroscopic characterization of a novel tetranuclear Fe cluster in an iron-sulfur protein isolated from Desulfovibrio desulfuricans. Biochemistry 1998, 37, 2830–2842. [Google Scholar] [CrossRef]
- Maiti, B.K. Cross-talk Between (Hydrogen)Sulfite and Metalloproteins: Impact on Human Health. Chem.-A Eur. J. 2022, 28, e202104342. [Google Scholar] [CrossRef]
- Reed, C.J.; Lam, Q.N.; Mirts, E.N.; Lu, Y. Molecular understanding of heteronuclear active sites in heme–copper oxidases, nitric oxide reductases, and sulfite reductases through biomimetic modelling. Chem. Soc. Rev. 2021, 50, 2486–2539. [Google Scholar] [CrossRef]
- Moura, I.; Pereira, A.S.; Tavares, P.; Moura, J.J.G. Simple and Complex Iron-Sulfur Proteins in Sulfate Reducing Bacteria. Adv. Inorg. Chem. 1999, 47, 361–419. [Google Scholar]
- Jeoung, J.-H.; Martins, B.M.; Dobbek, H. Double-Cubane [8Fe9S] Clusters: A Novel Nitrogenase-Related Cofactor in Biology. Chembiochem 2020, 21, 1710–1716. [Google Scholar] [CrossRef]
- Del Barrio, M.; Sensi, M.; Fradale, L.; Bruschi, M.; Greco, C.; de Gioia, L.; Bertini, L.; Fourmond, V.; Léger, C. Interaction of the H-Cluster of FeFe Hydrogenase with Halides. J. Am. Chem. Soc. 2018, 140, 5485–5492. [Google Scholar] [CrossRef] [PubMed]
- Volbeda, A.; Fontecilla-Camps, J.C. Structural bases for the catalytic mechanism of Ni-containing carbon monoxide dehydrogenases. Dalton Trans. 2005, 21, 3443–3450. [Google Scholar] [CrossRef]
- Jeoung, J.H.; Dobbek, H. ATP-dependent substrate reduction at an [Fe8S9] double-cubane cluster. Proc. Natl. Acad. Sci. USA 2018, 115, 2994–2999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, T.; Koch, J.; Ermler, U.; Shima, S. Methanogenic heterodisulfide reductase (HdrABC-MvhAGD) uses two noncubane [4Fe-4S] clusters for reduction. Science 2017, 357, 699–703. [Google Scholar] [CrossRef] [Green Version]
- Vervoort, J.; Heering, D.; Peelen, S.; van Berkel, W. Flavodoxins. Methods Enzymol. 1994, 243, 188–203. [Google Scholar] [PubMed]
- Wastl, J.; Sticht, H.; Maier, U.-G.; Rösch, P.; Hoffmann, S. Identification and characterization of a eukaryotically encoded rubredoxin in a cryptomonad alga. FEBS Lett. 2000, 471, 191–196. [Google Scholar] [CrossRef] [Green Version]
- Zauner, S.; Fraunholz, M.; Wastl, J.; Penny, S.; Beaton, M.; Cavalier-Smith, T.; Maier, U.G.; Douglas, S. Chloroplast protein and centrosomal genes, a tRNA intron, and odd telomeres in an unusually compact eukaryotic genome, the cryptomonad nucleomorph. Proc. Natl. Acad. Sci. USA 2000, 97, 200–205. [Google Scholar] [CrossRef] [Green Version]
- Moura, I.; Bruschi, M.; Legall, J.; Moura, J.J.G.; Xavier, A.V. Isolation and characterization of desulforedoxin, a new type of non-heme iron protein from Desulfovibrio gigas. Biochem. Biophys. Res. Commun. 1977, 75, 1037–1044. [Google Scholar] [CrossRef]
- Zanello, P. The competition between chemistry and biology in assembling iron–sulfur derivatives. Molecular structures and electrochemistry. Part I. {Fe(SγCys)4} proteins. Coord. Chem. Rev. 2013, 257, 1777–1805. [Google Scholar] [CrossRef]
- Hagelueken, G.; Wiehlmann, L.; Adams, T.M.; Kolmar, H.; Heinz, D.W.; Tümmler, B.; Schubert, W.D. Crystal structure of the electron transfer complex rubredoxin rubredoxin reductase of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2007, 104, 12276–12281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, K.S.; Hille, R.; Hemann, C.; Tabita, F.R. Rubredoxin from the green sulfur bacterium Chlorobium tepidum functions as an electron acceptor for pyruvate ferredoxin oxidoreductase. J. Biol. Chem. 1999, 274, 29772–29778. [Google Scholar] [CrossRef] [Green Version]
- Ragsdale, S.W.; Ljungdahl, L.G.; DerVartanian, D.V. Isolation of carbon monoxide dehydrogenase from Acetobacterium woodii and comparison of its properties with those of the Clostridium thermoaceticum enzyme. J. Bacteriol. 1983, 155, 1224–1237. [Google Scholar] [CrossRef] [Green Version]
- Lin, I.J.; Gebel, E.B.; Machonkin, T.E.; Westler, W.M.; Markley, J.L. Changes in hydrogen-bond strengths explain reduction potentials in 10 rubredoxin variants. Proc. Natl. Acad. Sci. USA 2005, 102, 14581–14586. [Google Scholar] [CrossRef] [Green Version]
- Sun, N.; Dey, A.; Xiao, Z.; Wedd, A.G.; Hodgson, K.O.; Hedman, B.; Solomon, E.I. Solvation effects on S K-edge XAS spectra of Fe-S proteins: Normal and inverse effects on WT and mutant rubredoxin. J. Am. Chem. Soc. 2010, 132, 12639–12647. [Google Scholar] [CrossRef] [Green Version]
- Min, T.; Ergenekan, C.E.; Eidsness, M.K.; Ichiye, T.; Kang, C. Leucine 41 is a gate for water entry in the reduction of Clostridium pasteurianum rubredoxin. Protein Sci. 2001, 10, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Jenney, F.E.; Adams, M.W.W. Rubredoxin from Pyrococcus furiosus. In Methods in Enzymology; Elsevier: Amsterdam, The Netherlands, 2001; Volume 334, pp. 45–55. [Google Scholar]
- Moura, I.; Teixeira, M.; LeGall, J.; Moura, J.J. Spectroscopic studies of cobalt and nickel substituted rubredoxin and desulforedoxin. J. Inorg. Biochem. 1991, 44, 127–139. [Google Scholar] [CrossRef]
- Xx Thapper, A.; Rizzi, A.C.; Brondino, C.D.; Wedd, A.G.; Pais, R.J.; Maiti, B.K.; Moura, I.; Pauleta, S.R.; Moura, J.J. Copper-substituted forms of the wild type and C42A variant of rubredoxin. J. Inorg. Biochem. 2013, 127, 232–237. [Google Scholar] [CrossRef]
- Maiti, B.K.; Maia, L.B.; Moro, A.J.; Lima, J.C.; Cordas, C.M.; Moura, I.; Moura, J.J.G. Unusual Reduction Mechanism of Copper in Cysteine-Rich Environment. Inorg. Chem. 2018, 57, 8078–8088. [Google Scholar] [CrossRef] [PubMed]
- Maiti, B.K.; Maia, L.B.; Silveira, C.M.; Todorovic, S.; Carreira, C.; Carepo, M.S.; Grazina, R.; Moura, I.; Pauleta, S.R.; Moura, J.J.G. Incorporation of molybdenum in rubredoxin: Models for mononuclear molybdenum enzymes. J. Biol. Inorg. Chem. 2015, 20, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Slater, J.W.; Marguet, S.C.; Gray, M.E.; Monaco, H.A.; Sotomayor, M.; Shafaat, H.S. Power of the Secondary Sphere: Modulating Hydrogenase Activity in Nickel-Substituted Rubredoxin. ACS Catal. 2019, 9, 8928–8942. [Google Scholar] [CrossRef]
- Shafaat, H.S.; Rüdiger, O.; Ogata, H.; Lubitz, W. [NiFe] hydrogenases: A common active site for hydrogen metabolism under diverse conditions. Biochim. Biophys. Acta BBA-Bioenerg. 2013, 1827, 986–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Volbeda, A.; Charon, M.H.; Piras, C.; Hatchikian, E.C.; Frey, M.; Fontecilla-Camps, J.C. Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature 1995, 373, 580–587. [Google Scholar] [CrossRef] [PubMed]
- Volbeda, A.; Garcin, E.; Piras, C.; de Lacey, A.L.; Fernandez, V.M.; Hatchikian, E.C.; Frey, M.; Fontecilla-Camps, J.C. Structure of the [NiFe] Hydrogenase Active Site: Evidence for Biologically Uncommon Fe Ligands. J. Am. Chem. Soc. 1996, 118, 12989–12996. [Google Scholar] [CrossRef]
- Ogata, H.; Nishikawa, K.; Lubitz, W. Hydrogens detected by subatomic resolution protein crystallography in a [NiFe] hydrogenase. Nature 2015, 520, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Gloaguen, F.; Rauchfuss, T.B. Small molecule mimics of hydrogenases: Hydrides and redox. Chem. Soc. Rev. 2009, 38, 100–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.; Madden, C.; Ghirlanda, G. Photo-induced hydrogen production in a helical peptide incorporating a [FeFe] hydrogenase active site mimic. Chem. Commun. 2012, 79, 9816–9818. [Google Scholar] [CrossRef] [PubMed]
- Utschig, L.M.; Silver, S.C.; Mulfort, K.L.; Tiede, D.M. Nature-driven photochemistry for catalytic solar hydrogen production: A Photosystem I-transition metal catalyst hybrid. J. Am. Chem. Soc. 2011, 133, 16334–16337. [Google Scholar] [CrossRef] [PubMed]
- Thoi, V.S.; Sun, Y.; Long, J.R.; Chang, C.J. Complexes of earth-abundant metals for catalytic electrochemical hydrogen generation under aqueous conditions. Chem. Soc. Rev. 2013, 42, 2388–2400. [Google Scholar] [CrossRef] [PubMed]
- Schilter, D.; Camara, J.M.; Huynh, M.T.; Hammes-Schiffer, S.; Rauchfuss, T.B. Hydrogenase Enzymes and Their Synthetic Models: The Role of Metal Hydrides. Chem. Rev. 2016, 116, 8693–8749. [Google Scholar] [CrossRef] [Green Version]
- Denny, J.A.; Darensbourg, M.Y. Metallodithiolates as ligands in coordination, bioinorganic, and organometallic chemistry. Chem. Rev. 2015, 115, 5248–5273. [Google Scholar] [CrossRef]
- Ogo, S.; Ichikawa, K.; Kishima, T.; Matsumoto, T.; Nakai, H.; Kusaka, K.; Ohhara, T. A functional [NiFe]hydrogenase mimic that catalyzes electron and hydride transfer from H2. Science 2013, 339, 682–684. [Google Scholar] [CrossRef] [PubMed]
- Slater, J.W.; Shafaat, H.S. Nickel-Substituted Rubredoxin as a Minimal Enzyme Model for Hydrogenase. J. Phys. Chem. Lett. 2015, 6, 3731–3736. [Google Scholar] [CrossRef]
- Slater, J.W.; Marguet, S.C.; Cirino, S.L.; Maugeri, P.T.; Shafaat, H.S. Experimental and DFT Investigations Reveal the Influence of the Outer Coordination Sphere on the Vibrational Spectra of Nickel-Substituted Rubredoxin, a Model Hydrogenase Enzyme. Inorg. Chem. 2017, 56, 3926–3938. [Google Scholar] [CrossRef] [Green Version]
- Stevenson, M.J.; Marguet, S.C.; Schneider, C.R.; Shafaat, H.S. Light-Driven Hydrogen Evolution by Nickel-Substituted Rubredoxin. Chem. Sustain. Chem. 2017, 10, 4424–4429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Treviño, R.E.; Slater, J.W.; Shafaat, H.S. Robust Carbon-Based Electrodes for Hydrogen Evolution through Site-Selective Covalent Attachment of an Artificial Metalloenzyme. ACS Appl. Energy Mater. 2020, 3, 11099–11112. [Google Scholar]
- Naughton, K.J.; Treviño, R.E.; Moore, P.J.; Wertz, A.E.; Dickson, J.A.; Shafaat, H.S. In Vivo Assembly of a Genetically Encoded Artificial Metalloenzyme for Hydrogen Production. ACS Synth. Biol. 2021, 10, 2116–2120. [Google Scholar] [CrossRef]
- Detz, R.J.; Sakai, K.; Spiccia, L.; Brudvig, G.W.; Sun, L.; Reek, J.N.H. Towards a Bioinspired-Systems Approach for Solar Fuel Devices. Chem. Plus Chem. 2016, 81, 1024–1027. [Google Scholar] [CrossRef] [PubMed]
- Utschig, L.M.; Soltau, S.R.; Tiede, D.M. Light-driven hydrogen production from Photosystem I-catalyst hybrids. Curr. Opin. Chem. Biol. 2015, 25, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nichols, E.M.; Gallagher, J.J.; Liu, C.; Su, Y.; Resasco, J.; Yu, Y.; Sun, Y.; Yang, P.; Chang, M.C.; Chang, C.J. Hybrid bioinorganic approach to solar-to-chemical conversion. Proc. Natl. Acad. Sci. USA 2015, 112, 11461–11466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reisner, E. Solar Hydrogen Evolution with Hydrogenases: From Natural to Hybrid Systems. Eur. J. Inorg. Chem. 2011, 2011, 1005–1016. [Google Scholar] [CrossRef]
- Marguet, S.C.; Stevenson, M.J.; Shafaat, H.S. Intramolecular Electron Transfer Governs Photoinduced Hydrogen Evolution by Nickel-Substituted Rubredoxin: Resolving Elementary Steps in Solar Fuel Generation. J. Phys. Chem. B 2019, 123, 9792–9800. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.J.; Meyer, J.; Achim, C.; Peterson, J.; Hendrich, M.P.; Münck, E. Mössbauer, EPR, and MCD studies of the C9S and C42S variants of Clostridium pasteurianum rubredoxin and MDC studies of the wild-type protein. J. Biol. Inorg. Chem. 2000, 5, 475–487. [Google Scholar] [CrossRef]
- Moura, I.; Tavares, P.; Moura, J.J.; Ravi, N.; Huynh, B.H.; Liu, M.Y.; LeGall, J. Purification and characterization of desulfoferrodoxin. A novel protein from Desulfovibrio desulfuricans (ATCC 27774) and from Desulfovibrio vulgaris (strain Hildenborough) that contains a distorted rubredoxin center and a mononuclear ferrous center. J. Biol. Chem. 1990, 265, 21596–21602. [Google Scholar] [CrossRef]
- Werth, M.T.; Kurtz, D.M., Jr.; Moura, I.; LeGall, J. Proton NMR spectra of rubredoxins: New resonances assignable to .alpha.-CH and beta-CH2 hydrogens of cysteinate ligands to iron(II). J. Am. Chem. Soc. 1987, 109, 273–275. [Google Scholar] [CrossRef]
- Almeida, R.M.; Pauleta, S.R.; Moura, I.; Moura, J.J. Rubredoxin as a paramagnetic relaxation-inducing probe. J. Inorg. Biochem. 2009, 103, 1245–1253. [Google Scholar] [CrossRef] [PubMed]
- Almeida, R.; Turano, P.; Moura, I.M.A.M.G.D.; Pauleta, S.R.; Moura, J.J.G.D. Superoxide Reductase: Different Interaction Modes with its Two Redox Partners. Chem. Bio Chem. 2013, 14, 1858–1866. [Google Scholar] [CrossRef]
- Lamosa, P.; Brennan, L.; Vis, H.; Turner, D.L.; Santos, H. NMR structure of Desulfovibrio gigas rubredoxin: A model for studying protein stabilization by compatible solutes. Extremophiles 2001, 5, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, B.J.; Nunes, S.G.; Rusnak, F.; Moura, I.; Ascenso, C.; Moura, J.J.; Volkman, B.F.; Markley, J.L. Zinc-substituted Desulfovibrio gigas desulforedoxins: Resolving subunit degeneracy with nonsymmetric pseudocontact shifts. Protein Sci. 2002, 11, 2464–2470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armitage, I.M.; Drakenberg, T.; Reilly, B. Use of 113Cd NMR to probe the native metal binding sites in metalloproteins: An overview. Met. Ions. Life. Sci. 2013, 11, 117–144. [Google Scholar] [PubMed] [Green Version]
- Vasak, M. Application of 113Cd NMR to metallothioneins. Biodegradation 1998, 9, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Summers, M.F. 113Cd NMR spectroscopy of coordination compounds and proteins. Coord. Chem. Rev. 1988, 86, 43–134. [Google Scholar] [CrossRef]
- Ayhan, M.; Xiao, Z.; Lavery, M.J.; Hamer, A.M.; Nugent, K.W.; Scrofani, S.D.B.; Guss, M.; Wedd, A.G. The Rubredoxin from Clostridium pasteurianum: Mutation of the Conserved Glycine Residues 10 and 43 to Alanine and Valine. Inorg. Chem. 1996, 35, 5902–5911. [Google Scholar] [CrossRef]
- Fukuyama, K.; Matsubara, H.; Tsukihara, T.; Katsube, Y. Structure of [4Fe-4S] ferredoxin from Bacillus thermoproteolyticus refined at 2.3 A resolution. Structural comparisons of bacterial ferredoxins. J. Mol. Biol. 1989, 210, 383–398. [Google Scholar] [CrossRef]
- Fukuyama, K. Handbook of Metalloproteins; Messerschmidt, A., Huber, R., Wieghardt, K., Poulos, T., Eds.; Wiley: New York, NY, USA, 2001; pp. 543–552. [Google Scholar]
- Johnson, D.C.; Dean, D.R.; Smith, A.D.; Johnson, M.K. Structure, function, and formation of biological iron-sulfur clusters. Annu. Rev. Biochem. 2005, 74, 247–281. [Google Scholar] [CrossRef] [PubMed]
- Py, B.; Barras, F. Building Fe-S proteins: Bacterial strategies. Nat. Rev. Microbiol. 2010, 8, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Mettert, E.L.; Kiley, P.J. Fe-S proteins that regulate gene expression. Biochim. Biophys. Acta (BBA)-Mol. Cell Res. 2015, 1853, 1284–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, M.K. Encyclopedia of Inorganic Chemistry; King, R.B., Ed.; Wiley: Oxford, UK, 1994; Volume 4, pp. 1896–1915. [Google Scholar]
- Cammack, R. Iron—Sulfur Clusters in Enzymes: Themes and Variations. Adv. Inorg. Chem. 1992, 38, 281–322. [Google Scholar]
- Duff, J.L.C.; Breton, J.L.J.; Butt, J.N.; Armstrong, F.A.; Thomson, A.J. Novel Redox Chemistry of [3Fe-4S] Clusters: Electrochemical Characterization of the All-Fe(II) Form of the [3Fe-4S] Cluster Generated Reversibly in Various Proteins and Its Spectroscopic Investigation in Sulfolobus acidocaldarius Ferredoxin. J. Am. Chem. Soc. 1996, 118, 8593–8603. [Google Scholar] [CrossRef]
- Smith, E.T.; Blamey, J.M.; Zhou, Z.H.; Adams, M.W.W. A Variable-Temperature Direct Electrochemical Study of Metalloproteins from Hyperthermophilic Microorganisms Involved in Hydrogen Production from Pyruvate. Biochemistry 1995, 34, 7161–7169. [Google Scholar] [CrossRef] [PubMed]
- Moreno, C.; Macedo, A.L.; Moura, I.; LeGall, J.; Moura, J.J. Redox properties of Desulfovibrio gigas [Fe3S4] and [Fe4S4] ferredoxins and heterometal cubane-type clusters formed within the [Fe3S4] core. Square wave voltammetric studies. J. Inorg. Biochem. 1994, 53, 219–234. [Google Scholar] [CrossRef]
- Tong, J.; Feinberg, B.A. Direct square-wave voltammetry of superoxidized [4Fe-4S]3+ aconitase and associated 3Fe/4Fe cluster interconversions. J. Biol. Chem. 1994, 269, 24920–24927. [Google Scholar] [CrossRef]
- Shen, B.; Martin, L.L.; Butt, J.N.; Armstrong, F.A.; Stout, C.D.; Jensen, G.M.; Stephens, P.J.; La Mar, G.N.; Gorst, C.M.; Burgess, B.K. Azotobacter vinelandii ferredoxin I. Aspartate 15 facilitates proton transfer to the reduced [3Fe-4S] cluster. J. Biol. Chem. 1993, 268, 25928–25939. [Google Scholar] [CrossRef]
- Butt, J.N.; Armstrong, F.A.; Breton, J.; George, S.J.; Thomson, A.J.; Hatchikian, E.C. Investigation of metal ion uptake reactivities of [3Fe-4S] clusters in proteins: Voltammetry of co-adsorbed ferredoxin-aminocyclitol films at graphite electrodes and spectroscopic identification of transformed clusters. J. Am. Chem. Soc. 1991, 113, 6663–6670. [Google Scholar] [CrossRef]
- Shirakawa, T.; Takahashi, Y.; Wada, K.; Hirota, J.; Takao, T.; Ohmori, D.; Fukuyama, K. Identification of variant molecules of Bacillus thermoproteolyticus ferredoxin: Crystal structure reveals bound coenzyme A and an unexpected [3Fe-4S] cluster associated with a canonical [4Fe-4S] ligand motif. Biochemistry 2005, 44, 12402–12410. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, M.C.; Kent, T.A.; Emptage, M.; Merkle, H.; Beinert, H.; Münck, E. Evidence for the formation of a linear [3Fe-4S] cluster in partially unfolded aconitase. J. Biol. Chem. 1984, 259, 14463–14471. [Google Scholar] [CrossRef]
- Conover, R.C.; Kowal, A.T.; Fu, W.G.; Park, J.B.; Aono, S.; Adams, M.W.; Johnson, M.K. Spectroscopic characterization of the novel iron-sulfur cluster in Pyrococcus furiosus ferredoxin. J. Biol. Chem. 1990, 265, 8533–8541. [Google Scholar] [CrossRef]
- Busch, J.L.H.; Breton, J.L.; Bartlett, B.M.; Armstrong, F.A.; James, R.; Andrew, J.; Thomson, A.J. [3Fe-4S]↔[4Fe-4S] cluster interconversion in Desulfovibrio africanus ferredoxin III: Properties of an Asp14→Cys mutant. Biochem. J. 1997, 323, 95–102. [Google Scholar] [CrossRef]
- Werst, M.M.; Kennedy, M.C.; Beinert, H.; Hoffman, B.M. Oxygen-17, proton, and deuterium electron nuclear double resonance characterization of solvent, substrate, and inhibitor binding to the iron-sulfur [4Fe-4S]+ cluster of aconitase. Biochemistry 1990, 29, 10526–10532. [Google Scholar] [CrossRef] [PubMed]
- Calzolai, L.; Gorst, C.M.; Zhao, Z.-H.; Teng, Q.; Adams, M.W.W.; Mar, G.N.L. 1H NMR Investigation of the Electronic and Molecular Structure of the Four-Iron Cluster Ferredoxin from the Hyperthermophile Pyrococcus furiosus. Identification of Asp 14 as a Cluster Ligand in Each of the Four Redox States. Biochemistry 1995, 34, 11373–11384. [Google Scholar] [CrossRef]
- Moura, I.; Moura, J.J.G.; Munck, E.; Papaefthymiou, V.; LeGall, J. Evidence for the formation of a cobalt-iron-sulfur (CoFe3S4) cluster in Desulfovibrio gigas ferredoxin II. J. Am. Chem. Soc. 1986, 108, 349–351. [Google Scholar] [CrossRef]
- Fu, W.; Telser, J.; Hoffman, B.M.; Smith, E.T.; Adams, M.W.W.; Johnson, M.K. Interaction of Tl+ and Cs+ with the [Fe3S4] Cluster of Pyrococcus furiosus Ferredoxin: Investigation by Resonance Raman, MCD, EPR, and ENDOR Spectroscopy. J. Am. Chem. Soc. 1994, 116, 5722–5729. [Google Scholar] [CrossRef]
- Finnegan, M.G.; Conover, R.C.; Park, J.-B.; Zhou, Z.H.; Adams, M.W.W.; Johnson, M.K. Electronic, Magnetic, Redox, and Ligand-Binding Properties of [MFe3S4] Clusters (M = Zn, Co, Mn) in Pyrococcus furiosus Ferredoxin. Inorg. Chem. 1994, 34, 5358–5369. [Google Scholar] [CrossRef]
- Staples, C.R.; Dhawan, I.K.; Finnegan, M.G.; Dwinell, D.A.; Zhou, Z.H.; Huang, H.; Verhagen, M.F.J.M.; Adams, M.W.W.; Johnson, M.K. Electronic, Magnetic, and Redox Properties of [MFe3S4] Clusters (M = Cd, Cu, Cr) in Pyrococcus furiosus Ferredoxin. Inorg. Chem. 1997, 36, 5740–5749. [Google Scholar] [CrossRef] [PubMed]
- Ensign, S.A. Reactivity of Carbon Monoxide Dehydrogenase from Rhodospirillum rubrum with Carbon Dioxide, Carbonyl Sulfide, and Carbon Disulfide. Biochemistry 1995, 34, 5372–5381. [Google Scholar] [CrossRef] [PubMed]
- Svetlitchnyi, V.; Peschel, C.; Acker, G.; Meyer, O. Two membrane-associated NiFeS-carbon monoxide dehydrogenases from the anaerobic carbon-monoxide-utilizing eubacterium Carboxydothermus hydrogenoformans. J. Bacteriol. 2001, 183, 5134–5144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobbek, H.; Gremer, L.; Meyer, O.; Huber, R. Crystal structure and mechanism of CO dehydrogenase, a molybdo iron-sulfur flavoprotein containing S-selanylcysteine. Proc. Natl. Acad. Sci. USA 1999, 96, 8884–8889. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, O.; Gremer, L.; Ferner, R.; Ferner, M.; Dobbek, H.; Gnida, M.; Meyer-Klaucke, W.; Huber, R. The role of Se, Mo and Fe in the structure and function of carbon monoxide dehydrogenase. Biol. Chem. 2000, 381, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Ferry, J.G. CO dehydrogenase. Annu. Rev. Microbiol. 1995, 49, 305–333. [Google Scholar] [CrossRef]
- Ermler, U.; Grabarse, W.; Shima, S.; Goubeaud, M.; Thauer, R.K. Active sites of transition-metal enzymes with a focus on nickel. Curr. Opin. Struct. Biol. 1998, 8, 749–758. [Google Scholar] [CrossRef]
- Can, M.; Armstrong, F.A.; Ragsdale, S.W. Structure, Function, and Mechanism of the Nickel Metalloenzymes, CO Dehydrogenase, and Acetyl-CoA Synthase. Chem. Rev. 2014, 114, 4149–4174. [Google Scholar] [CrossRef] [PubMed]
- Jeoung, J.H.; Dobbek, H. Carbon dioxide activation at the Ni,Fe-cluster of anaerobic carbon monoxide dehydrogenase. Science 2007, 318, 1461–1464. [Google Scholar] [CrossRef] [Green Version]
- Busse, S.C.; Mar, G.N.L.; Yu, L.P.; Howard, J.B.; Smith, E.T.; Zhou, Z.H.; Adams, M.W.W. Proton NMR investigation of the oxidized three-iron clusters in the ferredoxins from the hyperthermophilic archae Pyrococcus furiosus and Thermococcus litoralis. Biochemistry 1992, 31, 11952–11962. [Google Scholar] [CrossRef]
- Calzolai, L.; Zhou, Z.H.; Adams, M.W.W.; Mar, G.N.L. Role of Cluster-Ligated Aspartate in Gating Electron Transfer in the Four-Iron Ferredoxin from the Hyperthermophilic Archaeon Pyrococcus furiosus. J. Am. Chem. Soc. 1996, 118, 2513–2514. [Google Scholar] [CrossRef]
- Telser, J.; Smith, E.T.; Adams, M.W.W.; Conover, R.C.; Johnson, M.K.; Hoffman, B.M. Cyanide Binding to the Novel 4Fe Ferredoxin from Pyrococcus furiosus: Investigation by EPR and ENDOR Spectroscopy. J. Am. Chem. Soc. 1995, 117, 5133–5140. [Google Scholar] [CrossRef]
- Butt, J.N.; Niles, J.; Armstrong, F.A.; Breton, J.; Thomson, A.J. Formation and properties of a stable ‘high-potential’ copper-iron-sulphur cluster in a ferredoxin. J. Nat. Struct. Biol. 1994, 1, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, K.K.P.; Surerus, K.K.; Conover, R.C.; Johnson, M.K.; Park, J.B.; Adams, M.W.W.; Munck, E. Moessbauer study of zinc-iron-sulfur ZnFe3S4 and nickel-iron-sulfur NiFe3S4 clusters in Pyrococcus furiosus ferredoxin. Inorg. Chem. 1993, 32, 927–936. [Google Scholar] [CrossRef]
- Surerus, K.K.; Munck, E.; Moura, I.; Moura, J.J.G.; LeGall, J. Evidence for the formation of a ZnFe3S4 cluster in Desulfovibrio gigas ferredoxin II. J. Am. Chem. Soc. 1987, 109, 3805–3807. [Google Scholar] [CrossRef]
- Martic, M.; Jakab-Simon, I.N.; Haahr, L.T.; Hagen, W.R.; Christensen, H.E. Heterometallic [AgFe3S4] ferredoxin variants: Synthesis, characterization, and the first crystal structure of an engineered heterometallic iron-sulfur protein. J. Biol. Inorg. Chem. 2013, 18, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Fiévet, A.; My, L.; Cascales, E.; Ansaldi, M.; Pauleta, S.R.; Moura, I.; Dermoun, Z.; Bernard, C.S.; Dolla, A.; Aubert, C. The anaerobe-specific orange protein complex of Desulfovibrio vulgaris hildenborough is encoded by two divergent operons coregulated by σ54 and a cognate transcriptional regulator. J. Bacteriol. 2011, 193, 3207–3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maiti, B.K.; Avilés, T.; Carepo, M.S.P.; Moura, I.; Pauleta, S.R.; Moura, J.J.G. Rearrangement of Mo-Cu-S Cluster Reflects the Structural Instability of Orange Protein Cofactor. Z. Anorg. Und Allg. Chem. (ZAAC) 2013, 639, 1361–1364. [Google Scholar] [CrossRef]
- Najmudin, S.; Bonifácio, C.; Duarte, A.G.; Pauleta, S.R.; Moura, I.; Moura, J.J.; Romão, M.J. Crystallization and crystallographic analysis of the apo form of the orange protein (ORP) from Desulfovibrio gigas. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2009, 65, 730–732. [Google Scholar] [CrossRef]
- Carepo, M.S.; Pauleta, S.R.; Wedd, A.G.; Moura, J.J.; Moura, I. Mo-Cu metal cluster formation and binding in an orange protein isolated from Desulfovibrio gigas. J. Biol. Inorg. Chem. 2014, 19, 605–614. [Google Scholar] [CrossRef]
- Pauleta, S.R.; Duarte, A.G.; Carepo, M.S.; Pereira, A.S.; Tavares, P.; Moura, I.; Moura, J.J.G. NMR assignment of the apo-form of a Desulfovibrio gigas protein containing a novel Mo-Cu cluster. Biomol. NMR Assign. 2007, 1, 81–83. [Google Scholar] [CrossRef] [PubMed]
- Maiti, B.K.; Moura, J.J.G. Diverse biological roles of the tetrathiomolybdate anion. Coord. Chem. Rev. 2021, 429, 213635. [Google Scholar] [CrossRef]
- Maiti, B.K. A review on chemical and physical properties of tetrathiomolybdate (TTM) anion drug is useful for TTM treated diseases. J. Indian Chem. Soc. 2021, 98, 100117. [Google Scholar] [CrossRef]
- Maiti, B.K.; Pal, K.; Sarkar, S. A structural model of mixed metal sulfide cluster of molybdenum and copper present in the orange protein of Desulfovibrio gigas. Inorg. Chem. Commun. 2004, 7, 1027–1029. [Google Scholar] [CrossRef]
- Maiti, B.K.; Govil, N.; Kundu, T.; Moura, J.J.G. Designed metal-ATCUN derivatives: Redox-and non-redox-based applications relevant for chemistry, biology, and medicine. Iscience 2020, 23, 101792. [Google Scholar] [CrossRef]
- Maiti, B.K.; Avilés, T.; Matzapetakis, M.; Moura, I.; Pauleta, S.R.; Moura, J.J.G. Synthesis of [MoS4]2−–M (M = Cu and Cd) Clusters: Potential NMR Spectroscopic Structural Probes for the Orange Protein. Eur. J. Inorg. Chem. 2012, 2012, 4159–4166. [Google Scholar] [CrossRef]
- Maiti, B.K.; Avilés, T.; Moura, I.; Pauleta, S.R.; Moura, J.J.G. Synthesis and characterization of [S2MoS2Cu(n-SPhF)]2−(n = o, m, p) clusters: Potential 19F-NMR structural probes for Orange Protein. Inorg. Chem. Commun. 2014, 45, 97–100. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiti, B.K.; Moura, J.J.G. Native Protein Template Assisted Synthesis of Non-Native Metal-Sulfur Clusters. BioChem 2022, 2, 182-197. https://doi.org/10.3390/biochem2030013
Maiti BK, Moura JJG. Native Protein Template Assisted Synthesis of Non-Native Metal-Sulfur Clusters. BioChem. 2022; 2(3):182-197. https://doi.org/10.3390/biochem2030013
Chicago/Turabian StyleMaiti, Biplab K., and José J. G. Moura. 2022. "Native Protein Template Assisted Synthesis of Non-Native Metal-Sulfur Clusters" BioChem 2, no. 3: 182-197. https://doi.org/10.3390/biochem2030013