An Optimized CoBRA Method for the Microfluidic Electrophoresis Detection of Breast Cancer Associated RASSF1 Methylation



Abstract
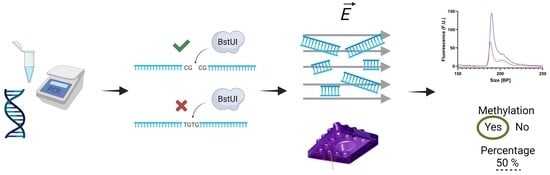
:
1. Introduction
2. Materials and Methods
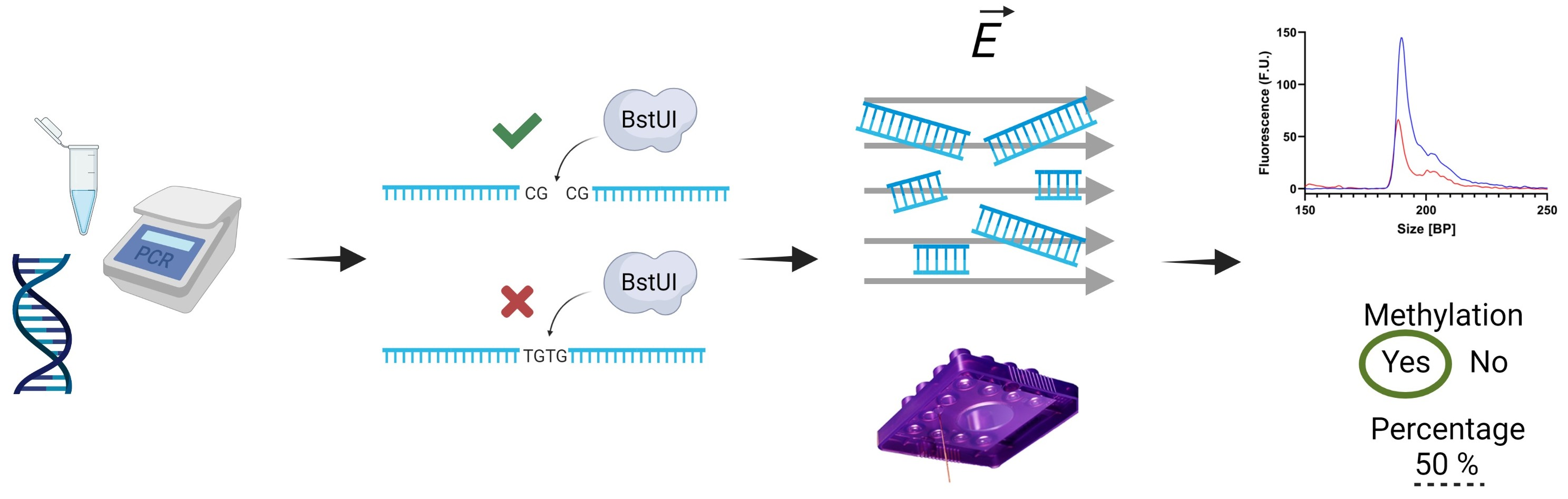
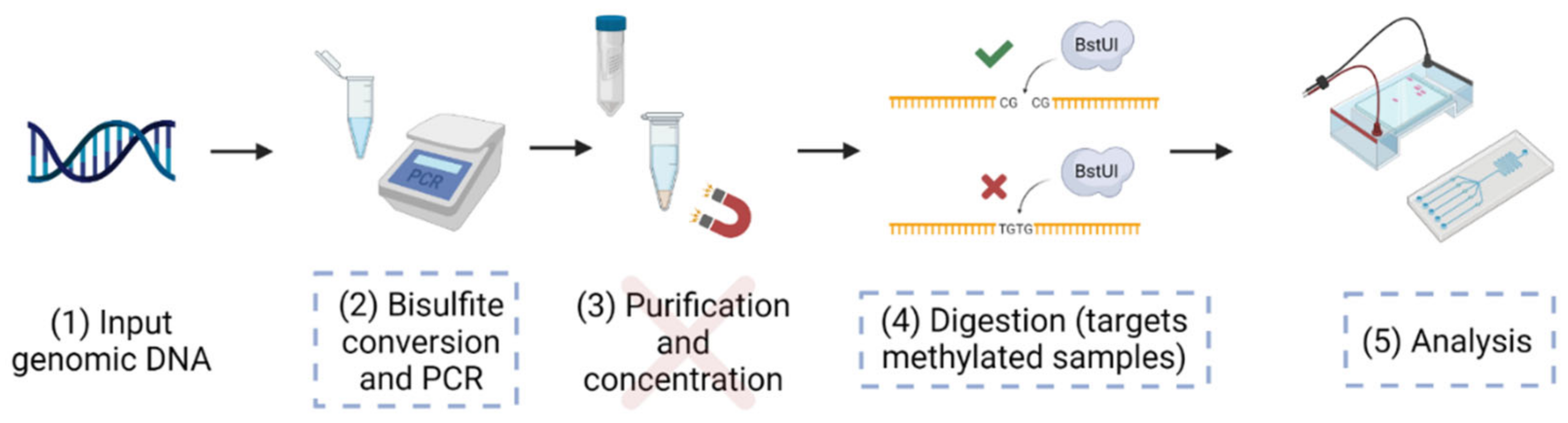
2.1. Bisulfite Treatment
2.2. Polymerase Chain Reaction (PCR)
2.3. Restriction Enzyme Digestion
2.4. Microfluidic Electrophoretic Analysis
3. Results
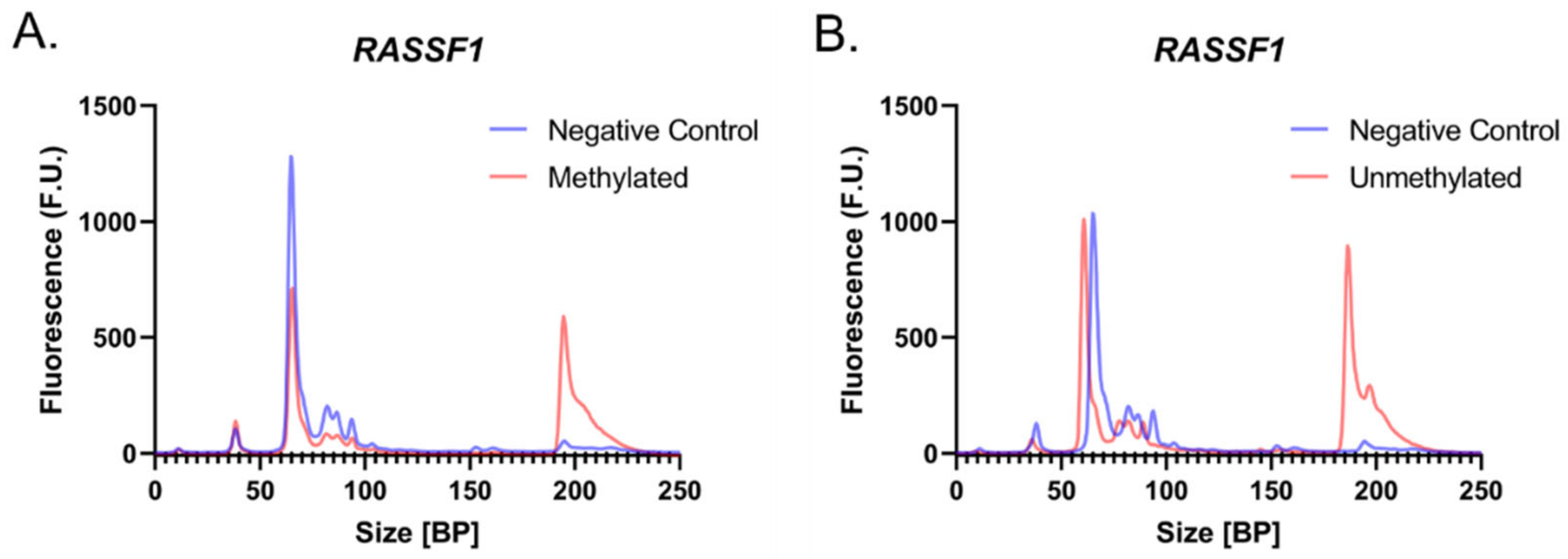
3.1. Primer Design and PCR Optimization for the Amplification of Fully Methylated and Fully Unmethylated Bisulfite Treated Samples
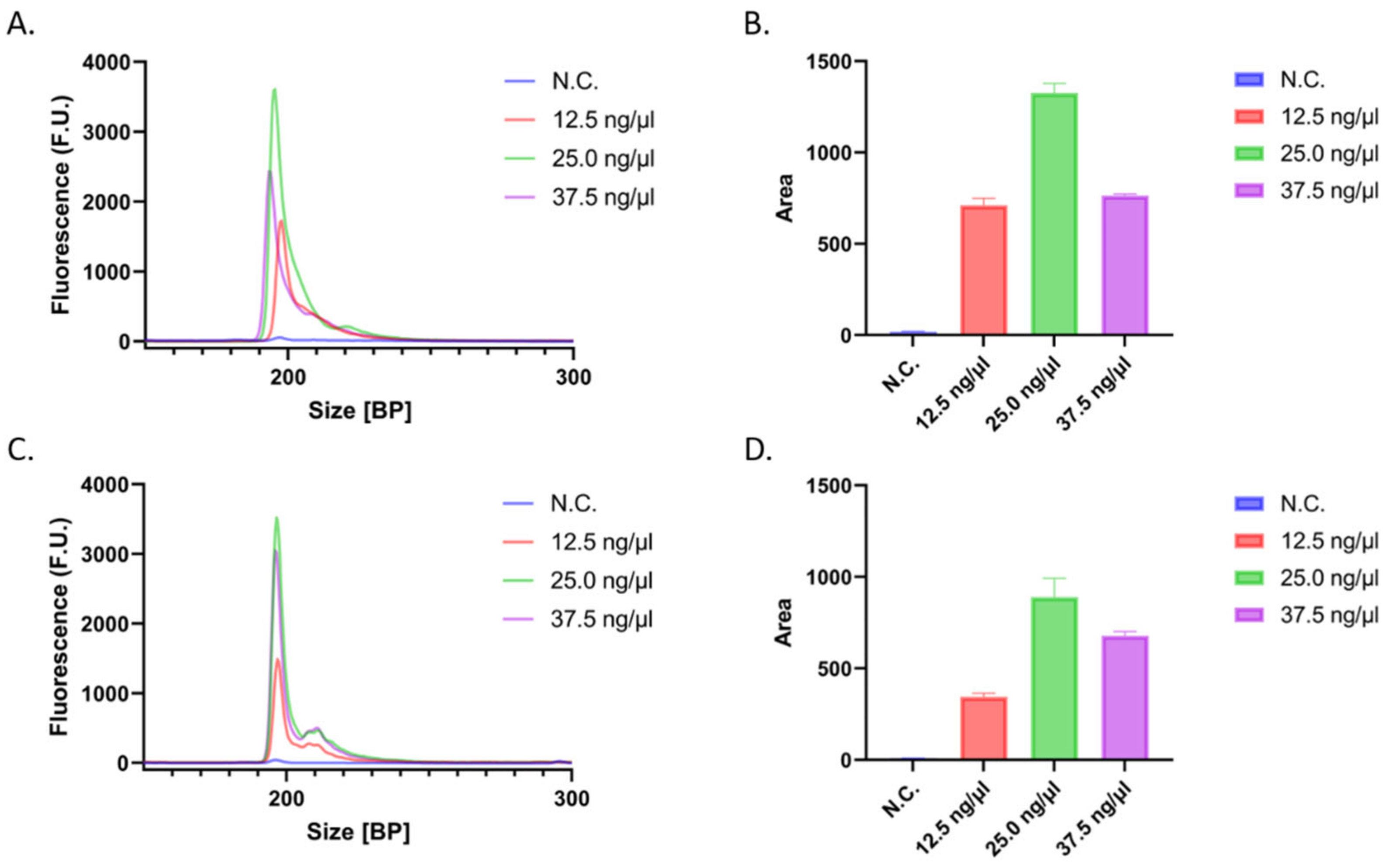
3.2. Initial Concentration Dependency of the System
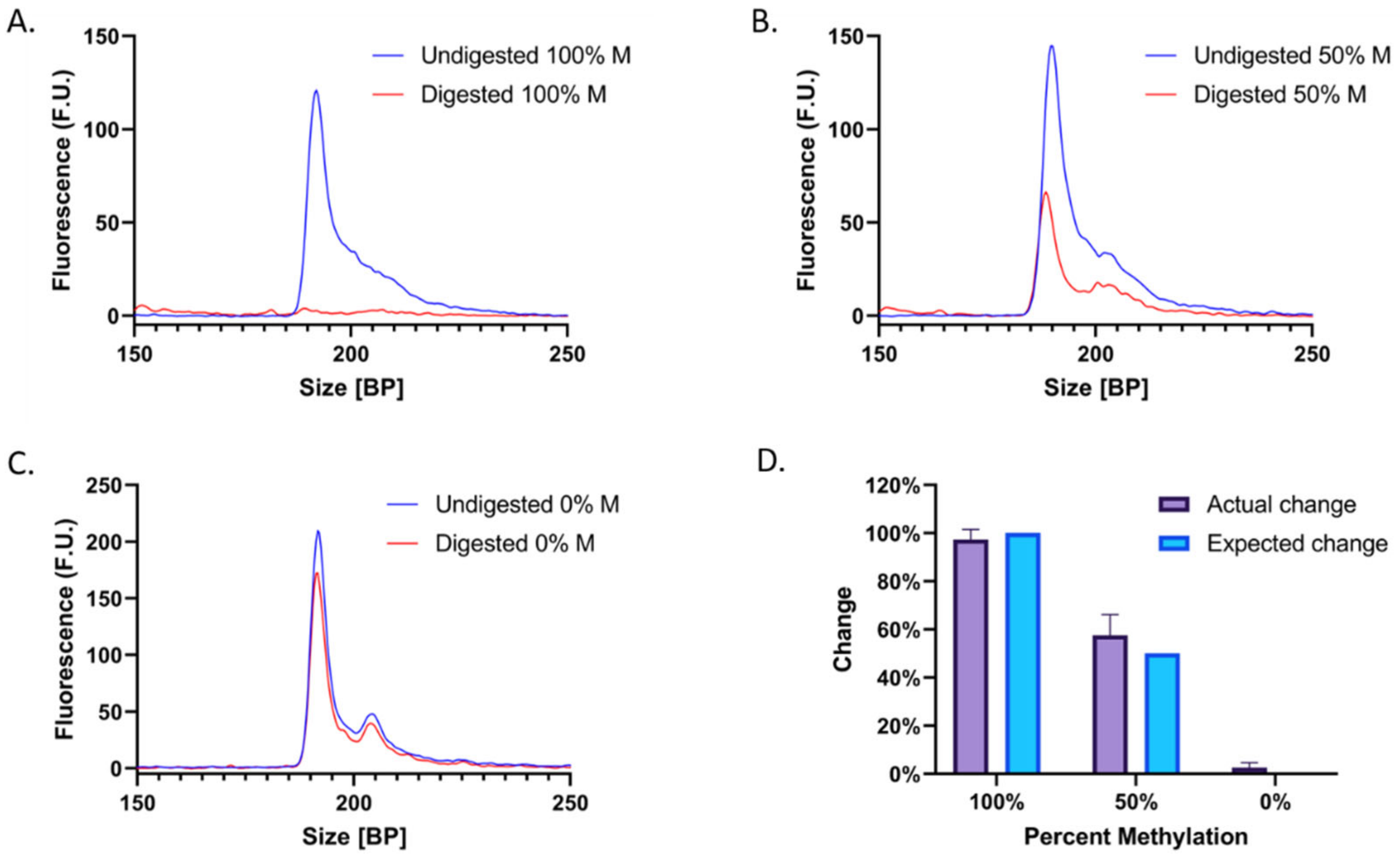
3.3. Methylation Quantification Accuracy
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, R.M.; Webb-Vargas, Y.; Wheeler, W.; Gail, M.H. Proportion of U.S. Trends in breast cancer incidence attributable to long-term changes in risk factor distributions. Cancer Epidemiol. Biomark. Prev. 2018, 27, 1214–1222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovelace, D.L.; McDaniel, L.R.; Golden, D. Long-Term Effects of Breast Cancer Surgery, Treatment, and Survivor Care. J. Midwifery Womens Health 2019, 64, 713–724. [Google Scholar] [CrossRef] [PubMed]
- Marmot, M.G.; Altman, D.G.; Cameron, D.A.; Dewar, J.A.; Thompson, S.G.; Wilcox, M. The benefits and harms of breast cancer screening: An independent review. Br. J. Cancer 2013, 108, 2205–2240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hortobagyi, G.N. Treatment of Breast Cancer. N. Engl. J. Med. 1998, 339, 974–984. [Google Scholar] [CrossRef]
- Bytnar, J.A.; Byrne, C.; Olsen, C.; Witkop, C.; Martin, M.B.; Banaag, A.; Koehlmoos, T. The Impact of Mammography Screening Guideline Changes in a Universally Insured Population. J. Womens Health 2021, 30, 1720–1728. [Google Scholar] [CrossRef]
- Pace, L.E. False-Positive Results of Mammography Screening in the Era of Digital Breast Tomosynthesis. JAMA Netw. Open 2022, 5, e222445. [Google Scholar] [CrossRef]
- Tsuruda, K.M.; Larsen, M.; Román, M.; Hofvind, S. Cumulative risk of a false-positive screening result: A retrospective cohort study using empirical data from 10 biennial screening rounds in Breast Screen Norway. Cancer 2022, 128, 1373–1380. [Google Scholar] [CrossRef]
- Ellisen, L.W.; Haber, D.A. Hereditary Breast Cancer. Annu. Rev. Med. 1998, 49, 425–436. [Google Scholar] [CrossRef]
- Moore, L.D.; Le, T.; Fan, G. DNA Methylation and Its Basic Function. Neuropsychopharmacology 2012, 38, 23–38. [Google Scholar] [CrossRef]
- Szyf, M. DNA methylation signatures for breast cancer classification and prognosis. Genome Med. 2012, 4, 26. [Google Scholar] [CrossRef]
- Cervera, R.; Ramos, A.; Lluch, A.; Climent, J. DNA Methylation in Breast Cancer. In Epigenetic Biomarkers and Diagnostics; Elsevier: Amsterdam, The Netherlands, 2016; pp. 297–312. [Google Scholar] [CrossRef]
- Buyru, N.; Altinisik, J.; Ozdemir, F.; Demokan, S.; Dalay, N. Methylation profiles in breast cancer. Cancer Investig. 2009, 27, 307–312. [Google Scholar] [CrossRef]
- Shinozaki, M.; Hoon, D.S.; Giuliano, A.E.; Hansen, N.M.; Wang, H.J.; Turner, R.; Taback, B. Distinct Hypermethylation Profile of Primary Breast Cancer Is Associated with Sentinel Lymph Node Metastasis. Clin. Cancer Res. 2005, 11, 2156–2162. [Google Scholar] [CrossRef] [Green Version]
- Salta, S.; Nunes, S.P.; Fontes-Sousa, M.; Lopes, P.; Freitas, M.; Caldas, M.; Antunes, L.; Castro, F.; Antunes, P.; Palma de Sousa, S.; et al. A DNA Methylation-Based Test for Breast Cancer Detection in Circulating Cell-Free DNA. J. Clin. Med. 2018, 7, 420. [Google Scholar] [CrossRef] [Green Version]
- Yuan, J.; Luo, R.Z.; Fujii, S.; Wang, L.; Hu, W.; Andreeff, M.; Pan, Y.; Kadota, M.; Oshimura, M.; Sahin, A.A.; et al. Aberrant Methylation and Silencing of ARHI, an Imprinted Tumor Suppressor Gene in which the Function Is Lost in Breast Cancers. Cancer Res. 2003, 63, 4174–4180. [Google Scholar]
- Esteller, M.; Silva, J.M.; Dominguez, G.; Bonilla, F.; Matias-Guiu, X.; Lerma, E.; Bussaglia, E.; Prat, J.; Harkes, I.C.; Repasky, E.A.; et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl. Cancer Inst. 2000, 92, 564–569. [Google Scholar] [CrossRef] [Green Version]
- Herman, J.G.; Merlo, A.; Mao, L.I.; Lapidus, R.G.; Issa, J.P.J.; Davidson, N.E.; Sidransky, D.; Baylin, S.B. Inactivation of the CDKN2/p16/MTS1 Gene Is Frequently Associated with Aberrant DNA Methylation in All Common Human Cancers. Cancer Res. 1995, 55, 4525–4530. [Google Scholar]
- Graff, J.R.; Herman, J.G.; Lapidus, R.G.; Chopra, H.; Xu, R.; Jarrard, D.F.; Isaacs, W.B.; Pitha, P.M.; Davidson, N.E.; Baylin, S.B. E-Cadherin Expression Is Silenced by DNA Hypermethylation in Human Breast and Prostate Carcinomas. Cancer Res. 1995, 55, 5195–5199. [Google Scholar]
- Lapidus, R.G.; Ferguson, A.T.; Ottaviano, Y.L.; Parl, F.F.; Smith, H.S.; Weitzman, S.A.; Baylin, S.B.; Issa, J.P.; Davidson, N.E. Methylation of estrogen and progesterone receptor gene 5′ CpG islands correlates with lack of estrogen and progesterone receptor gene expression in breast tumors. Clin. Cancer Res. 1996, 2, 805–810. [Google Scholar]
- Hagrass, H.A.; Pasha, H.F.; Ali, A.M. Estrogen receptor alpha (ERα) promoter methylation status in tumor and serum DNA in Egyptian breast cancer patients. Gene 2014, 552, 81–86. [Google Scholar] [CrossRef]
- Esteller, M.; Corn, P.G.; Urena, J.M.; Gabrielson, E.; Baylin, S.B.; Herman, J.G. Inactivation of Glutathione S-Transferase P1 Gene by Promoter Hypermethylation in Human Neoplasia. Cancer Res. 1998, 58, 4515–4518. [Google Scholar] [PubMed]
- Sirchia, S.M.; Ferguson, A.T.; Sironi, E.; Subramanyan, S.; Orlandi, R.; Sukumar, S.; Sacchi, N. Evidence of epigenetic changes affecting the chromatin state of the retinoic acid receptor β2 promoter in breast cancer cells. Oncogene 2000, 19, 1556–1563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kioulafa, M.; Kaklamanis, L.; Mavroudis, D.; Georgoulias, V.; Lianidou, E.S. Prognostic significance of RASSF1A promoter methylation in operable breast cancer. Clin. Biochem. 2009, 42, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Dammann, R.; Yang, G.; Pfeifer, G.P. Hypermethylation of the CpG Island of Ras Association Domain Family 1A (RASSF1A), a Putative Tumor Suppressor Gene from the 3p21.3 Locus, Occurs in a Large Percentage of Human Breast Cancers. Cancer Res. 2001, 61, 3105–3109. [Google Scholar] [PubMed]
- Feng, W.; Shen, L.; Wen, S.; Rosen, D.G.; Jelinek, J.; Hu, X.; Huan, S.; Huang, M.; Liu, J.; Sahin, A.A.; et al. Correlation between CpG methylation profiles and hormone receptor status in breast cancers. Breast Cancer Res. 2007, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Dallol, A.; Da Silva, N.F.; Viacava, P.; Minna, J.D.; Bieche, I.; Maher, E.R.; Latif, F. SLIT2, a Human Homologue of the Drosophila Slit2 Gene, Has Tumor Suppressor Activity and Is Frequently Inactivated in Lung and Breast Cancers. Cancer Res. 2002, 62, 5874–5880. [Google Scholar]
- Dickinson, R.E.; Dallol, A.; Bieche, I.; Krex, D.; Morton, D.; Maher, E.R.; Latif, F. Epigenetic inactivation of SLIT3 and SLIT1 genes in human cancers. Br. J. Cancer 2004, 91, 2071. [Google Scholar] [CrossRef] [Green Version]
- Dallol, A.; Forgacs, E.; Martinez, A.; Sekido, Y.; Walker, R.; Kishida, T.; Rabbitts, P.; Maher, E.R.; Minna, J.D.; Latif, F. Tumour specific promoter region methylation of the human homologue of the Drosophila Roundabout gene DUTT1 (ROBO1) in human cancers. Oncogene 2002, 21, 3020–3028. [Google Scholar] [CrossRef] [Green Version]
- Bachman, K.E.; Herman, J.G.; Corn, P.G.; Merlo, A.; Costello, J.F.; Cavenee, W.K.; Baylin, S.B.; Graff, J.R. Methylation-associated Silencing of the Tissue Inhibitor of Metalloproteinase-3 Gene Suggests a Suppressor Role in Kidney, Brain, and Other Human Cancers. Cancer Res. 1999, 59, 798–802. [Google Scholar]
- Umbricht, C.B.; Evron, E.; Gabrielson, E.; Ferguson, A.; Marks, J.; Sukumar, S. Hypermethylation of 14-3-3 σ (stratifin) is an early event in breast cancer. Oncogene 2001, 20, 3348–3353. [Google Scholar] [CrossRef] [Green Version]
- AFerguson, A.T.; Evron, E.; Umbricht, C.B.; Pandita, T.K.; Chan, T.A.; Hermeking, H.; Marks, J.R.; Lambers, A.R.; Futreal, P.A.; Stampfer, M.R.; et al. High frequency of hypermethylation at the 14-3-3 σ locus leads to gene silencing in breast cancer. Proc. Natl. Acad. Sci. USA 2000, 97, 6049. [Google Scholar] [CrossRef] [Green Version]
- Kurdyukov, S.; Bullock, M. DNA Methylation Analysis: Choosing the Right Method. Biology 2016, 5, 3. [Google Scholar] [CrossRef]
- Phillips, K.A.; Pletcher, M.J.; Ladabaum, U. Is the $1000 Genome really $1000? Understanding the full benefits and costs of genomic sequencing. Technol. Health Care 2015, 23, 373–379. [Google Scholar] [CrossRef] [Green Version]
- Pruneri, G.; De Braud, F.; Sapino, A.; Aglietta, M.; Vecchione, A.; Giusti, R.; Marchiò, C.; Scarpino, S.; Baggi, A.; Bonetti, G.; et al. Next-Generation Sequencing in Clinical Practice: Is It a Cost-Saving Alternative to a Single-Gene Testing Approach? Pharm.-Open 2021, 5, 285–298. [Google Scholar] [CrossRef]
- MedlinePlus. What Is the Cost of Genetic Testing, and How Long Does It Take to Get the Results? 2020. Available online: https://medlineplus.gov/ (accessed on 20 August 2022).
- Xiong, Z.; Laird, P.W. COBRA: A sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997, 25, 2532–2534. [Google Scholar] [CrossRef]
- Brena, R.M.; Auer, H.; Kornacker, K.; Plass, C. Quantification of DNA methylation in electrofluidics chips (Bio-COBRA). Nat. Protoc. 2006, 1, 52–58. [Google Scholar] [CrossRef]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [Green Version]
- Gardiner-Garden, M.; Frommer, M. CpG Islands in vertebrate genomes. J. Mol. Biol. 1987, 196, 261–282. [Google Scholar] [CrossRef]
- Walter, R.F.H.; Rozynek, P.; Casjens, S.; Werner, R.; Mairinger, F.D.; Speel, E.J.M.; Zur Hausen, A.; Meier, S.; Wohlschlaeger, J.; Theegarten, D.; et al. Methylation of L1RE1, RARB, and RASSF1 function as possible biomarkers for the differential diagnosis of lung cancer. PLoS ONE 2018, 13, e0195716. [Google Scholar] [CrossRef]
- Cohen, Y.; Singer, G.; Lavie, O.; Dong, M.; Beller, U.; Sidransky, D. The RASSF1A Tumor Suppressor Gene Is Commonly Inactivated in Adenocarcinoma of the Uterine Cervix. Clin. Cancer Res. 2003, 9, 2981–2984. [Google Scholar] [PubMed]
- Daniunaite, K.; Jarmalaite, S.; Kalinauskaite, N.; Petroska, D.; Laurinavicius, A.; Lazutka, J.R.; Jankevicius, F. Prognostic Value of RASSF1 Promoter Methylation in Prostate Cancer. J. Urol. 2014, 192, 1849–1855. [Google Scholar] [CrossRef] [PubMed]
- Kaminsky, Z.; Tochigi, M.; Jia, P.; Pal, M.; Mill, J.; Kwan, A.; Ioshikhes, I.; Vincent, J.B.; Kennedy, J.L.; Strauss, J.; et al. A multi-tissue analysis identifies HLA complex group 9 gene methylation differences in bipolar disorder. Mol. Psychiatry 2012, 17, 728–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andraos, C.; Koorsen, G.; Knight, J.C.; Bornman, L. Vitamin D receptor gene methylation is associated with ethnicity, tuberculosis, and Taq I polymorphism. HIM 2011, 72, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Huang, H.; Huang, R.; Zhang, W.; Zhou, G.; Wu, Z.; Lv, C.; Han, X.; Jiang, L.; Li, Y.; et al. SEPT9 Gene Methylation as a Noninvasive Marker for Hepatocellular Carcinoma. Dis. Markers 2020, 2020, 6289063. [Google Scholar] [CrossRef]
- Chen, D.; Meng, L.; Pei, F.; Zheng, Y.; Leng, J. A review of DNA methylation in depression. J. Clin. Neurosci. 2017, 43, 39–46. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Incidence (%) | Source(s) |
|---|---|---|
| APC | 10, 49, 51 | [13,14,15] |
| ARHI | 15–20 | [16] |
| BRCA1 | 5, 13 | [13,17] |
| CDH1 | 53 | [14] |
| CDKN2A | 31 | [18] |
| DAPK1 | 14 | [13] |
| E-cadherin | >50 | [19] |
| ERα | 25, 79 | [20,21] |
| FOXA1 | 62 | [15] |
| GSTP1 | 21, 30, 40 | [13,14,22] |
| PR | 46 | [20] |
| RARβ2 | 4, 24, 25 | [13,14,23] |
| RASSF1 | 48, 57, 58, 62, 79, 81 | [13,14,15,24,25,26] |
| SLIT2 | 43 | [27] |
| SLIT3 | 16 | [28] |
| ROBO1 | 19 | [29] |
| TIMP-3 | 5, 27 | [13,30] |
| TWIST | 48 | [14] |
| 14-3-3σ | 38–96, 91 | [31,32] |
| Bisulfite-Sequencing [33] | Standard CoBRA [37] | Bio-CoBRA [38] | Our Method | |
|---|---|---|---|---|
| Analysis method | Sequencing | PAGE gel electrophoresis | Microfluidics (BioAnalyzer) | Microfluidics (LabChip) |
| Requires sequencing | ✓ | ✕ | ✕ | ✕ |
| Requires concentration step | ✓ | ✓ | ✓ | ✕ |
| Requires PCR purification step | ✓ | ✓ | ✓ | ✕ |
| Number of CpG sites analyzed | Many | One | Not specified | Two |
| Enzyme digestion time | Variable | ≥4 h | ≥2 h | 15 min |
| Automation/high throughput | Barcoded samples needed, 2 days, low throughput | Manual 10 min/sample (20 samples), medium throughput | Automated 175 s/sample (12 samples), medium throughput |
Automated
94 s/sample (96 samples), high throughput |
| Gene | Sequence, Fwd. (5′-3′) | Sequence, Rev. (5′-3′) | Expected Amplicon Length (bp) | Annealing Temperature (°C) |
|---|---|---|---|---|
| RASSF1 | GGG TTT TAT AGT TTT TGT ATT TAG GTT TTT ATT | CCG CAA CTC AAT AAA CTC AAA CT | 204 | 56 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aibel, C.; Coll De Peña, A.; Tripathi, A. An Optimized CoBRA Method for the Microfluidic Electrophoresis Detection of Breast Cancer Associated RASSF1 Methylation. BioTech 2023, 12, 7. https://doi.org/10.3390/biotech12010007
Aibel C, Coll De Peña A, Tripathi A. An Optimized CoBRA Method for the Microfluidic Electrophoresis Detection of Breast Cancer Associated RASSF1 Methylation. BioTech. 2023; 12(1):7. https://doi.org/10.3390/biotech12010007
Chicago/Turabian StyleAibel, Claire, Adriana Coll De Peña, and Anubhav Tripathi. 2023. "An Optimized CoBRA Method for the Microfluidic Electrophoresis Detection of Breast Cancer Associated RASSF1 Methylation" BioTech 12, no. 1: 7. https://doi.org/10.3390/biotech12010007