High-Level Production of Soluble Cross-Reacting Material 197 in Escherichia coli Cytoplasm Due to Fine Tuning of the Target Gene’s mRNA Structure

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Generation of the CRM197-Encoding Gene
2.2. Molecular Cloning
2.3. Mutagenesis
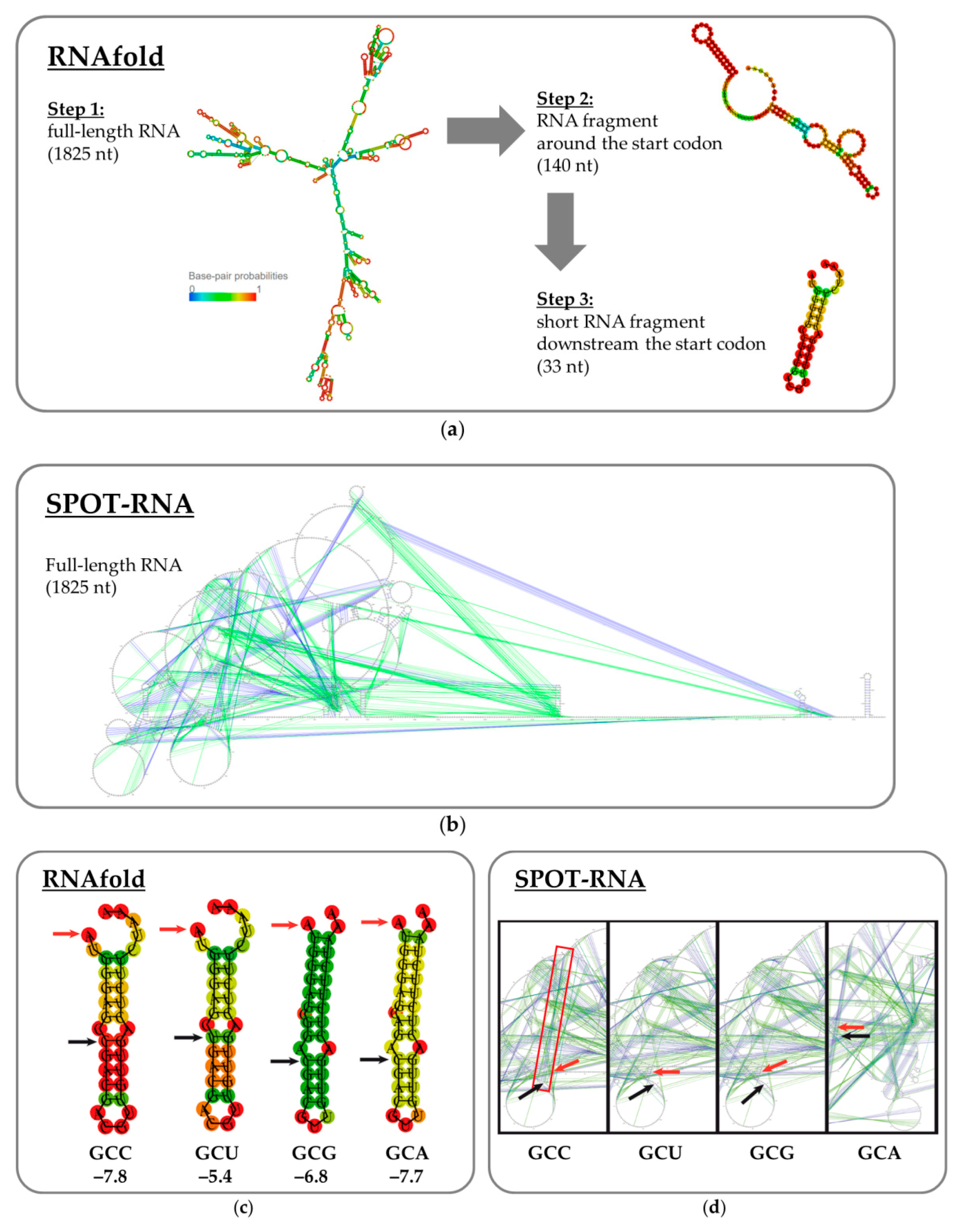
2.4. RNA Secondary Structure Analysis
2.5. Analytical Gene Expression
2.6. Kinetics of Gene Expression at Different Temperatures
2.7. Cell Cultivation in a Fermenter
2.8. Refolding of the Inclusion Body Fraction
2.9. Isolation of Periplasmic Proteins
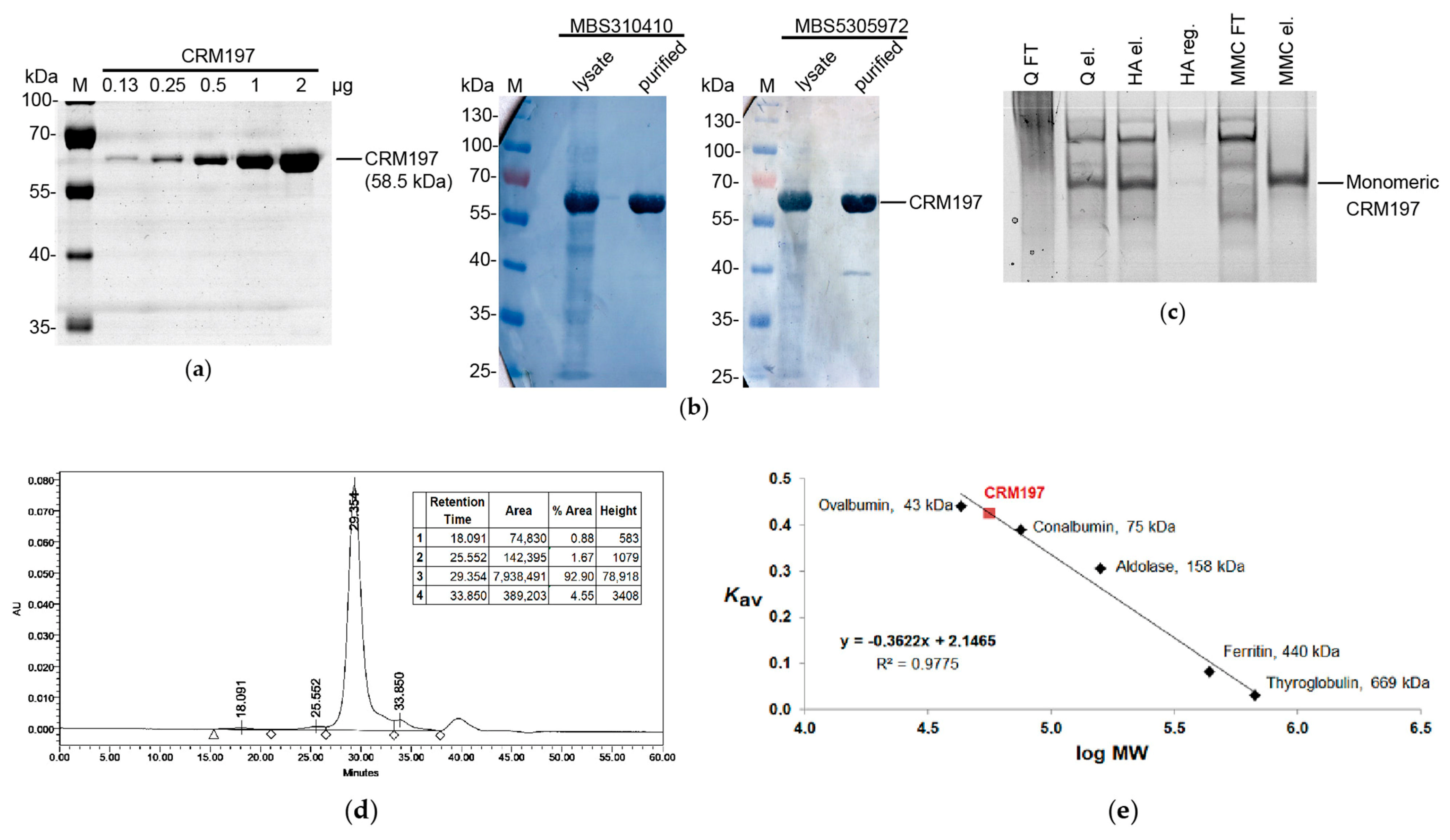
2.10. CRM197 Purification
2.11. Laemmli Electrophoresis
2.12. Western Blotting
2.13. Native Gel Electrophoresis
2.14. Analytical Size-Exclusion Chromatography
2.15. N-Terminal Protein Sequencing
3. Results
3.1. Gene and Vector Construction
3.2. Refolding of CRM197 from Inclusion Bodies
3.3. Periplasmic Expression of CRM197
3.4. Choice of Expression Strain and Vector
3.5. Optimization of Expression Temperature
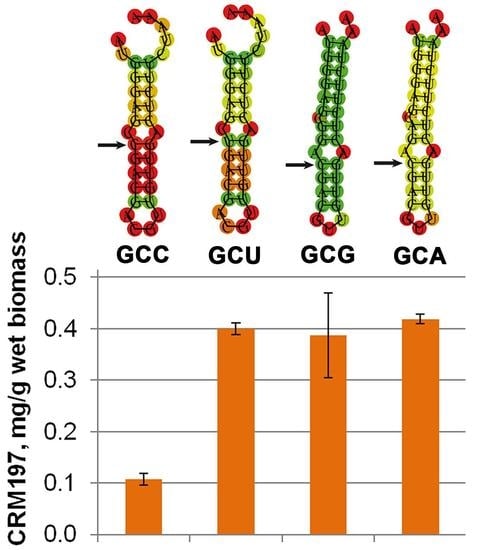
3.6. Mutagenesis of mRNA
3.7. Large-Scale CRM197 Expression and Purification
3.8. Analysis of the Purified CRM197
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Uchida, T.; Pappenheimer, A.M., Jr.; Greany, R. Diphtheria toxin and related proteins. I. Isolation and properties of mutant proteins serologically related to diphtheria toxin. J. Biol. Chem. 1973, 248, 3838–3844. [Google Scholar] [CrossRef]
- Malito, E.; Bursulaya, B.; Chen, C.; Lo Surdo, P.; Picchianti, M.; Balducci, E.; Biancucci, M.; Brock, A.; Berti, F.; Bottomley, M.J.; et al. Structural basis for lack of toxicity of the diphtheria toxin mutant CRM197. Proc. Natl. Acad. Sci. USA 2012, 109, 5229–5234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinefield, H.R. Overview of the development and current use of CRM(197) conjugate vaccines for pediatric use. Vaccine 2010, 28, 4335–4339. [Google Scholar] [CrossRef] [PubMed]
- Bröker, M.; Costantino, P.; DeTora, L.; McIntosh, E.D.; Rappuoli, R. Biochemical and biological characteristics of cross-reacting material 197 (CRM197), a non-toxic mutant of diphtheria toxin: Use as a conjugation protein in vaccines and other potential clinical applications. Biologicals 2011, 39, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Pichichero, M.E. Protein carriers of conjugate vaccines: Characteristics, development, and clinical trials. Hum. Vaccin. Immunother. 2013, 9, 2505–2523. [Google Scholar] [CrossRef] [Green Version]
- McCarthy, P.C.; Sharyan, A.; Sheikhi Moghaddam, L. Meningococcal vaccines: Current status and emerging strategies. Vaccines 2018, 6, 12. [Google Scholar] [CrossRef] [Green Version]
- McVernon, J.; MacLennan, J.; Clutterbuck, E.; Buttery, J.; Moxon, E.R. Effect of infant immunisation with meningococcus serogroup C-CRM(197) conjugate vaccine on diphtheria immunity and reactogenicity in pre-school aged children. Vaccine 2003, 21, 2573–2579. [Google Scholar] [CrossRef]
- Huo, Z.; Sinha, R.; McNeela, E.A.; Borrow, R.; Giemza, R.; Cosgrove, C.; Heath, P.T.; Mills, K.H.; Rappuoli, R.; Griffin, G.E.; et al. Induction of protective serum meningococcal bactericidal and diphtheria-neutralizing antibodies and mucosal immunoglobulin A in volunteers by nasal insufflations of the Neisseria meningitidis serogroup C polysaccharide-CRM197 conjugate vaccine mixed with chitosan. Infect. Immun. 2005, 73, 8256–8265. [Google Scholar] [CrossRef] [Green Version]
- Stickings, P.; Peyre, M.; Coombes, L.; Muller, S.; Rappuoli, R.; Del Giudice, G.; Partidos, C.D.; Sesardic, D. Transcutaneous immunization with cross-reacting material CRM(197) of diphtheria toxin boosts functional antibody levels in mice primed parenterally with adsorbed diphtheria toxoid vaccine. Infect. Immun. 2008, 76, 1766–1773. [Google Scholar] [CrossRef] [Green Version]
- Rappuoli, R.; Perugini, M.; Marsili, I.; Fabbiani, S. Rapid purification of diphtheria toxin by phenyl sepharose and DEAE-cellulose chromatography. J. Chromatogr. A 1983, 268, 543–548. [Google Scholar] [CrossRef]
- Stefan, A.; Conti, M.; Rubboli, D.; Ravagli, L.; Presta, E.; Hochkoeppler, A. Overexpression and purification of the recombinant diphtheria toxin variant CRM197 in Escherichia coli. J. Biotechnol. 2011, 156, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Mahamad, P.; Boonchird, C.; Panbangred, W. High level accumulation of soluble diphtheria toxin mutant (CRM197) with co-expression of chaperones in recombinant Escherichia coli. Appl. Microbial. Biotechnol. 2016, 100, 6319–6330. [Google Scholar] [CrossRef]
- Uthailak, N.; Mahamad, P.; Chittavanich, P.; Yanarojana, S.; Wijagkanalan, W.; Petre, J.; Panbangred, W. Molecular cloning, structural modeling and the production of soluble triple-mutated diphtheria toxoid (K51E/G52E/E148K) co-expressed with molecular chaperones in recombinant Escherichia coli. Mol. Biotechnol. 2017, 59, 117–127. [Google Scholar] [CrossRef]
- Goffin, P.; Dewerchin, M.; De Rop, P.; Blais, N.; Dehottay, P. High-yield production of recombinant CRM197, a non-toxic mutant of diphtheria toxin, in the periplasm of Escherichia coli. Biotechnol. J. 2017, 12, 1700168. [Google Scholar] [CrossRef] [PubMed]
- Roth, R.; van Zyl, P.; Tsekoa, T.; Stoychev, S.; Mamputha, S.; Buthelezi, S.; Crampton, M. Co-expression of sulphydryl oxidase and protein disulphide isomerase in Escherichia coli allows for production of soluble CRM197. J. Appl. Microbiol. 2017, 122, 1402–1411. [Google Scholar] [CrossRef]
- Park, A.R.; Jang, S.W.; Kim, J.S.; Park, Y.G.; Koo, B.S.; Lee, H.C. Efficient recovery of recombinant CRM197 expressed as inclusion bodies in E. coli. PLoS ONE 2018, 13, e0201060. [Google Scholar] [CrossRef]
- Mishra, R.P.N.; Yadav, R.S.P.; Jones, C.; Nocadello, S.; Minasov, G.; Shuvalova, L.A.; Anderson, W.F.; Goel, A. Structural and immunological characterization of E. coli derived recombinant CRM197 protein used as carrier in conjugate vaccines. Biosci. Rep. 2018, 38, BSR20180238. [Google Scholar] [CrossRef] [Green Version]
- Tarahomjoo, S.; Bandehpour, M.; Aghaebrahimian, M.; Ahangaran, S. Soluble diphtheria toxin variant, CRM 197 was obtained in Escherichia coli at high productivity using SUMO fusion and an adjusted expression strategy. Protein Pept. Lett. 2022, 29, 350–359. [Google Scholar] [CrossRef]
- Oganesyan, N.; Lees, A. Expression and Purification of CRM197 and Related Proteins. WO2015117093A1, 31 January 2014. [Google Scholar]
- Blattner, C.R.; Frisch, D.A.; Novy, R.E.; Henker, T.M.; Steffen, E.A.; Blattner, F.R.; Choi, H.; Posfai, G.; Landry, C.F. Enhanced Production of Recombinant CRM197 in E. coli. U.S. Patent US10479820B2, 3 March 2014. [Google Scholar]
- Zhou, J.; Petracca, R. Secretory expression of recombinant diphtheria toxin mutants in B. subtilis. J. Tongji Med. Univ. 1999, 19, 253–256. [Google Scholar] [CrossRef]
- Puppala, L.; Mudili, V. Nucleic Acid Encoding CRM197 and Process for Improved Expression Thereof. WO2019043593A1, 29 August 2018. [Google Scholar]
- Retallack, D.M.; Chew, L.; Jin, H. High Level Expression of Recombinant CRM197. WO2011123139A1, 30 March 2010. [Google Scholar]
- Orr, N.; Galen, J.E.; Levine, M.M. Expression and immunogenicity of a mutant diphtheria toxin molecule, CRM(197), and its fragments in Salmonella typhi vaccine strain CVD 908-htrA. Infect. Immun. 1999, 67, 4290–4294. [Google Scholar] [CrossRef]
- Aw, R.; Ashik, M.R.; Islam, A.; Khan, I.; Mainuddin, M.; Islam, M.A.; Ahasan, M.M.; Polizzi, K.M. Production and purification of an active CRM197 in Pichia pastoris and its immunological characterization using a Vi-typhoid antigen vaccine. Vaccine 2021, 39, 7379–7386. [Google Scholar] [CrossRef] [PubMed]
- Brodzik, R.; Spitsin, S.; Pogrebnyak, N.; Bandurska, K.; Portocarrero, C.; Andryszak, K.; Koprowski, H.; Golovkin, M. Generation of plant-derived recombinant DTP subunit vaccine. Vaccine 2009, 27, 3730–3734. [Google Scholar] [CrossRef]
- Hickey, J.M.; Toprani, V.M.; Kaur, K.; Mishra, R.; Goel, A.; Oganesyan, N.; Lees, A.; Sitrin, R.; Joshi, S.B.; Volkin, D.B. Analytical comparability assessments of 5 recombinant CRM197 proteins from different manufacturers and expression systems. J. Pharm. Sci. 2018, 107, 1806–1819. [Google Scholar] [CrossRef] [Green Version]
- Hashemzadeh, M.S.; Mohammadi, M.; Ghaleh, H.; Sharti, M.; Choopani, A.; Panda, A.K. Expression, solubilization, refolding and final purification of recombinant proteins as expressed in the form of “classical inclusion bodies” in E. coli. Protein Pept. Lett. 2021, 28, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Orlova, N.A.; Vorobiev, I.I. Plasmid Vector pHYP with High Segregation Stability for Expression of Recombinant Protein, Bacterium—Producent of Precursor of Recombinant Protein and Method to Produce Recombinant Protein. RU2496877C2, 15 December 2011. [Google Scholar]
- Gerdes, K.; Larsen, J.E.; Molin, S. Stable inheritance of plasmid R1 requires two different loci. J. Bacteriol. 1985, 161, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Hayes, F. Toxins-antitoxins: Plasmid maintenance, programmed cell death, and cell cycle arrest. Science 2003, 301, 1496–1499. [Google Scholar] [CrossRef] [PubMed]
- Studier, F.W.; Rosenberg, A.H.; Dunn, J.J.; Dubendorff, J.W. Use of T7 RNA polymerase to direct expression of cloned genes. Methods Enzymol. 1990, 185, 60–89. [Google Scholar] [CrossRef]
- Lobstein, J.; Emrich, C.A.; Jeans, C.; Faulkner, M.; Riggs, P.; Berkmen, M. SHuffle, a novel Escherichia coli protein expression strain capable of correctly folding disulfide bonded proteins in its cytoplasm. Microb. Cell Fact. 2012, 11, 56. [Google Scholar] [CrossRef] [Green Version]
- Froger, A.; Hall, J.E. Transformation of plasmid DNA into E. coli using the heat shock method. J. Vis. Exp. 2007, 6, e253. [Google Scholar] [CrossRef] [Green Version]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Singh, J.; Hanson, J.; Paliwal, K.; Zhou, Y. RNA secondary structure prediction using an ensemble of two-dimensional deep neural networks and transfer learning. Nat. Commun. 2019, 10, 5407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunelle, J.L.; Green, R. One-dimensional SDS-polyacrylamide gel electrophoresis (1D SDS-PAGE). Methods Enzymol. 2014, 541, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Arndt, C.; Koristka, S.; Feldmann, A.; Bergmann, R.; Bachmann, M. Coomassie Brilliant Blue staining of polyacrylamide gels. Methods Mol. Biol. 2018, 1853, 27–30. [Google Scholar] [CrossRef] [PubMed]
- Scopsi, L.; Larsson, L.I. Increased sensitivity in peroxidase immunocytochemistry. A comparative study of a number of peroxidase visualization methods employing a model system. Histochemistry 1986, 84, 221–230. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Ke, N.; Berkmen, M. Use of the SHuffle strains in production of proteins. Curr. Protoc. Protein. Sci. 2016, 85, 5.26.1–5.26.21. [Google Scholar] [CrossRef]
- Ma, Z.; Chen, B.; Zhang, F.; Yu, M.; Liu, T.; Liu, L. Increasing the expression levels of papillomavirus major capsid protein in Escherichia coli by N-terminal deletion. Protein Expr. Purif. 2007, 56, 72–79. [Google Scholar] [CrossRef]
- Wei, M.; Wang, D.; Li, Z.; Song, S.; Kong, X.; Mo, X.; Yang, Y.; He, M.; Li, Z.; Huang, B.; et al. N-terminal truncations on L1 proteins of human papillomaviruses promote their soluble expression in Escherichia coli and self-assembly in vitro. Emerg. Microbes Infect. 2018, 7, 160. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Texada, D.E. Low-usage codons and rare codons of Escherichia coli. Gene Ther. Mol. Biol. 2006, 10, 1–12. [Google Scholar]
- Meinnel, T.; Mechulam, Y.; Blanquet, S. Methionine as translation start signal: A review of the enzymes of the pathway in Escherichia coli. Biochimie 1993, 75, 1061–1075. [Google Scholar] [CrossRef]
- Daly, R.; Hearn, M.T. Expression of heterologous proteins in Pichia pastoris: A useful experimental tool in protein engineering and production. J. Mol. Recognit. 2005, 18, 119–138. [Google Scholar] [CrossRef]
- Berger, M.; Kaup, M.; Blanchard, V. Protein glycosylation and its impact on biotechnology. Adv. Biochem. Eng. Biotechnol. 2012, 127, 165–185. [Google Scholar] [CrossRef]
- Thak, E.J.; Yoo, S.J.; Moon, H.Y.; Kang, H.A. Yeast synthetic biology for designed cell factories producing secretory recombinant proteins. FEMS Yeast Res. 2020, 20, foaa009. [Google Scholar] [CrossRef] [PubMed]
- Derman, A.I.; Prinz, W.A.; Belin, D.; Beckwith, J. Mutations that allow disulfide bond formation in the cytoplasm of Escherichia coli. Science 1993, 262, 1744–1747. [Google Scholar] [CrossRef] [PubMed]
- Bessette, P.H.; Aslund, F.; Beckwith, J.; Georgiou, G. Efficient folding of proteins with multiple disulfide bonds in the Escherichia coli cytoplasm. Proc. Natl. Acad. Sci. USA 1999, 96, 13703–13708. [Google Scholar] [CrossRef] [Green Version]
- Ritz, D.; Lim, J.; Reynolds, C.M.; Poole, L.B.; Beckwith, J. Conversion of a peroxiredoxin into a disulfide reductase by a triplet repeat expansion. Science 2001, 294, 158–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrer, M.; Chernikova, T.N.; Yakimov, M.M.; Golyshin, P.N.; Timmis, K.N. Chaperonins govern growth of Escherichia coli at low temperatures. Nat. Biotechnol. 2003, 21, 1266–1267. [Google Scholar] [CrossRef]
- Schein, C. Production of soluble recombinant proteins in bacteria. Nat. Biotechnol. 1989, 7, 1141–1149. [Google Scholar] [CrossRef]
- Sørensen, H.P.; Mortensen, K.K. Soluble expression of recombinant proteins in the cytoplasm of Escherichia coli. Microb. Cell Fact. 2005, 4, 1. [Google Scholar] [CrossRef] [Green Version]
- Bhatwa, A.; Wang, W.; Hassan, Y.I.; Abraham, N.; Li, X.Z.; Zhou, T. Challenges associated with the formation of recombinant protein inclusion bodies in Escherichia coli and strategies to address them for industrial applications. Front. Bioeng. Biotechnol. 2021, 9, 630551. [Google Scholar] [CrossRef]
- Kiefhaber, T.; Rudolph, R.; Kohler, H.H.; Buchner, J. Protein aggregation in vitro and in vivo: A quantitative model of the kinetic competition between folding and aggregation. Biotechnology 1991, 9, 825–829. [Google Scholar] [CrossRef]





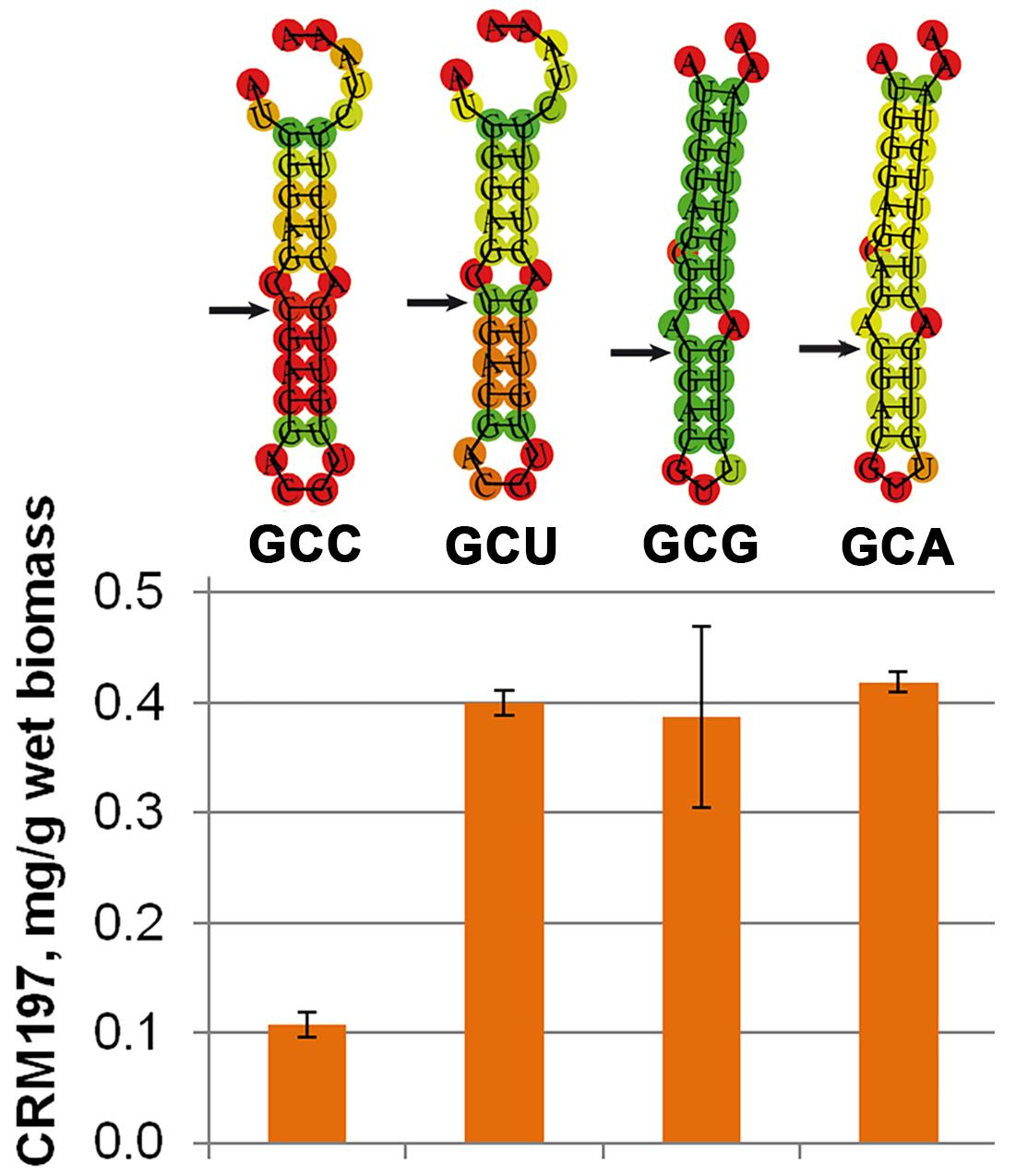{kind=link}
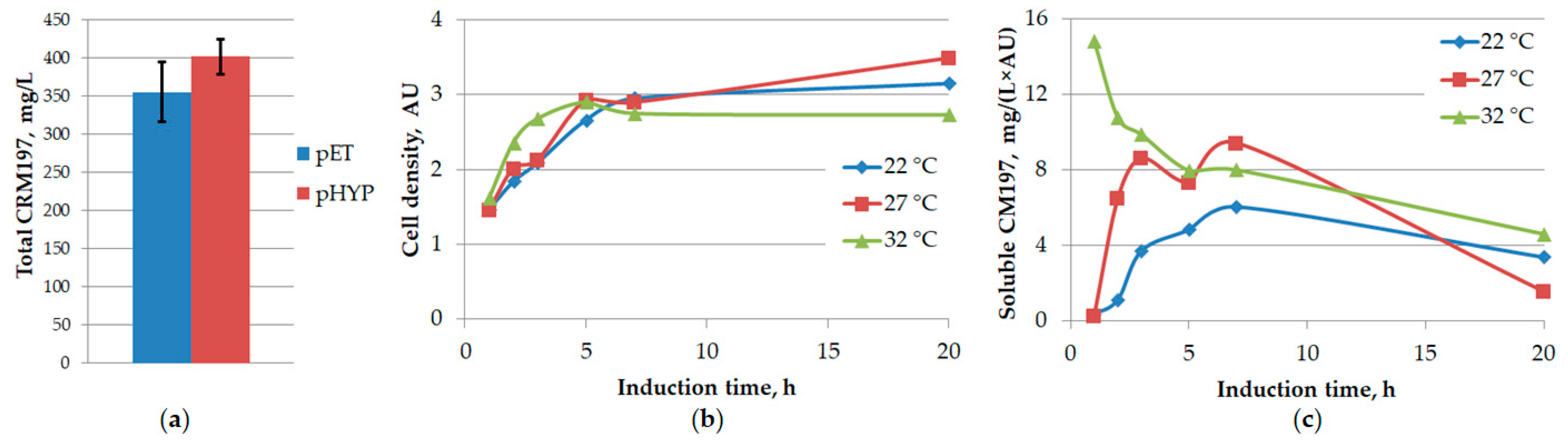{kind=link}
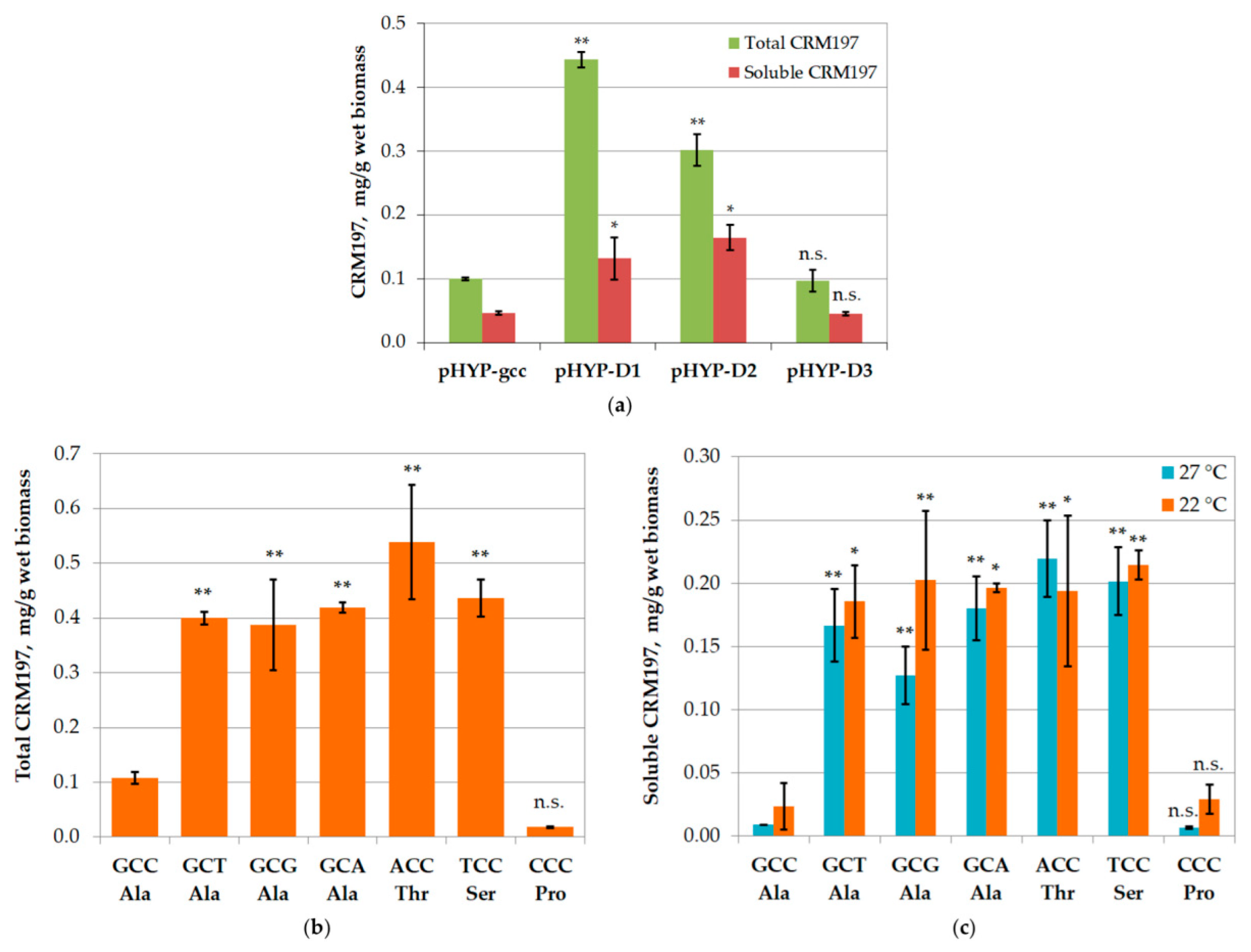{kind=link}
{kind=link}
{kind=link}
| Vaccine | Marketing Authorization Holder | Bacterial Polysaccharides | CRM197 Amount in One Dose, µg |
|---|---|---|---|
| Menveo | GSK Vaccines S.r.l. | Neisseria meningitidis polysaccharides, serogroups A, C, Y, and W-135 | 32.7–64.1 |
| Menjugate | GSK Vaccines S.r.l. | Neisseria meningitidis polysaccharides, serogroup C (strain C11) | 12.5–25.0 |
| Prevenar 13 | Pfizer Europe MA EEIG | Streptococcus pneumoniae polysaccharides, serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, and 23F | 32 |
| Vaxneuvance | Merck Sharp & Dohme B.V. | Streptococcus pneumoniae polysaccharides, serotypes 1, 3, 4, 5, 6A, 6B, 7F, 9V, 14, 18C, 19A, 19F, 22F, 23F, and 33F | 30 |
| Year | Expression Host | Compartmentalization | Solubility | Refolding Required | Protein Processing Required | Fermentation Scale | Cell Density | Protein Yield, mg/L | Reference |
|---|---|---|---|---|---|---|---|---|---|
| 1983 | Corynebacterium diphtheriae | Secreted | Soluble | No | No | 5-L fermenter | Low | 175–250 3 | [10] |
| 1999 | Bacillus subtilis | Secreted | Soluble | No | No | Shake flask | Low | 7.1 3 | [21] |
| 2011 | Escherichia coli | Cytoplasmic | Insoluble (inclusion bodies) | Yes | Yes 1 | Shake flask | Low | 250 ± 50 4 | [11] |
| 2016 | E. coli | Cytoplasmic | Soluble + insoluble | No | Yes 1 | Shake flask | Low | 154 ± 13 3 | [12] |
| 2017 | E. coli | Periplasmic | Soluble (?) 5 | No | No | 150-L fermenter | High | 3200 4,5 | [14] |
| 2017 | E. coli | Cytoplasmic | Soluble + insoluble | No | No | 2-L fermenter | High | 106 ± 1.5 3 | [15] |
| 2018 | E. coli | Cytoplasmic | Insoluble (inclusion bodies) | Yes | Yes 1 | Shake flask | Low | 196 4 | [16] |
| 2018 | E. coli | Cytoplasmic | Insoluble (inclusion bodies) | Yes | No | 20-L fermenter | High | Not specified | [17] |
| 2021 | Pichia pastoris | Secreted | Soluble | No | No | 16-L bioreactor | High | 113 6 | [25] |
| 2022 | E. coli | Cytoplasmic | Soluble + insoluble | No | Yes 2 | Shake flask | Low | 130 4 | [18] |
| 2022 | E. coli | Cytoplasmic | Soluble | No | No | 16-L fermenter | High | 150–270 6 | This work |
| Plasmid Name | Original Vector | Mutation Type | The Initial Codons of the Target Gene | The Initial Amino Acid Residues of the Target Protein |
|---|---|---|---|---|
| pET28a-CRM197-gcc | pET28a | None | ATGGGAGCCGACGAC | MGADD |
| pHYP-CRM197-gcc | pHYP | None | ATGGGAGCCGACGAC | MGADD |
| pHYP-CRM197-peri | pHYP | Leader peptide insertion | ATGATTAAATTTCTCTCTGCATTAATTCTTCTACTGGTCACGACGGCGGCTCAGGCTGGAGCCGACGAC | MIKFLSALILLLVTTAAQAGADD |
| pHYP-D1 | pHYP | Deletion | ATG---GCCGACGAC | M-ADD |
| pHYP-D2 | pHYP | Deletion | ATG------GACGAC | M--DD |
| pHYP-D3 | pHYP | Deletion | ATG---------GAC | M---D |
| pHYP-CRM197-gca | pHYP | Synonymous substitution | ATGGGAGCAGACGAC | MGADD |
| pHYP-CRM197-gcg | pHYP | Synonymous substitution | ATGGGAGCGGACGAC | MGADD |
| pHYP-CRM197-gct | pHYP | Synonymous substitution | ATGGGAGCTGACGAC | MGADD |
| pHYP-CRM197-acc | pHYP | Nonsynonymous substitution | ATGGGAACCGACGAC | MGTDD |
| pHYP-CRM197-tcc | pHYP | Nonsynonymous substitution | ATGGGATCCGACGAC | MGSDD |
| pHYP-CRM197-ccc | pHYP | Nonsynonymous substitution | ATGGGACCCGACGAC | MGPDD |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khodak, Y.A.; Ryazanova, A.Y.; Vorobiev, I.I.; Kovalchuk, A.L.; Ovechko, N.N.; Aparin, P.G. High-Level Production of Soluble Cross-Reacting Material 197 in Escherichia coli Cytoplasm Due to Fine Tuning of the Target Gene’s mRNA Structure. BioTech 2023, 12, 9. https://doi.org/10.3390/biotech12010009
Khodak YA, Ryazanova AY, Vorobiev II, Kovalchuk AL, Ovechko NN, Aparin PG. High-Level Production of Soluble Cross-Reacting Material 197 in Escherichia coli Cytoplasm Due to Fine Tuning of the Target Gene’s mRNA Structure. BioTech. 2023; 12(1):9. https://doi.org/10.3390/biotech12010009
Chicago/Turabian StyleKhodak, Yulia Alexandrovna, Alexandra Yurievna Ryazanova, Ivan Ivanovich Vorobiev, Alexander Leonidovich Kovalchuk, Nikolay Nikolaevich Ovechko, and Petr Gennadievich Aparin. 2023. "High-Level Production of Soluble Cross-Reacting Material 197 in Escherichia coli Cytoplasm Due to Fine Tuning of the Target Gene’s mRNA Structure" BioTech 12, no. 1: 9. https://doi.org/10.3390/biotech12010009