
Classification of B-Cell Lymphomas and Immunodeficiency-Related Lymphoproliferations: What’s New?

Abstract
:1. Introduction
2. Follicular Lymphomas
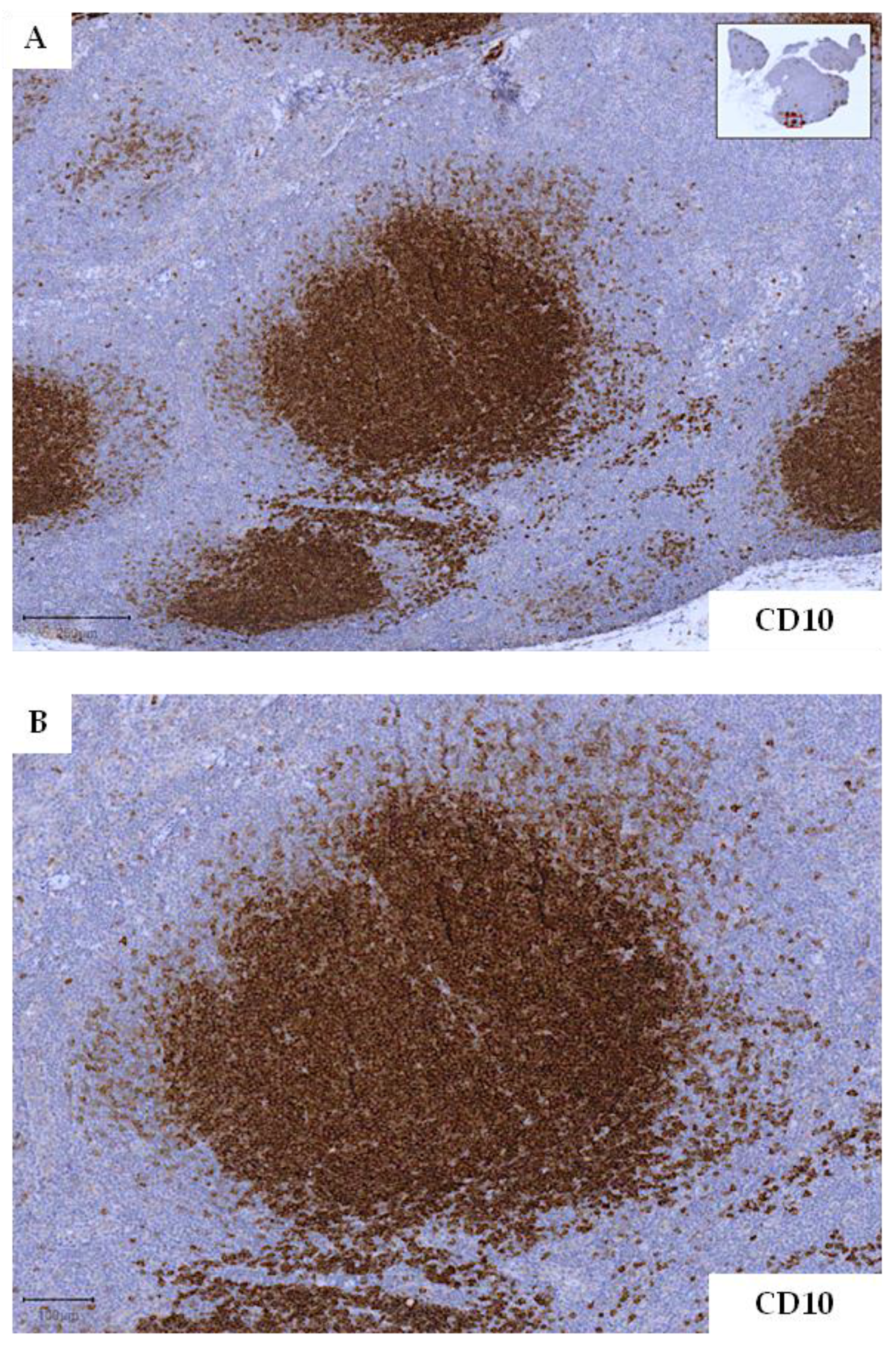
2.1. Incipient/Early Lesions of Follicular Lymphoma
2.2. Follicular Lymphoma
2.3. Lymphomas with a Follicular Pattern Occurring Preferentially in Pediatric and Young Adults
3. Cutaneous Follicle Center Lymphoma
4. Transformation of Indolent B-Cell Lymphomas
5. Large B-Cell Lymphomas
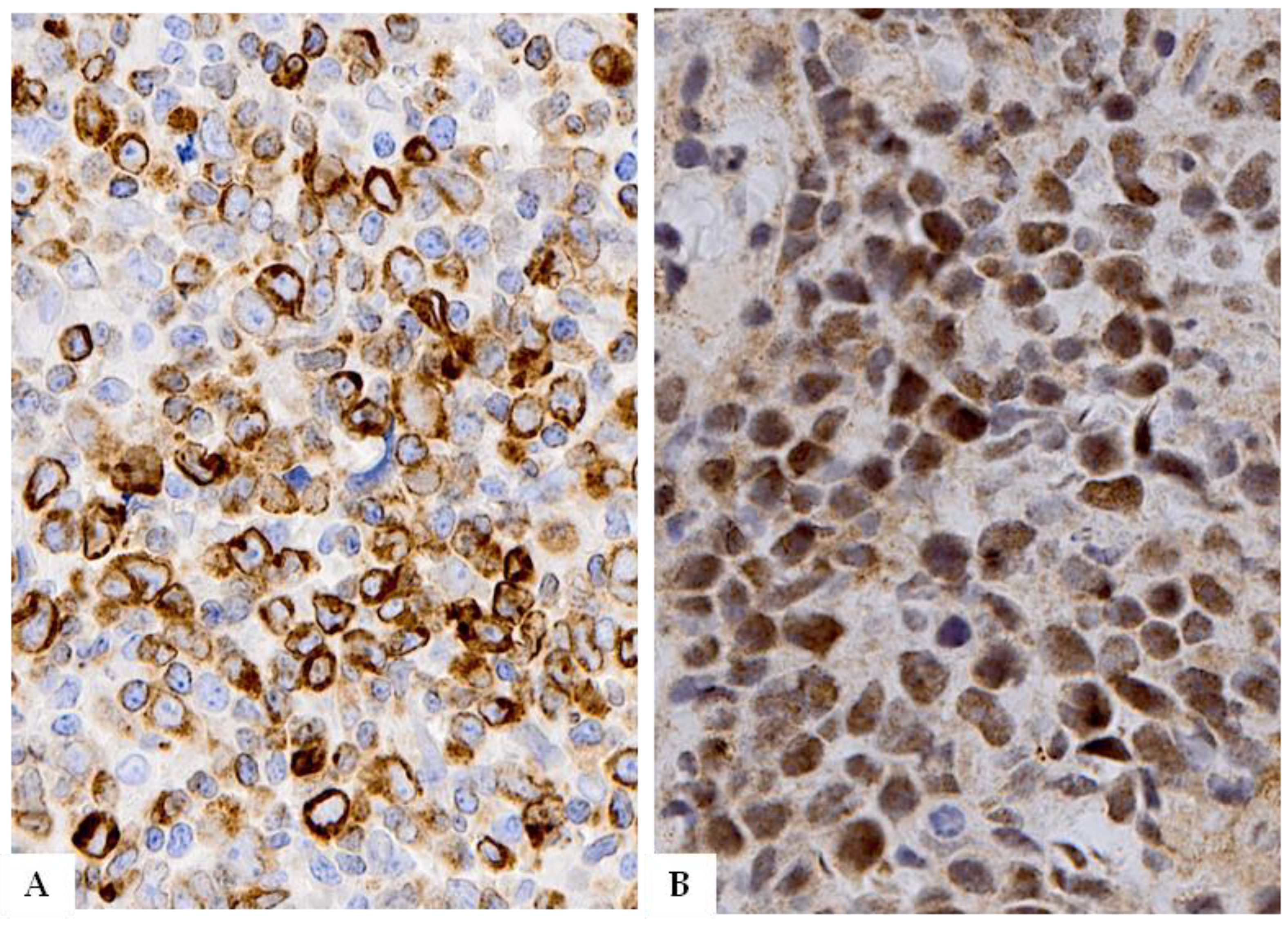
5.1. Large B-Cell Lymphoma with IRF4 Rearrangement
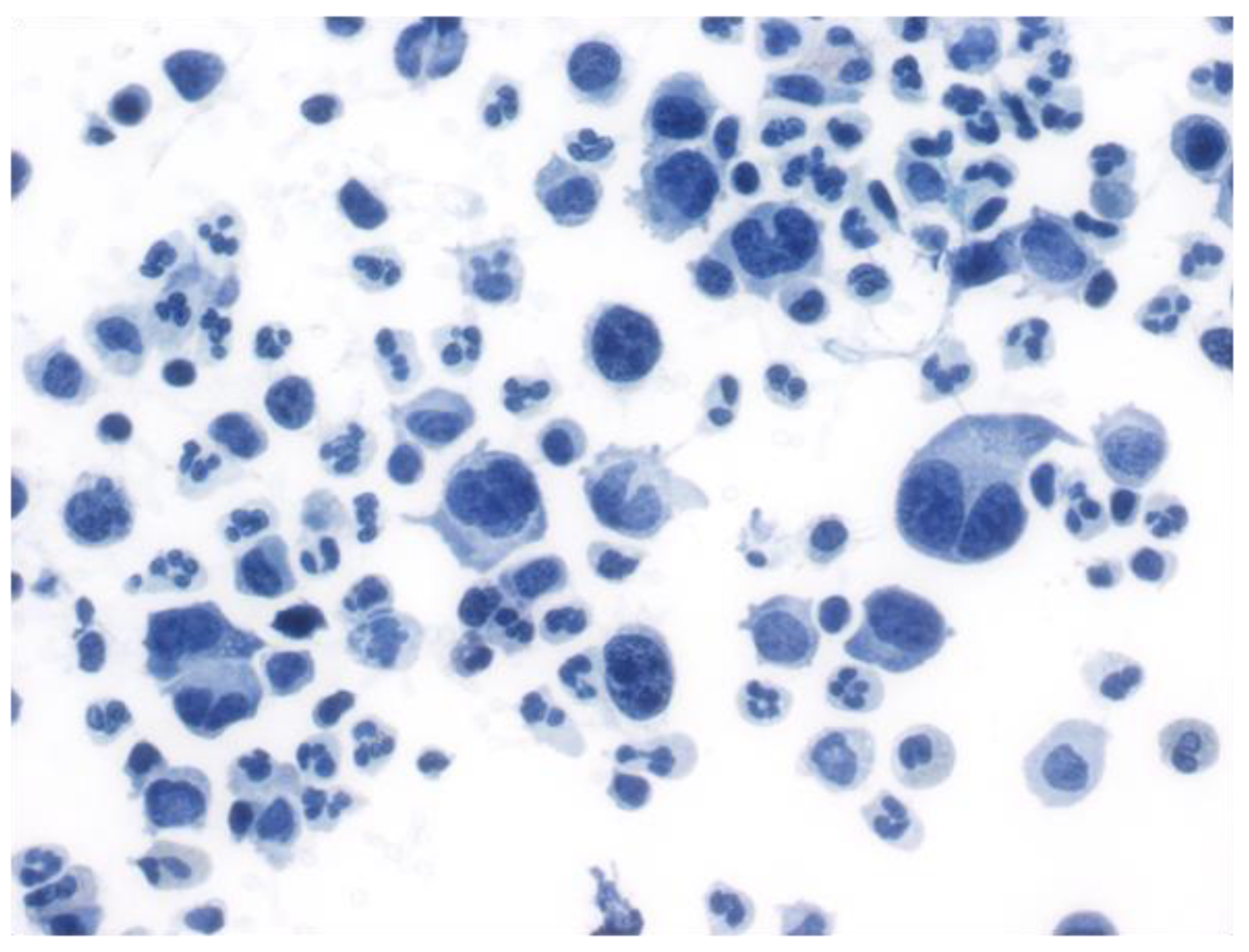
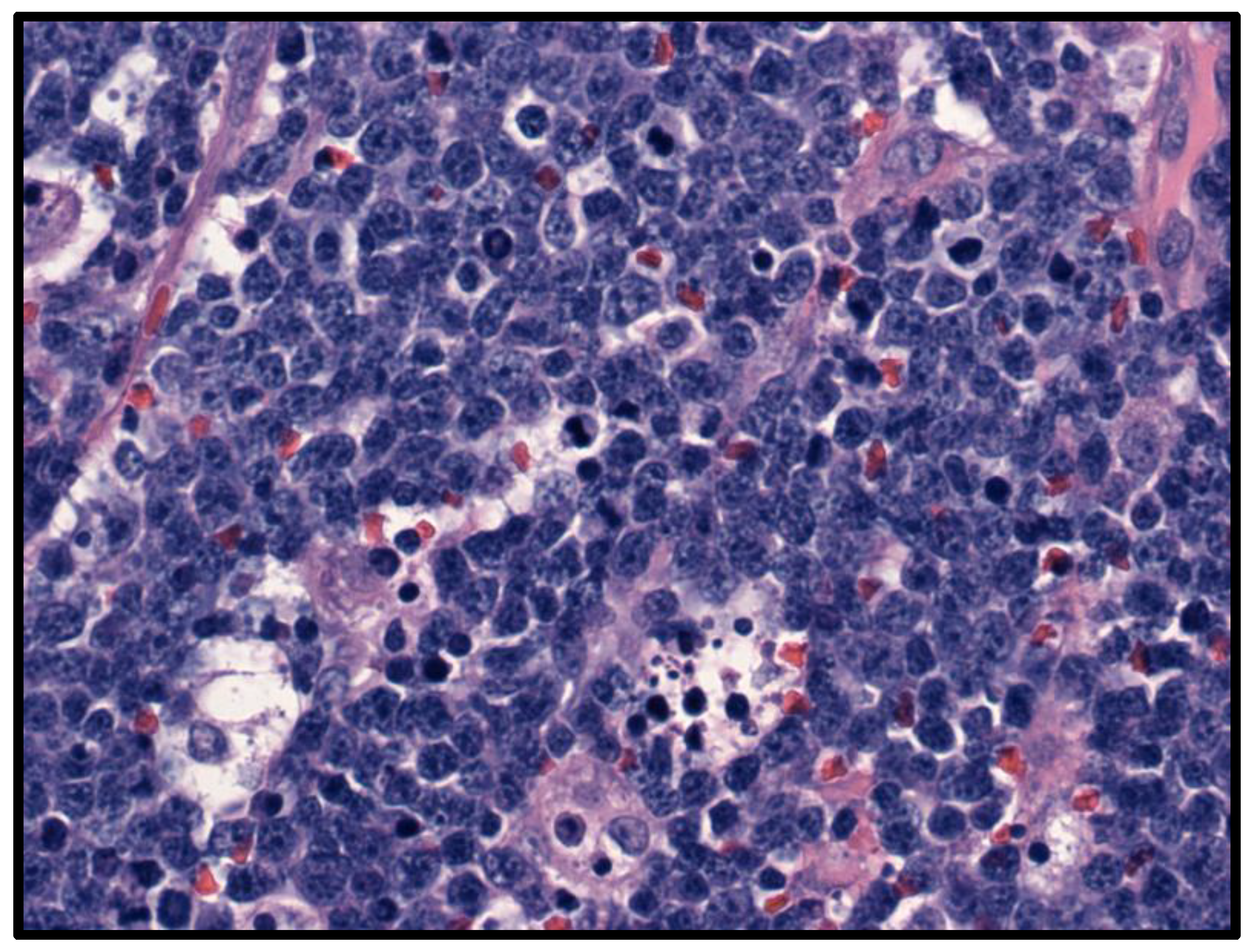
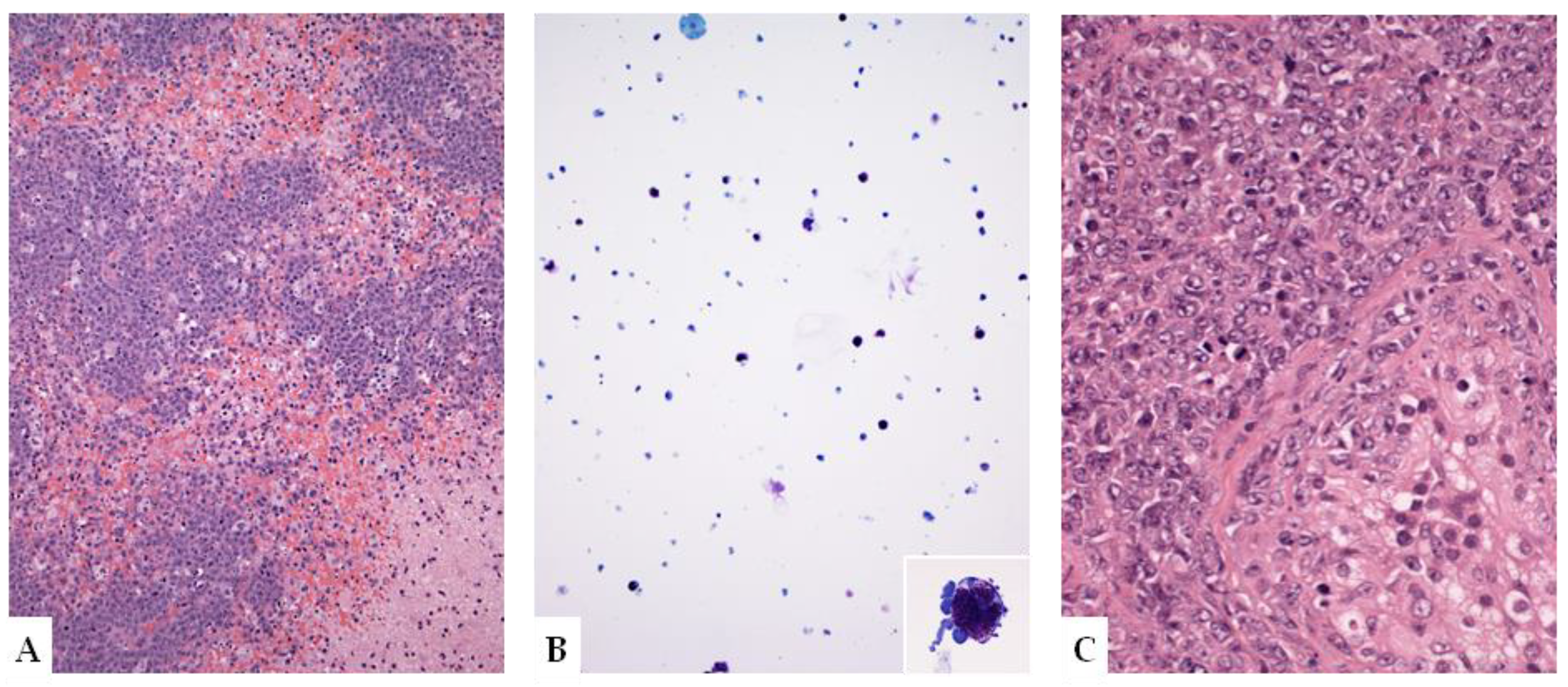
5.2. Fluid Overload-Associated Large B-Cell Lymphoma (WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, 5th Edition; WHO-HAEM5)/HHV8 and EBV-Negative Primary Effusion-Based Lymphoma (International Consensus Classification; ICC)
5.3. High-Grade B-Cell Lymphoma with 11q Aberration (WHO-HAEM5)/Large B-Cell Lymphoma with 11q Aberration (ICC)
5.4. Double-Hit Lymphoma
5.5. Large B-Cell Lymphomas (LBCL) of Immune-Privileged Sites
5.6. Mediastinal Grey Zone Lymphoma and Primary Mediastinal Large B-Cell Lymphoma
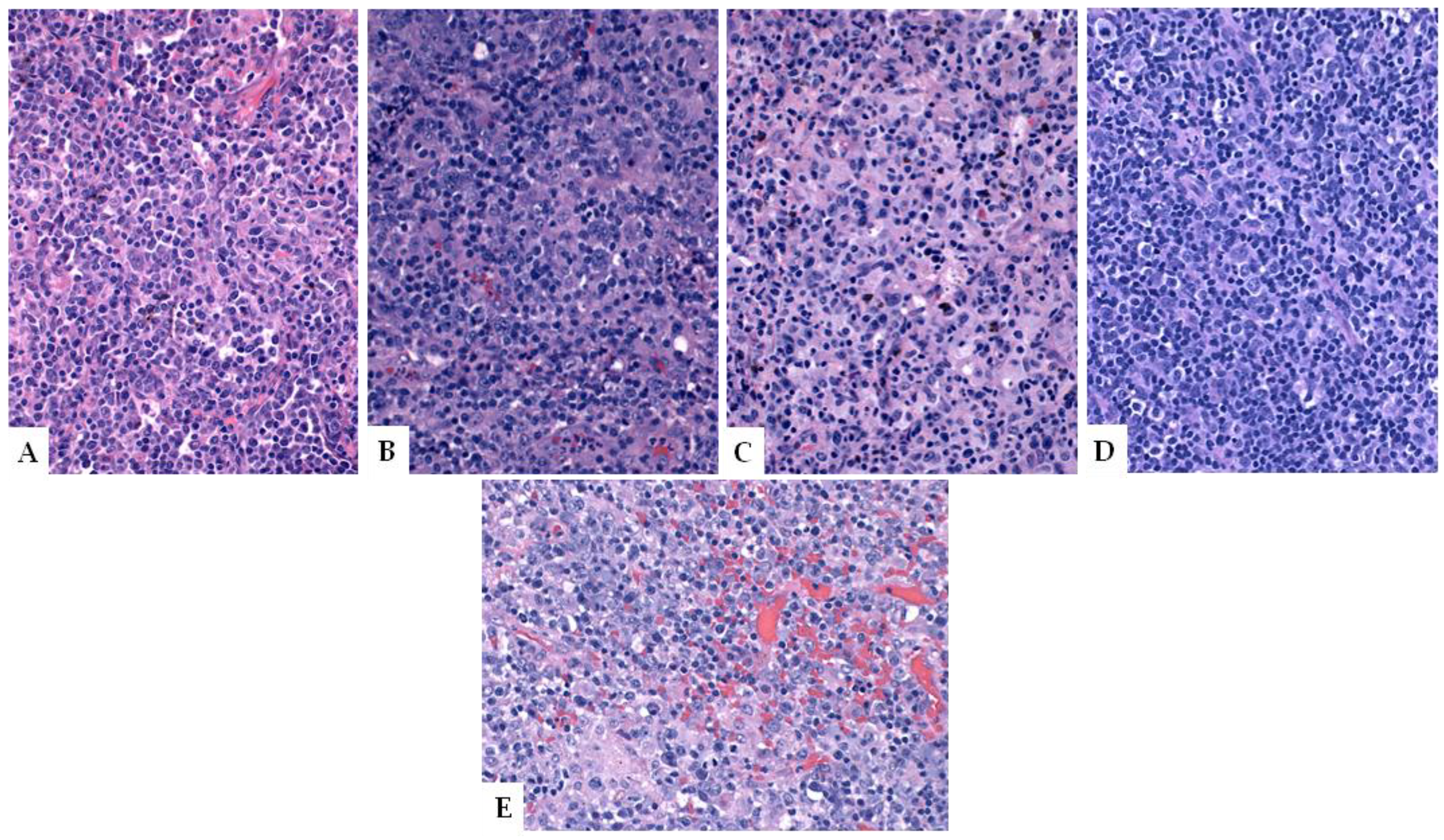
6. Immunodeficiency-Related Lymphoproliferative Disorders
7. Viral-Associated Lymphoproliferative Disorders
7.1. Epstein–Barr Virus Lesions (EBV)
7.2. Kaposi Sarcoma Herpesvirus/Human Herpesvirus 8 (KSHV/HHV8)
8. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Pathology and Genetics of Tumours of Haematopoietic and Lymphoid Tissues, 3rd ed.; IARC: Lyon, France, 2001. [Google Scholar]
- World Health Organization. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; IARC: Lyon, France, 2008. [Google Scholar]
- World Health Organization. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised 4th ed.; IARC: Lyon, France, 2017. [Google Scholar]
- Campo, E.; Swerdlow, S.H.; Harris, N.L.; Pileri, S.; Stein, H.; Jaffe, E.S. The 2008 WHO classification of lymphoid neoplasms and beyond: Evolving concepts and practical applications. Blood J. Am. Soc. Hematol. 2011, 117, 5019–5032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood J. Am. Soc. Hematol. 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Campo, E.; Jaffe, E.S.; Cook, J.R.; Quintanilla-Martinez, L.; Swerdlow, S.H.; Anderson, K.C.; Brousset, P.; Cerroni, L.; de Leval, L.; Dirnhofer, S.; et al. The International Consensus Classification of Mature Lymphoid Neoplasms: A Report from the Clinical Advisory Committee. Blood J. Am. Soc. Hematol. 2022, 140, 1229–1253. [Google Scholar] [CrossRef]
- Alaggio, R.; Amador, C.; Anagnostopoulos, I.; Attygalle, A.D.; Araujo, I.B.D.O.; Berti, E.; Bhagat, G.; Borges, A.M.; Boyer, D.; Calaminici, M.; et al. The 5th edition of the World Health Organization Classification of Haematolymphoid Tumours: Lymphoid Neoplasms. Leukemia 2022, 36, 1720–1748. [Google Scholar] [CrossRef]
- Bower, M.; Carbone, A. KSHV/HHV8-Associated Lymphoproliferative Disorders: Lessons Learnt from People Living with HIV. Hemato 2021, 2, 703–712. [Google Scholar] [CrossRef]
- Carbone, A.; Roulland, S.; Gloghini, A.; Younes, A.; von Keudell, G.; López-Guillermo, A.; Fitzgibbon, J. Follicular lymphoma. Nat. Rev. Dis. Prim. 2019, 5, 83. [Google Scholar] [CrossRef] [PubMed]
- Campo, E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J.; Vardiman, J.W. Follicular lymphoma. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; IARC Press: Lyon, France, 2017; pp. 266–277. [Google Scholar]
- Jaffe, E.S.; Quintanilla-Martinez, L. t(14;18)-positive B cells: Is it seed or soil? Blood J. Am. Soc. Hematol. 2018, 132, 1631–1632. [Google Scholar] [CrossRef]
- Mamessier, E.; Song, J.Y.; Eberle, F.C.; Pack, S.; Drevet, C.; Chetaille, B.; Abdullaev, Z.; Adelaïde, J.; Birnbaum, D.; Chaffanet, M.; et al. Early lesions of follicular lymphoma: A genetic perspective. Haematologica 2014, 99, 481–488. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.; Ramis-Zaldivar, J.E.; Bonzheim, I.; Steinhilber, J.; Müller, I.; Haake, A.; Yu, S.C.; Raffeld, M.; Fend, F.; Salaverria, I.; et al. CREBBP gene mutations are frequently detected in in situ follicular neoplasia. Blood J. Am. Soc. Hematol. 2018, 132, 2687–2690. [Google Scholar] [CrossRef] [Green Version]
- Nann, D.; Ramis-Zaldivar, J.E.; Müller, I.; Gonzalez-Farre, B.; Schmidt, J.; Egan, C.; Salmeron-Villalobos, J.; Clot, G.; Mattern, S.; Otto, F.; et al. Follicular lymphoma t(14;18)-negative is genetically a heterogeneous disease. Blood Adv. 2020, 4, 5652–5665. [Google Scholar] [CrossRef]
- Liu, Q.; Salaverria, I.; Pittaluga, S.; Jegalian, A.G.; Xi, L.; Siebert, R.; Raffeld, M.; Hewitt, S.M.; Jaffe, E.S. Follicular lymphomas in children and young adults: A comparison of the pediatric variant with usual follicular lymphoma. Am. J. Surg. Pathol. 2013, 37, 333–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lucioni, M.; Fraticelli, S.; Neri, G.; Feltri, M.; Ferrario, G.; Riboni, R.; Paulli, M. Primary Cutaneous B-Cell Lymphoma: An Update on Pathologic and Molecular Features. Hemato 2022, 3, 318–340. [Google Scholar] [CrossRef]
- Bhavsar, S.; Liu, Y.C.; Gibson, S.E.; Moore, E.M.; Swerdlow, S.H. Mutational Landscape of TdT+ Large B-cell Lymphomas Supports Their Distinction From B-lymphoblastic Neoplasms: A Multiparameter Study of a Rare and Aggressive Entity. Am. J. Surg. Pathol. 2022, 46, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.S.; Carbone, A. Evolution in the Definition of Follicular Lymphoma and Diffuse Large B-Cell Lymphoma: A Model for the Future of Personalized Medicine. Hemato 2022, 3, 466–474. [Google Scholar] [CrossRef]
- Pasqualucci, L.; Dalla-Favera, R. The genetic landscape of diffuse large B-cell lymphoma. Semin. Hematol. 2015, 52, 67–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Said, J.; Compo, E. Primary effusion lymphoma. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC: Lyon, France, 2017; pp. 323–324. [Google Scholar]
- Gisriel, S.D.; Yuan, J.; Braunberger, R.C.; Maracaja, D.L.; Chen, X.; Wu, X.; McCracken, J.; Chen, M.; Xie, Y.; Brown, L.E.; et al. Human herpesvirus 8-negative effusion-based large B-cell lymphoma: A distinct entity with unique clinicopathologic characteristics. Mod. Pathol. 2022, 35, 1411–1422. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; An, J.; Yoon, S.O.; Kim, M.; An, J.; Yoon, S.O.; Yong, S.H.; Kim, J.S.; Yang, W.I.; Leem, A.Y.; et al. Human herpesvirus 8-negative effusion-based lymphoma with indolent clinical behavior in an elderly patient: A case report and literature review. Oncol. Lett. 2020, 20, 343. [Google Scholar] [CrossRef]
- Chan, J.K.C.A.K.; Gaulard, P. Diffuse large B-cell lymphoma associated with chronic inflammation. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC: Lyon, France, 2017; pp. 309–311. [Google Scholar]
- Leoncini, L.C.E.; Stein, H.; Harris, N.L.; Jaffe, E.S.; Kluin, P.M. Burkitt-like lymphoma with 11q aberration. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC: Lyon, France, 2017; p. 334. [Google Scholar]
- Wagener, R.; Seufert, J.; Raimondi, F.; Bens, S.; Kleinheinz, K.; Nagel, I.; Altmüller, J.; Thiele, H.; Hübschmann, D.; Kohler, C.W.; et al. The mutational landscape of Burkitt-like lymphoma with 11q aberration is distinct from that of Burkitt lymphoma. Blood J. Am. Soc. Hematol. 2019, 133, 962–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Farre, B.; Ramis-Zaldivar, J.E.; Salmeron-Villalobos, J.; Balagué, O.; Celis, V.; Verdu-Amoros, J.; Nadeu, F.; Sábado, C.; Ferrández, A.; Garrido, M.; et al. Burkitt-like lymphoma with 11q aberration: A germinal center-derived lymphoma genetically unrelated to Burkitt lymphoma. Haematologica 2019, 104, 1822–1829. [Google Scholar] [CrossRef]
- Cucco, F.; Barrans, S.; Sha, C.; Clipson, A.; Crouch, S.; Dobson, R.; Chen, Z.; Thompson, J.S.; Care, M.A.; Cummin, T.; et al. Distinct genetic changes reveal evolutionary history and heterogeneous molecular grade of DLBCL with MYC/BCL2 double-hit. Leukemia 2020, 34, 1329–1341. [Google Scholar] [CrossRef]
- Evrard, S.M.; Péricart, S.; Grand, D.; Amara, N.; Escudié, F.; Gilhodes, J.; Bories, P.; Traverse-Glehen, A.; Dubois, R.; Brousset, P.; et al. Targeted next generation sequencing reveals high mutation frequency of CREBBP, BCL2 and KMT2D in high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements. Haematologica 2019, 104, e154–e157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landsburg, D.J.; Petrich, A.M.; Abramson, J.S.; Sohani, A.R.; Press, O.; Cassaday, R.; Chavez, J.C.; Song, K.; Zelenetz, A.D.; Gandhi, M.; et al. Impact of oncogene rearrangement patterns on outcomes in patients with double-hit non-Hodgkin lymphoma. Cancer 2016, 122, 559–564. [Google Scholar] [CrossRef]
- Li, S.; Desai, P.; Lin, P.; Yin, C.C.; Tang, G.; Wang, X.J.; Konoplev, S.N.; Khoury, J.D.; Bueso-Ramos, C.E.; Medeiros, L.J. MYC/BCL6 double-hit lymphoma (DHL): A tumour associated with an aggressive clinical course and poor prognosis. Histopathology 2016, 68, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Collinge, B.; Ben-Neriah, S.; Chong, L.; Boyle, M.; Jiang, A.; Miyata-Takata, T.; Farinha, P.; Craig, J.W.; Slack, G.W.; Ennishi, D.; et al. The impact of MYC and BCL2 structural variants in tumors of DLBCL morphology and mechanisms of false-negative MYC IHC. Blood 2021, 137, 2196–2208. [Google Scholar] [CrossRef] [PubMed]
- Rosenwald, A.; Bens, S.; Advani, R.; Barrans, S.; Copie-Bergman, C.; Elsensohn, M.H.; Natkunam, Y.; Calaminici, M.; Sander, B.; Baia, M.; et al. Prognostic Significance of MYC Rearrangement and Translocation Partner in Diffuse Large B-Cell Lymphoma: A Study by the Lunenburg Lymphoma Biomarker Consortium. J. Clin. Oncol. 2019, 37, 3359–3368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copie-Bergman, C.; Cuillière-Dartigues, P.; Baia, M.; Briere, J.; Delarue, R.; Canioni, D.; Salles, G.; Parrens, M.; Belhadj, K.; Fabiani, B.; et al. MYC-IG rearrangements are negative predictors of survival in DLBCL patients treated with immunochemotherapy: A GELA/LYSA study. Blood J. Am. Soc. Hematol. 2015, 126, 2466–2474. [Google Scholar] [CrossRef] [Green Version]
- McPhail, E.D.; Maurer, M.J.; Macon, W.R.; Feldman, A.L.; Kurtin, P.J.; Ketterling, R.P.; Vaidya, R.; Cerhan, J.R.; Ansell, S.M.; Porrata, L.F.; et al. Inferior survival in high-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements is not associated with MYC/IG gene rearrangements. Haematologica 2018, 103, 1899–1907. [Google Scholar] [CrossRef] [Green Version]
- Bonzheim, I.; Giese, S.; Deuter, C.; Süsskind, D.; Zierhut, M.; Waizel, M.; Szurman, P.; Federmann, B.; Schmidt, J.; Quintanilla-Martinez, L.; et al. High frequency of MYD88 mutations in vitreoretinal B-cell lymphoma: A valuable tool to improve diagnostic yield of vitreous aspirates. Blood J. Am. Soc. Hematol. 2015, 126, 76–79. [Google Scholar] [CrossRef] [Green Version]
- Lacy, S.E.; Barrans, S.L.; Beer, P.A.; Painter, D.; Smith, A.G.; Roman, E.; Cooke, S.L.; Ruiz, C.; Glover, P.; Van Hoppe, S.J.; et al. Targeted sequencing in DLBCL, molecular subtypes, and outcomes: A Haematological Malignancy Research Network report. Blood 2020, 135, 1759–1771. [Google Scholar] [CrossRef] [PubMed]
- Wright, G.W.; Phelan, J.D.; Coulibaly, Z.A.; Roulland, S.; Young, R.M.; Wang, J.Q.; Schmitz, R.; Morin, R.D.; Tang, J.; Jiang, A.; et al. A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 2020, 37, 551–568. [Google Scholar] [CrossRef]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Kamburov, A.; Redd, R.A.; Lawrence, M.S.; Roemer, M.G.; Li, A.J.; Ziepert, M.; et al. Author Correction: Molecular subtypes of diffuse large B cell lymphoma are associated with distinct pathogenic mechanisms and outcomes. Nat. Med. 2018, 24, 1290–1291. [Google Scholar] [CrossRef]
- Chapuy, B.; Roemer, M.G.; Stewart, C.; Tan, Y.; Abo, R.P.; Zhang, L.; Dunford, A.J.; Meredith, D.M.; Thorner, A.R.; Jordanova, E.S.; et al. Targetable genetic features of primary testicular and primary central nervous system lymphomas. Blood J. Am. Soc. Hematol. 2016, 127, 869–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, R.L.; Goodlad, J.R.; Calaminici, M.; Dotlic, S.; Montes-Moreno, S.; Oschlies, I.; Ponzoni, M.; Traverse-Glehen, A.; Ott, G.; Ferry, J.A. Lymphomas arising in immune-privileged sites: Insights into biology, diagnosis, and pathogenesis. Virchows Arch. 2020, 476, 647–665. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Schips, J.; Granai, M.; Quintanilla-Martinez, L.; Fend, F. The Grey Zones of Classic Hodgkin Lymphoma. Cancers 2022, 14, 742. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, E.S.; Stein, H.; Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L. B-cell lymphoma, unclassifiable, with features Intermediate between diffuse large B-cell lymphoma and classic Hodgkin lymphoma. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC: Lyon, France, 2017; pp. 342–344. [Google Scholar]
- Gaulard, P.; Kovrigina, A.M.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Moller, P.; Stein, H.; Gascoyne, R.D. Primary mediastinal large B-cell lymphoma. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC: Lyon, France, 2017; pp. 314–316. [Google Scholar]
- Gascoyne, R.D.; Chan, J.K.C.; Campo, E.; Rosenwald, A.; Jaffe, E.S.; Stein, H.; Chan, W.C.; Swerdlow, S.H. Diffuse large B-cell lymphoma, NOS. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues; IARC: Lyon, France, 2017; pp. 291–297. [Google Scholar]
- Lamarre, L.; Jacobson, J.O.; Aisenberg, A.C.; Harris, N.L. Primary large cell lymphoma of the mediastinum. A histologic and immunophenotypic study of 29 cases. Am. J. Surg. Pathol. 1989, 13, 730–739. [Google Scholar] [CrossRef]
- Pileri, S.A.; Gaidano, G.; Zinzani, P.L.; Falini, B.; Gaulard, P.; Zucca, E.; Pieri, F.; Berra, E.; Sabattini, E.; Ascani, S.; et al. Primary mediastinal B-cell lymphoma: High frequency of BCL-6 mutations and consistent expression of the transcription factors OCT-2, BOB.1, and PU.1 in the absence of immunoglobulins. Am. J. Pathol. 2003, 162, 243–253. [Google Scholar] [CrossRef]
- Wilson, W.H.; Pittaluga, S.; Nicolae, A.; Camphausen, K.; Shovlin, M.; Steinberg, S.M.; Roschewski, M.; Staudt, L.M.; Jaffe, E.S.; Dunleavy, K. A prospective study of mediastinal gray-zone lymphoma. Blood J. Am. Soc. Hematol. 2014, 124, 1563–1569. [Google Scholar] [CrossRef] [Green Version]
- Pittaluga, S.; Nicolae, A.; Wright, G.W.; Melani, C.; Roschewski, M.; Steinberg, S.; Huang, D.; Staudt, L.M.; Jaffe, E.S.; Wilson, W.H. Gene Expression Profiling of Mediastinal Gray Zone Lymphoma and Its Relationship to Primary Mediastinal B-cell Lymphoma and Classical Hodgkin Lymphoma. Blood Cancer Discov. 2020, 1, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Chapuy, B.; Stewart, C.; Dunford, A.J.; Kim, J.; Wienand, K.; Kamburov, A.; Griffin, G.K.; Chen, P.H.; Lako, A.; Redd, R.A.; et al. Genomic analyses of PMBL reveal new drivers and mechanisms of sensitivity to PD-1 blockade. Blood J. Am. Soc. Hematol. 2019, 134, 2369–2382. [Google Scholar] [CrossRef]
- Barth, T.F.; Leithäuser, F.; Möller, P. Mediastinal B-cell lymphoma, a lymphoma type with several characteristics unique among diffuse large B-cell lymphomas. Ann. Hematol. 2001, 80 (Suppl. S3), B49-53. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A.; Aiello, A.; Testi, A.; Cabras, A. B-cell lymphomas with features intermediate between distinct pathologic entities. From pathogenesis to pathology. Hum. Pathol. 2010, 41, 621–631. [Google Scholar]
- Van Krieken, J.H.O.M.; Elenitoba-Johnson, K.S.J.; Jaffee, E.S. Lymphoproliferative disease asspciated with primary immune disorders. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC: Lyon, France, 2017; pp. 444–448. [Google Scholar]
- Said, J.; Campo, E.; Rosenwald, A.; Harris, N.L. Lymphomas associated with HIV infection. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC: Lyon, France, 2017; pp. 449–452. [Google Scholar]
- Swerdlow, S.H.W.S.; Chadburn, A.; Ferry, J.A. Post-transplant lymphoproliferative disorders. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC: Lyon, France, 2017; pp. 453–462. [Google Scholar]
- Gaulard, P.S.S.; Harris, N.L.; Sundstrom, C.; Jaffe, E.S. Other iatrogenic immunodeficiency-associated lymphoproliferative disorders. In WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Revised 4th ed.; Swerdlow, S.H., Campo, E., Harris, N.L., Jaffe, E.S., Pileri, S.A., Stein, H., Thiele, J., Eds.; IARC: Lyon, France, 2017; pp. 462–464. [Google Scholar]
- Carbone, A.; Vaccher, E.; Gloghini, A. Hematologic cancers in individuals infected by HIV. Blood 2022, 139, 995–1012. [Google Scholar] [CrossRef] [PubMed]
- Natkunam, Y.; Gratzinger, D.; Chadburn, A.; Goodlad, J.R.; Chan, J.K.; Said, J.; Jaffe, E.S.; de Jong, D. Immunodeficiency-associated lymphoproliferative disorders: Time for reappraisal? Blood J. Am. Soc. Hematol. 2018, 132, 1871–1878. [Google Scholar] [CrossRef] [Green Version]
- Natkunam, Y.; Gratzinger, D.; de Jong, D.; Chadburn, A.; Goodlad, J.R.; Chan, J.K.; Said, J.; Jaffe, E.S. Immunodeficiency and Dysregulation: Report of the 2015 Workshop of the Society for Hematopathology/European Association for Haematopathology. Am. J. Clin. Pathol. 2017, 147, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Nador, R.G.; Chadburn, A.; Gundappa, G.; Cesarman, E.; Said, J.W.; Knowles, D.M. Human immunodeficiency virus (HIV)-associated polymorphic lymphoproliferative disorders. Am. J. Surg. Pathol. 2003, 27, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Tokuhira, M.; Tamaru, J.I.; Kizaki, M. Clinical management for other iatrogenic immunodeficiency-associated lymphoproliferative disorders. J. Clin. Exp. Hematop. 2019, 59, 72–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pina-Oviedo, S.; Miranda, R.N.; Medeiros, L.J. Cancer Therapy-associated Lymphoproliferative Disorders: An Under-recognized Type of Immunodeficiency-associated Lymphoproliferative Disorder. Am. J. Surg. Pathol. 2018, 42, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Gong, S.; Crane, G.M.; McCall, C.M.; Xiao, W.; Ganapathi, K.A.; Cuka, N.; Davies-Hill, T.; Xi, L.; Raffeld, M.; Pittaluga, S.; et al. Expanding the Spectrum of EBV-positive Marginal Zone Lymphomas: A Lesion Associated with Diverse Immunodeficiency Settings. Am. J. Surg. Pathol. 2018, 42, 1306–1316. [Google Scholar] [CrossRef]
- Knowles, D.M.; Cesarman, E.; Chadburn, A.; Frizzera, G.; Chen, J.; Rose, E.A.; Michler, R.E. Correlative morphologic and molecular genetic analysis demonstrates three distinct categories of posttransplantation lymphoproliferative disorders. Blood 1995, 85, 552–565. [Google Scholar] [CrossRef] [Green Version]
- Dojcinov, S.D.; Venkataraman, G.; Pittaluga, S.; Wlodarska, I.; Schrager, J.A.; Raffeld, M.; Hills, R.K.; Jaffe, E.S. Age-related EBV-associated lymphoproliferative disorders in the Western population: A spectrum of reactive lymphoid hyperplasia and lymphoma. Blood J. Am. Soc. Hematol. 2011, 117, 4726–4735. [Google Scholar] [CrossRef] [Green Version]
- Cesarman, E.; Chadburn, A.; Rubinstein, P.G. KSHV/HHV8-mediated hematologic diseases. Blood 2022, 139, 1013–1025. [Google Scholar] [CrossRef] [PubMed]
- Zanelli, M.; Sanguedolce, F.; Zizzo, M.; Palicelli, A.; Bassi, M.C.; Santandrea, G.; Martino, G.; Soriano, A.; Caprera, C.; Corsi, M.; et al. Primary effusion lymphoma occurring in the setting of transplanted patients: A systematic review of a rare, life-threatening post-transplantation occurrence. BMC Cancer 2021, 21, 468. [Google Scholar] [CrossRef] [PubMed]
- Nador, R.G.; Cesarman, E.; Chadburn, A.; Dawson, D.B.; Ansari, M.Q.; Sald, J.; Knowles, D.M. Primary effusion lymphoma: A distinct clinicopathologic entity associated with the Kaposi’s sarcoma-associated herpes virus. Blood 1996, 88, 645–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rossi, G.; Cozzi, I.; Della Starza, I.; De Novi, L.A.; De Propris, M.S.; Gaeta, A.; Petrucci, L.; Pulsoni, A.; Pulvirenti, F.; Ascoli, V. Human herpesvirus-8-positive primary effusion lymphoma in HIV-negative patients: Single institution case series with a multidisciplinary characterization. Cancer Cytopathol. 2021, 129, 62–74. [Google Scholar] [CrossRef]
- Zanelli, M.; Zizzo, M.; Bisagni, A.; Froio, E.; De Marco, L.; Valli, R.; Filosa, A.; Luminari, S.; Martino, G.; Massaro, F.; et al. Germinotropic lymphoproliferative disorder: A systematic review. Ann. Hematol. 2020, 99, 2243–2253. [Google Scholar] [CrossRef] [PubMed]
- Chadburn, A.; Said, J.; Gratzinger, D.; Chan, J.K.; de Jong, D.; Jaffe, E.S.; Natkunam, Y.; Goodlad, J.R. HHV8/KSHV-Positive Lymphoproliferative Disorders and the Spectrum of Plasmablastic and Plasma Cell Neoplasms: 2015 SH/EAHP Workshop Report-Part 3. Am. J. Clin. Pathol. 2017, 147, 171–187. [Google Scholar] [CrossRef] [Green Version]
- Fajgenbaum, D.C.; Uldrick, T.S.; Bagg, A.; Frank, D.; Wu, D.; Srkalovic, G.; Simpson, D.; Liu, A.Y.; Menke, D.; Chandrakasan, S.; et al. International, evidence-based consensus diagnostic criteria for HHV-8-negative/idiopathic multicentric Castleman disease. Blood J. Am. Soc. Hematol. 2017, 129, 1646–1657. [Google Scholar] [CrossRef] [Green Version]
- Carbone, A.; Borok, M.; Damania, B.; Gloghini, A.; Polizzotto, M.N.; Jayanthan, R.K.; Fajgenbaum, D.C.; Bower, M. Castleman disease. Nat. Rev. Dis. Prim. 2021, 7, 84. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WHO Classification, 5th Edition | WHO Classification, Revised 4th Edition |
|---|---|
| Follicular lymphoma | |
| In situ follicular B-cell neoplasm | In situ follicular neoplasia |
| Follicular lymphoma | (Same) |
| Pediatric-type follicular lymphoma | (Same) |
| Duodenal-type follicular lymphoma | (Same) |
| Cutaneous follicle center lymphoma | |
| Primary cutaneous follicle center lymphoma | (Same) |
| Transformations of indolent B-cell lymphomas | |
| Transformations of indolent B-cell lymphomas | Not previously included |
| Large B-cell lymphomas | |
| Diffuse large B-cell lymphoma, NOS | (Same) |
| T-cell/histiocyte-rich large B-cell lymphoma | (Same) |
| Diffuse large B-cell lymphoma/high grade B-cell lymphoma with MYC and BCL2 rearrangements | High-grade B-cell lymphoma with MYC and BCL2 and/or BCL6 rearrangements |
| ALK-positive large B-cell lymphoma | (Same) |
| Large B-cell lymphoma with IRF4 rearrangement | (Same) |
| High-grade B-cell lymphoma with 11q aberrations | Burkitt-like lymphoma with 11q aberration |
| Lymphomatoid granulomatosis | (Same) |
| EBV-positive diffuse large B-cell lymphoma | EBV-positive diffuse large B-cell lymphoma, NOS |
| Diffuse large B-cell lymphoma associated with chronic inflammation | (Same) |
| Fibrin-associated large B-cell lymphoma | Not previously included (previously considered a subtype of diffuse large B-cell lymphoma associated with chronic inflammation) |
| Fluid overload-associated large B-cell lymphoma | Not previously included |
| Plasmablastic lymphoma | (Same) |
| Primary large B-cell lymphoma of immune-privileged sites | Not previously included, encompassing primary diffuse large B-cell lymphoma of the CNS in revised 4th edition (plus primary large B-cell lymphoma of the vitreoretina and primary large B-cell lymphoma of the testis) |
| Primary cutaneous diffuse large B-cell lymphoma, leg type | (Same) |
| Intravascular large B-cell lymphoma | (Same) |
| Primary mediastinal large B-cell lymphoma | (Same) |
| Mediastinal grey zone lymphoma | B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and classic Hodgkin lymphoma |
| High-grade B-cell lymphoma, NOS | (Same) |
| Burkitt lymphoma | |
| Burkitt lymphoma | (Same) |
| KSHV/HHV8-associated B-cell lymphoid proliferations and lymphomas | |
| Primary effusion lymphoma | (Same) |
| KSHV/HHV8-positive diffuse large B-cell lymphoma | HHV8-positive diffuse large B-cell lymphoma, NOS |
| KSHV/HHV8-positive germinotropic lymphoproliferative disorder | HHV8-positive germinotropic lymphoproliferative disorder |
| Lymphoid proliferations and lymphomas associated with immune deficiency and dysregulation | |
| Hyperplasias arising in immune deficiency/dysregulation | Not previously included, encompassing non-destructive post-transplant lymphoproliferative disorders, among others |
| Polymorphic lymphoproliferative disorders arising in immune deficiency/dysregulation | Not previously included, encompassing polymorphic posttransplant lymphoproliferative disorders, other iatrogenic immunodeficiency-associated lymphoproliferative disorders, among others |
| EBV-positive mucocutaneous ulcer | (Same) |
| Lymphomas arising in immune deficiency/dysregulation | Not previously included, encompassing monomorphic posttransplant lymphoproliferative disorders, classic Hodgkin lymphoma posttransplant lymphoproliferative disorders, lymphomas associated with HIV infection, among others |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chadburn, A.; Gloghini, A.; Carbone, A. Classification of B-Cell Lymphomas and Immunodeficiency-Related Lymphoproliferations: What’s New? Hemato 2023, 4, 26-41. https://doi.org/10.3390/hemato4010003
Chadburn A, Gloghini A, Carbone A. Classification of B-Cell Lymphomas and Immunodeficiency-Related Lymphoproliferations: What’s New? Hemato. 2023; 4(1):26-41. https://doi.org/10.3390/hemato4010003
Chicago/Turabian StyleChadburn, Amy, Annunziata Gloghini, and Antonino Carbone. 2023. "Classification of B-Cell Lymphomas and Immunodeficiency-Related Lymphoproliferations: What’s New?" Hemato 4, no. 1: 26-41. https://doi.org/10.3390/hemato4010003