
Cell Immortalization: In Vivo Molecular Bases and In Vitro Techniques for Obtention
 ,
,  and
and
Abstract
:1. Introduction
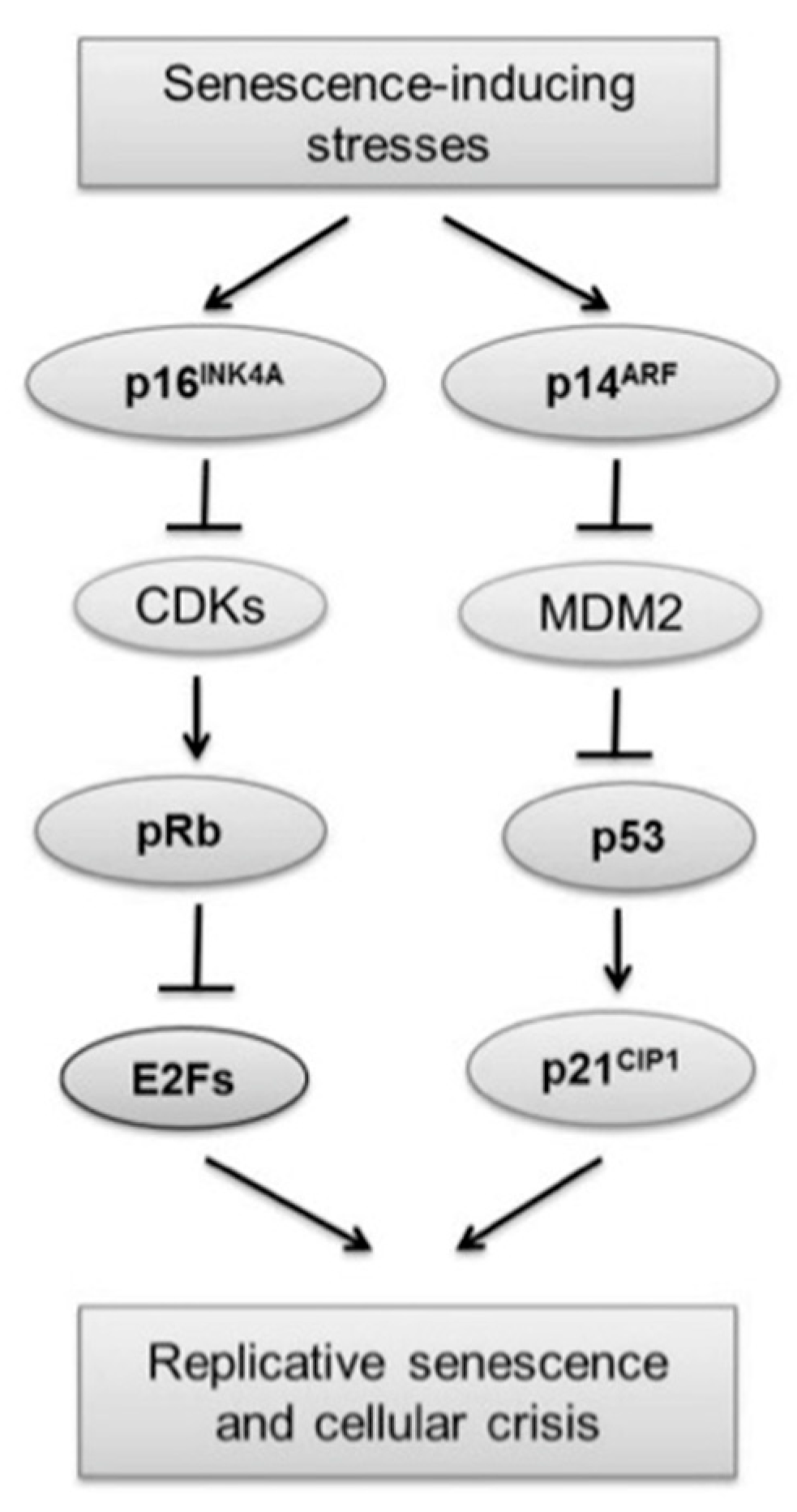
1.1. Telomeres
1.2. Mechanism Involved in Lengthening of Telomeres
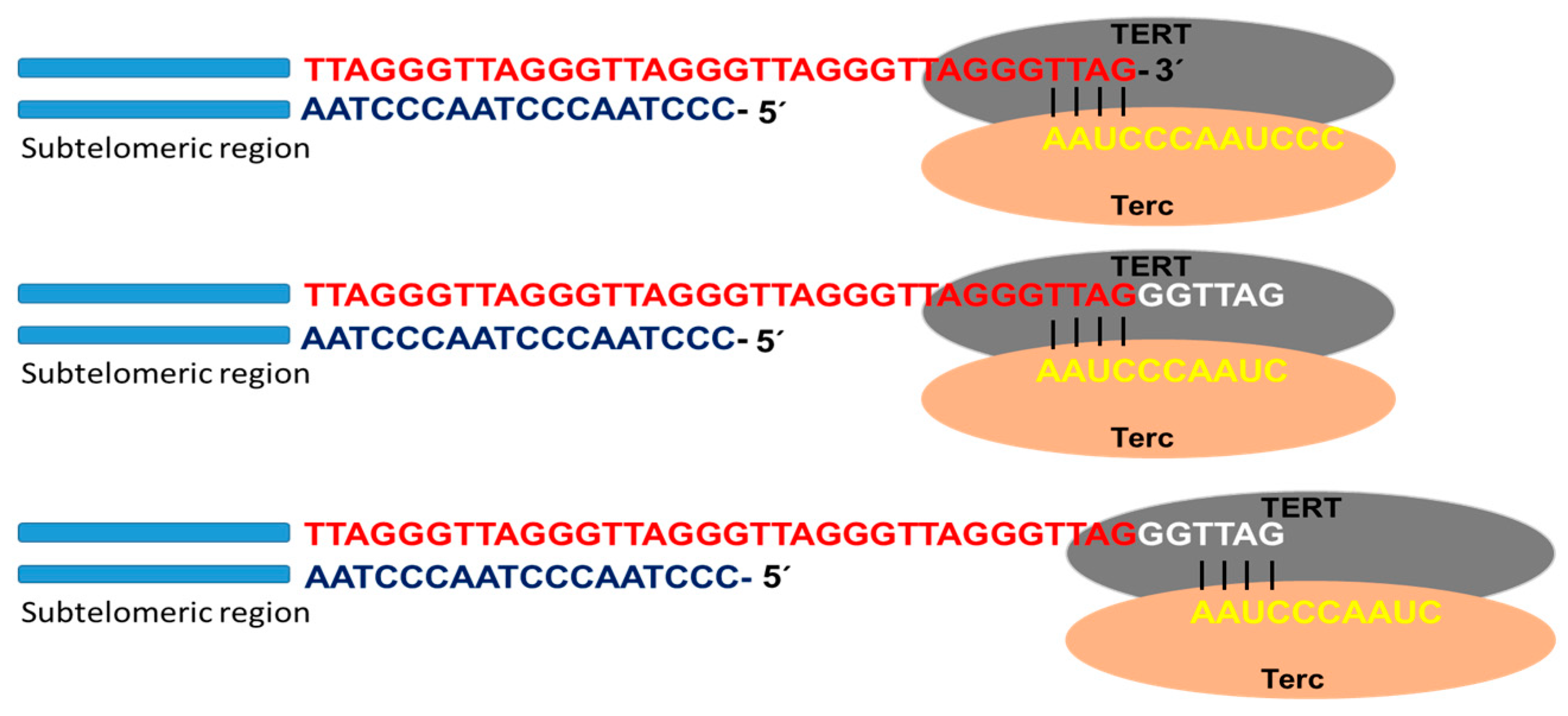
Telomerase
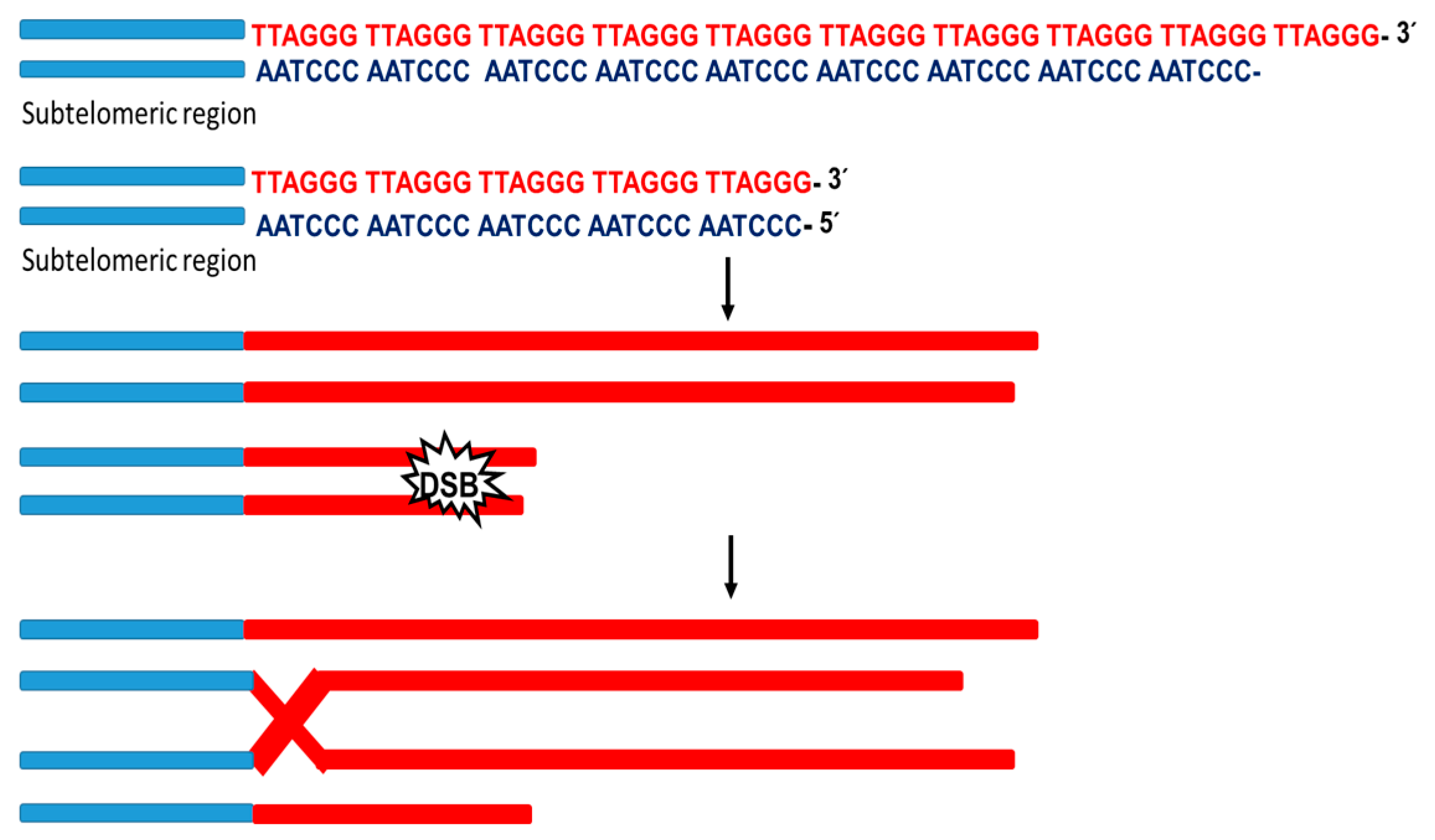
2. Alternative Telomere Lengthening (ALT)
2.1. Phenotypic Markers of ALT Cells
2.2. Activation of the ALT Pathway
3. Coexistence of Telomerase and the ALT Pathway
3.1. In Vitro Cell Immortalization
3.2. Techniques for In Vitro Cell Immortalization
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hayflick, L.; Moorhead, P.S. The Serial Cultivation of Human Diploid Cell Strains. Exp. Cell Res. 1961, 1, 585–621. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L. How and why we age. Exp. Gerontol. 1998, 33, 377. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L. The limited in vitro lifetime of human diploid cell strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, E.H. Telomeres—No end in sight. Cell 1994, 77, 621–623. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W.; Bacchetti, S. A survey of telomerase activity in human cancer. Eur. J. Cancer 1997, 33, 787–791. [Google Scholar] [CrossRef]
- Heaphy, C.M.; Subhawong, A.P.; Hong, S.M.; Goggins, M.G.; Montgomery, E.A.; Gabrielson, E.; Heaphy, C.M.; Subhawong, A.P.; Hong, S.M.; Goggins, M.G.; et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 2011, 179, 1608–1615. [Google Scholar] [CrossRef]
- Arnoult, N.; Karlseder, J. Complex interactions between the DNA-damage response and mammalian telomeres. Nat. Struct. Mol. Biol. 2015, 22, 859–866. [Google Scholar] [CrossRef] [Green Version]
- Denchi, E.L.; de Lange, T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature 2007, 448, 1068–1071. [Google Scholar] [CrossRef]
- Harley, C.B.; Futcher, A.B.; Greider, C.W. Telomeres shorten during ageing of human fibroblasts. Nature 1990, 345, 458–460. [Google Scholar] [CrossRef]
- Doksani, Y.; Wu, J.Y.; de Lange, T.; Zhuang, X. Super-resolution fluorescence imaging of telomeres reveals TRF2-dependent T-loop formation. Cell 2013, 155, 345–356. [Google Scholar] [CrossRef]
- Rai, R.; Zheng, H.; He, H.; Luo, Y.; Multani, A.; Carpenter, P.B.; Chang, S. The function of classical and alternative non-homologous end-joining pathways in the fusion of dysfunctional telomeres. EMBO J. 2010, 29, 2598–2610. [Google Scholar] [CrossRef] [Green Version]
- Okamoto, K.; Bartocci, C.; Ouzounov, I.; Diedrich, J.K.; Yates, J.R.; Denchi, E.L. A two-step mechanism for TRF2- mediated chromosome-end protection. Nature 2013, 494, 502–505. [Google Scholar] [CrossRef] [Green Version]
- Maciejowski, J.; de Lange, T. Telomeres in Cancer: Tumour suppression and genome instability. Nat. Rev. 2017, 18, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Shay, J.W. Role of telomeres and telomerase in aging and cancer. Cancer Discov. 2016, 6, 584–593. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.B.; Graham, M.E.; Lovrecz, G.O.; Bache, N.; Robinson, P.J.; Reddel, R.R. Protein composition of catalytically active human telomerase from immortal cells. Science 2007, 315, 1850–1853. [Google Scholar] [CrossRef]
- Blackburn, E.H. Switching and signaling at the telomere. Cell 2001, 106, 661–673. [Google Scholar] [CrossRef] [Green Version]
- Greider, C.W.; Blackburn, E.H. Identification of a specific telomere terminal transferase activity in Tetrahymena extracts. Cell 1985, 43, 405–413. [Google Scholar] [CrossRef]
- De Vitis, M.; Berardinelli, F.; Sgura, A. Telomere lenght maintenence in Cancer: AT the Crossroad between Telomerase and Alternative Lenthening of Telomeres (ALT). Int. J. Mol. Sci. 2018, 19, 606. [Google Scholar] [CrossRef] [Green Version]
- Harrington, L. Does the reservoir for self-renewal stem from the ends? Oncogene 2004, 23, 7283–7289. [Google Scholar] [CrossRef] [Green Version]
- Gunes, C.; Lenhard, R.K. The role of the telomeres in stem cells and cancer. Cell 2013, 152, 390–393. [Google Scholar] [CrossRef]
- Sperka, T.; Wang, J.; Rudolph, K.L. DNA damage checkpoints in stem cells, ageing and cancer. Nat. Rev. Mol. Cell Biol. 2012, 13, 579–590. [Google Scholar] [CrossRef]
- Okamoto, K.; Seimiya, H. Revisiting telomeres shortening in Cancer. Cells 2019, 8, 107. [Google Scholar] [CrossRef] [Green Version]
- Begus-Nahrmann, Y.; Hartmman, D.; Kraus, J.; Eshragi, P.; Scheffold, A.; Grieb, M.; Rasche, V.; Schirmacher, P.; Lee, H.W.; Kestler, H.A.; et al. Transient telomere dysfunction induces chromosomal instability and promotes carcinogenesis. J. Clin. Investig. 2012, 122, 2283–2288. [Google Scholar] [CrossRef]
- Barthel, F.P.; Wei, W.; Tang, M.; Martinez-Ledesma, E.; Hu, X.; Amin, S.B.; Akdemir, K.C.; Seth, S.; Song, X.; Wang, Q.; et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat. Genet. 2017, 49, 349–357. [Google Scholar] [CrossRef]
- Bell, R.J.; Rube, H.T.; Kreig, A.; Mancini, A.; Fouse, S.D.; Nagarajan, R.P.; Choi, S.; Hong, C.; He, D.; Pekmezci, M.; et al. Cancer. The transcription factor GABP selectively binds and activates the mutant TERT promoter in cancer. Science 2015, 348, 1036–1039. [Google Scholar] [CrossRef] [Green Version]
- Stern, J.L.; Theodorescu, D.; Vogelstein, B.; Papadopoulos, N.; Cech, T.R. Mutation of the TERT promoter, switch to active chromatin and monoallelic TERT expression in multiple cancers. Genes Dev. 2015, 29, 2219–2224. [Google Scholar] [CrossRef] [Green Version]
- Weinhold, N.; Jacobsen, A.; Schultz, N.; Sander, C.; Lee, W. Genome-wide analysis of noncoding regulatory mutations in cancer. Nat. Genet. 2014, 46, 1160–1165. [Google Scholar] [CrossRef]
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180. [Google Scholar] [CrossRef]
- Labussière, M.; Boisselier, B.; Mokhtari, K.; DiStefano, A.L.; Rahimian, A.; Rossetto, M.; Ciccarino, P.; Saulnier, O.; Paterra, R.; Marie, Y.; et al. Combined analysis of TERT, EGFR, and IDH status defines distinct prognostic glioblastoma classes. Neurology 2014, 83, 1200–1206. [Google Scholar] [CrossRef]
- Spiegl-Kreinecker, S.; Lötsch, D.; Neumayer, K.; Kastler, L.; Gojo, J.; Pirker, C.; Pichler, J.; Weis, S.; Kumar, R.; Webersinke, G.; et al. TERT promoter mutations are associated with poor prognosis and cell immortalization in meningioma. Neuro Oncol. 2018, 20, 1584–1593. [Google Scholar] [CrossRef]
- Sahm, F.; Schrimpf, D.; Olar, A.; Koelsche, C.; Reuss, D.; Bissel, J.; Kratz, A.; Capper, D.; Schefzyk, S.; Hielscher, T.; et al. TERT promoter mutations and risk of recurrence in meningioma. J. Natl. Cancer Inst. 2016, 108, djv370. [Google Scholar] [CrossRef]
- Lee, D.D.; Leão, R.; Komosa, M.; Gallo, M.; Zhang, C.H.; Lipman, T.; Remke, M.; Heidari, A.; Nunes, N.M.; Apolónio, J.; et al. DNA hypermethylation within TERT promoter upregulates TERT expression in cancer. J. Clin. Investig. 2019, 129, 223–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dilley, R.L.; Greenberg, R.A. Alternative telomere maintenance and cancer. Trends Cancer 2015, 1, 145–156. [Google Scholar] [CrossRef] [Green Version]
- Henson, J.D.; Hannay, J.A.; McCarthy, S.W.; Royds, J.A.; Yeager, T.R.; Robinson, R.A.; Wharton, S.B.; Jellinek, D.A.; Arbuckle, S.M.; Yoo, J.; et al. A robust assay for alternative lengthening of telomeres in tumors shows the significance of alternative lengthening of telomeres in sarcomas and astrocytomas. Clin. Cancer Res. 2005, 11, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Liau, J.Y.; Lee, J.C.; Tsai, J.H.; Yang, C.Y.; Liu, T.L.; Ke, Z.L.; Hsu, H.H.; Jeng, Y.M. Comprehensive screening of alternative lengthening of telomeres phenotype and loss of telomeres phenotype and loss of ATRX expression in sarcomas. Mod. Pathol. 2015, 28, 1545–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.C.; Jeng, Y.M.; Liau, J.Y.; Tsai, J.H.; Hsu, H.H.; Yang, C.Y. Alternative lengthening of telomeres and loss of ATRX are frequent events in pleomorphic and dedifferentiated liposarcomas. Mod. Pathol. 2015, 28, 1064–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, A.A.; Watson, C.M.; Noble, J.R.; Pickett, H.A.; Tam, P.P.; Reddel, R.R. Alternative lengthening of telomeres in normal mammalian somatic cells. Genes Dev. 2013, 27, 18–23. [Google Scholar] [CrossRef] [Green Version]
- Coluzzi, E.; Buonsante, R.; Leone, S.; Asmar, A.J.; Miller, K.L.; Cimini, D.; Sgura, A. Transient ALT activation protects human primary cells from chromosome instability induced by low chronic oxidative stress. Sci. Rep. 2017, 7, 43309. [Google Scholar] [CrossRef]
- De Vitis, M.; Berardinelli, F.; Coluzzi, E.; Marinaccio, J.; O’Sullivan, R.J.; Sgura, A.A. X-rays activate telomeric homologous recombination mediated repair in primary cells. Cells 2019, 8, 708. [Google Scholar] [CrossRef] [Green Version]
- Pezzolo, A.; Pistorio, A.; Gambini, C.; Haupt, R.; Ferraro, M.; Erminio, G.; De Bernardi, B.; Garaventa, A.; Pistoia, V. Intratumoral diversity of telomere length in individual neuroblastoma tumors. Oncotarget 2015, 6, 7493–7503. [Google Scholar] [CrossRef]
- Gocha, A.R.; Nuovo, G.; Iwenofu, O.H.; Groden, J. Human sarcomas are mosaic for telomerase-dependent and telomerase-independent telomere maintenance mechanisms: Implications for telomere-based therapies. Am. J. Pathol. 2013, 182, 41–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Liang, P.; Liu, D.; Huang, J.; Songyang, Z. Telomere regulation in pluripotent stem cells. Protein Cell 2014, 5, 194–202. [Google Scholar] [CrossRef] [Green Version]
- Liu, L. Linking Telomere Regulation to Stem Cell Pluripotency. Trends Genet. 2017, 33, 16–33. [Google Scholar] [CrossRef] [PubMed]
- Cesare, A.J.; Reddel, R.R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Amorim, J.P.; Santos, G.; Vinagre, J.; Soares, P. The Role of ATRX in the Alternative Lengthening of Telomeres (ALT) Phenotype. Genes 2016, 7, 66. [Google Scholar] [CrossRef]
- Osterwald, S.; Deeg, K.I.; Chung, I.; Parisotto, D.; Worz, S.; Rohr, K.; Erfle, H.; Rippe, K. PML induces compaction, TRF2 depletion and DNA damage signaling at telomeres and promotes their alternative lengthening. J. Cell Sci. 2015, 128, 1887–1990. [Google Scholar] [CrossRef] [Green Version]
- Acharya, S.; Kaul, Z.; Gocha, A.S.; Martinez, A.R.; Harris, J.; Parvin, J.D.; Groden, J. Association of BLM and BRCA1 during telomere maintenance in ALT cells. PLoS ONE 2014, 9, e103819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nabetani, A.; Ishikawa, F. Alternative lengthening of telomeres pathway: Recombination-mediated telomere maintenance mechanism in human cells. J. Biochem. 2011, 149, 5–14. [Google Scholar] [CrossRef] [Green Version]
- Mender, I.; Shay, J.W. Telomere Dysfunction Induced Foci (TIF) Analysis. Bio. Protoc. 2015, 5, e1656. [Google Scholar] [CrossRef] [Green Version]
- Roumelioti, F.M.; Sotiriou, S.K.; Katsini, V.; Chiourea, M.; Halazonetis, T.D. Alternative lengthening of human telomeres is a conservative DNA replication process with features of break-induced replication. EMBO Rep. 2016, 17, 1731–1737. [Google Scholar] [CrossRef]
- Cox, K.E.; Marechal, A.; Flynn, R.L. SMARCAL1 Resolves Replication Stress at ALT Telomeres. Cell Rep. 2016, 14, 1032–1040. [Google Scholar] [CrossRef] [Green Version]
- Fallet, E.; Jolivet, P.; Soudet, J.; Lisby, M.; Gilson, E.; Teixeira, M.T. Length-dependent processing of telomeres in the absence of telomerase. Nucleic Acids Res. 2014, 42, 3648–3665. [Google Scholar] [CrossRef] [PubMed]
- Tacconi, E.M.; Tarsounas, M. How homologous recombination maintains telomere integrity. Chromosoma 2015, 124, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Xie, Y.; Zhang, Z.; Mao, P.; Liu, J.; Ma, W.; Zhao, Y. Telomeric Recombination Induced by DNA Damage Results in Telomere Extension and Length Heterogeneity. Neoplasia 2018, 20, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.M.; Yadav, T.; Ouyang, J.; Lan, L.; Zou, L. Alternative Lengthening of Telomeres through Two Distinct Break-Induced Replication Pathways. Cells Rep. 2019, 26, 955–968. [Google Scholar] [CrossRef] [Green Version]
- Verma, P.; Dilley, R.L.; Zhang, T.; Gyparaki, M.T.; Li, Y.; Greenberg, R.A. RAD52 and SLX4 act nonepistatically to ensure telomere stability during alternative telomere lengthening. Genes Dev. 2019, 33, 221–235. [Google Scholar] [CrossRef] [Green Version]
- Benson, F.E.; Baumann, P.; West, S.C. Synergistic actions of Rad51 and Rad52 in recombination and DNA repair. Nature 1998, 391, 401–404. [Google Scholar] [CrossRef]
- Bhowmick, R.; Minocherhomji, S.; Hickson, I.D. RAD52 Facilitates Mitotic DNA Synthesis Following Replication Stress. Mol. Cell 2016, 64, 1117–1126. [Google Scholar] [CrossRef] [Green Version]
- Sobino, A.P.; Allen, J.A.; Neumann, A.A.; Yang, S.F.; Walsh, M.E.; Henson, J.D. BLM and SLX4 play opposing roles in recombination-dependent replication at human telomeres. EMBO J. 2017, 36, 2907–2919. [Google Scholar] [CrossRef]
- Perrem, K.; Colgin, L.M.; Neumann, A.A.; Yeager, T.R.; Reddel, R.R. Coexistence of alternative lengthening of telomeres and telomerase in hTERT- transfected GM847 cells. Mol. Cell Biol. 2001, 21, 3862–3875. [Google Scholar] [CrossRef]
- Episkopou, H.; Draskovic, I.; VanBeneden, A.; Tilman, G.; Mattiussi, M.; Gobin, M.; Arnoult, N.; Londoño-Vallejo, A.; Decottignies, A. Decottignies A: Alternative lengthening of telomeres is characterized by reduced compaction of telomeric chromatin. Nucleic Acids Res. 2014, 42, 4391–4405. [Google Scholar] [CrossRef]
- Episkopou, H.; Diman, A.; Claude, E.; Viceconte, N.; Decottignies, A. TSPYL5 depletion induces specific death of ALT cells through USP7-dependent proteasomal degradation of POT1. Mol. Cell 2019, 75, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Tilman, G.; Loriot, A.; VanBeneden, A.; Arnoult, N.; Londono-Vallejo, J.A.; DeSmet, C.; Decottignies, A. Subtelomeric DNA hypomethylation is not required for telomeric sister chromatid exchanges in ALT cells. Oncogene 2009, 28, 1682–1693. [Google Scholar] [CrossRef] [Green Version]
- Bechter, O.E.; Zou, Y.; Walker, W.; Wright, W.E.; Shay, J.W. Telomeric recombination in mismatch repair deficient human colon cancer cells after telomerase inhibition. Cancer Res. 2004, 64, 3444–3451. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, S.; Yan, Z.; Pei, M. A prospect of cell immortalization combined with matrix microenvironmental optimization strategy for tissue engineering and regeneration. Cell Biosci. 2019, 9, 7. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.; Lee, C. Immortalization of Primary Keratinocytes and Its Application to Skin Research. Biomol. Ther. 2015, 23, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Bryan, T.M.; Reddel, R.R. SV40-induced immortalization of human cells. Crit. Rev. Oncog. 1994, 5, 331–357. [Google Scholar] [CrossRef]
- Foddis, R.; DeRienzo, A.; Broccoli, D.; Bocchetta, M.; Stekala, E.; Rizzo, P.; Tosolini, A.; Grobelny, J.V.; Jhanwar, S.C.; Pass, H.I.; et al. SV40 infection induces telomerase activity in human mesothelial cells. Oncogene 2002, 21, 1434–1442. [Google Scholar] [CrossRef] [Green Version]
- Matsumura, T.; Takesue, M.; Westerman, K.A.; Okitsu, T.; Sakaguchi, M.; Fukazawa, T.; Totsugawa, T.; Noguchi, H.; Yamamoto, S.; Stolz, D.B.; et al. Establishment of an immortalized human-liver endothelial cell line with SV40T and hTERT. Transplantation 2004, 77, 1357–1365. [Google Scholar] [CrossRef]
- Orosz, D.; Resnick, M.; Wehland, J.; Douglas, J.; Edwards, J.; Jacobberger, J.; Choo, C.K.; Yau, C.F.; Chan, K.W.; Resnick, M.I.; et al. Growth, immortalization, and differentiation potential of normal adult human proximal tubule cells. In Vitro Cell. Dev. Biol. Anim. 2004, 40, 22–34. [Google Scholar] [CrossRef]
- Garcia-Mesa, Y.; Jay, T.; Checkley, M.A.; Luttge, B.; Dobrowolski, C.; Valadkhan, S.; Landreth, G.E.; Karn, J.; Alvarez-Carbonell, D. Immortalization of primary microglia: A new platform to study HIV regulation in the central nervous system. J. Neurovirol. 2017, 23, 47–66. [Google Scholar] [CrossRef] [Green Version]
- Darimont, C.; Mace, K. Immortalization of human preadipocytes. Biochimie 2003, 85, 1231–1233. [Google Scholar] [CrossRef]
- Trakarnsanga, K.; Griffiths, R.E.; Wilson, M.C.; Blair, A.; Satchwell, T.J.; Meinders, M. An immortalized adult human erythroid line facilitates sustainable and scalable generation of functional red cells. Nat. Commun. 2017, 8, 14750. [Google Scholar] [CrossRef] [Green Version]
- Shin, H.Y.; Yang, W.; Lee, E.j.; Han, G.H.; Cho, H.; Chay, D.B.; Kim, J.H. Establishment of five immortalized human ovarian surface epithelial cell lines via SV40 T antigen or HPV E6/E7 expression. PLoS ONE 2018, 13. [Google Scholar] [CrossRef]
- Schutze, D.M.; Krijgsman, O.; Snijders, P.; Ylstra, B.; Weischenfeldt, J.; Mardin, B.R.; Stütz, A.M.; Korbel, J.O.; de Winter, J.P.; Meijer, C.J. Immortalization capacity of HPV types is inversely related to chromosomal instability. Oncotarget 2016, 7, 37608–37621. [Google Scholar] [CrossRef]
- Marinkovic, D.; Marinkovic, T. c-Myc misregulation triggers complex process of genomic instability. Genetika 2018, 50, 731–745. [Google Scholar] [CrossRef] [Green Version]
- Vafa, O.; Wade, M.; Kern, S.; Beeche, M.; Pandita, T.K.; Wahl, G. c-Myc can induce DNA damage, increase reactive oxygen species, and mitigate p53 function: A mechanism for oncogene-induced genetic instability. Mol. Cell 2002, 9, 1031–1044. [Google Scholar] [CrossRef]
- Klapproth, K.S.; Sander, D.; Marinkovic, B.; Baumann, T. The IKK2/NF-{kappa} B pathway suppresses MYC-induced lymphomagenesis. Blood 2009, 114, 2448–2458. [Google Scholar] [CrossRef] [Green Version]
- De Filippis, L.; Ferrari, D.; Nodari, L.R.; Amati, B.; Snyder, E.; Vescovi, A.L. Immortalization of Human Neural Stem Cells with the c-Myc Mutant T58A. PLoS ONE 2008, 3, e3310. [Google Scholar] [CrossRef]
- Li, Z.; Oganesyan, D.; Mooney, R.; Rong, X.; Christensen, M.J.; Shahmanyan, D. L-MYC Expression Maintains Self-Renewal and Prolongs Multipotency of Primary Human Neural Stem Cells. Stem Cell Rep. 2016, 7, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, M.; Takizawa, N.; Narita, M.; Ichisaka, T.; Yamanaka, S. Promotion of direct reprogramming by transformation deficient Myc. Proc. Natl. Acad. Sci. USA 2010, 107, 14152–14157. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Zakaria, S.M.; Tabor, V.H.; Singh, M.; Tronnersjö, S.; Goodwin, J.; Selivanova, G.; Bartek, J.; Castell, A.; Larsson, L.G. MYC and RAS are unable to cooperate in overcoming cellular senescence and apoptosis in normal human fibroblasts. Cell Cycle 2018, 17, 2697–2715. [Google Scholar] [CrossRef] [Green Version]
- Hung, C.; Maiuri, T.; Bowie, L.E.; Gotesman, R.; Son, S.; Falcone, M.; Giordano, J.V.; Gillis, T.; Mattis, V.; Lau, T.; et al. A patient-derived cellular model for Huntington’s disease reveals phenotypes at clinically relevant CAG lengths. Mol. Bio. Cell 2018, 29, 2809–2820. [Google Scholar] [CrossRef]
- Bodnar, A.G.; Ouellette, M.; Frolkis, M.; Holt, S.E.; Chiu, C.P.; Morin, G.B.; Harley, C.B.; Shay, J.W.; Lichtsteiner, S.; Wright, W.E. Extension of life-span by introduction of telomerase into normal human cells. Science 1998, 279, 349–352. [Google Scholar] [CrossRef] [Green Version]
- Simonsen, J.L.; Rosada, C.; Serakinci, N.; Justesen, J.; Stenderup, K.; Rattan, S.I.; Jensen, T.G.; Kassem, M. Telomerase expression extends the proliferative life-span and maintains the osteogenic potential of human bone marrow stromal cells. Nat. Biotechnol. 2002, 20, 592–596. [Google Scholar] [CrossRef]
- Piera-Velazquez, S.; Jimenez, S.A.; Stokes, D. Increased life span of human osteoarthritic chondrocytes by exogenous expression of telomerase. Arthritis Rheum. 2002, 46, 683–693. [Google Scholar] [CrossRef]
- Siska, E.K.; Weisman, I.; Romano, J.; Ivics, Z.; IzsvaÂk, Z.; Barkai, U.; Petrakis, S.; Koliakos, G. Generation of an immortalized mesenchymal stem cell line producing a secreted biosensor protein for glucose monitoring. PLoS ONE 2017, 12, e0185498. [Google Scholar] [CrossRef] [Green Version]
- Qin, S.Q.; Kusuma, G.D.; Al-Sowayan, B.; Pace, R.S.; Isenmann, S.; Pertile, M.D.; Gronthos, S.; Abumaree, M.H.; Brennecke, S.P.; Kalionis, B. Establishment and characterization of fetal and maternal mesenchymal stem/stromal cell lines from the human term placenta. Placenta 2016, 39, 134–146. [Google Scholar] [CrossRef]
- Villard, O.; Armanet, M.; Couderc, G.; Bony, C.; Moreaux, J.; Noël, D.; De Vos, J.; Klein, B.; Veyrune, J.L.; Wojtusciszyn, A. Characterization of immortalized human islet stromal cells reveals a MSC-like profile with pancreatic features. Stem Cell Res. Ther. 2020, 11, 190. [Google Scholar] [CrossRef]
- Serakinci, N.; Guldberg, P.; Burns, J.S.; Abdallah, B.; Schrodder, H.; Jensen, T.; Kassem, M. Adult human mesenchymal stem cell as a target for neoplastic transformation. Oncogene 2004, 23, 5095–5098. [Google Scholar] [CrossRef]
- Burns, J.S.; Abdallah, B.M.; Guldberg, P.; Rygaard, J.; Schroder, H.D.; Kassem, M. Tumorigenic heterogeneity in cancer stem cells evolved from long term cultures of telomerase-immortalized human mesenchymal stem cells. Cancer Res. 2005, 65, 3126–3135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noble, J.R.; Zhong, Z.H.; Neumann, A.A.; Melki, J.R.; Clark, S.J.; Reddel, R.R. Alterations in the p16 (INK4a) and p53 tumor suppressor genes of hTERT immortalized human fibroblasts. Oncogene 2004, 23, 3116–3121. [Google Scholar] [CrossRef] [Green Version]
- Hung, C.J.; Yao, C.L.; Cheng, F.C.; Wu, M.L.; Wang, T.H.; Hwang, S.M. Establishment of immortalized mesenchymal stromal cells with red fluorescence protein expression for in vivo transplantation and tracing in the rat model with traumatic brain injury. Cytotherapy 2010, 12, 455–465. [Google Scholar] [CrossRef]
- Wall, I.; Santiago Toledo, G.; Jat, P. Recent advances in conditional cell immortalization technology. Cell Gene Ther. Insights 2016, 2, 339–355. [Google Scholar] [CrossRef]
- Littlewood, T.D.; Hancock, D.C.; Danielian, P.S.; Parker, M.G.; Evan, G.I. A modified oestrogen receptor ligand-binding domain as an improved switch for the regulation of heterologous proteins. Nucleic Acids Res. 1995, 23, 1686–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danielian, P.S.; White, R.; Hoare, S.A.; Fawell, S.E.; Parker, M.G. Identification of residues in the estrogen receptor that confer differential sensitivity to estrogen and hydroxytamoxifen. Mol. Endocrinol. 1993, 7, 232–240. [Google Scholar]
- Tegtmeyer, P. Function of simian virus 40 gene an in transforming infection. J. Virol. 1975, 15, 613–618. [Google Scholar] [CrossRef] [Green Version]
- Frederiksen, K.; Jat, P.S.; Valtz, N.; Levy, D.; McKay, R. Immortalization of precursor cells from the mammalian CNS. Neuron 1988, 1, 439–448. [Google Scholar] [CrossRef]
- Shay, J.W.; Wright, W.E.; Werbin, H. Defining the molecular mechanisms of human cell immortalization. Biochim. Biophys. Acta 1991, 1072, 1–7. [Google Scholar] [CrossRef]
- Kawabata, K.; Sakurai, F.; Koizumi, N.; Hayakawa, T.; Mizuguchi, H. Adenovirus vector-mediated gene transfer into stem cells. Mol. Pharm. 2006, 3, 95–103. [Google Scholar] [CrossRef]
- Paillard, F. Reversible cell immortalization with the Cre-lox system. Hum. Gene 1999, 10, 1597–1598. [Google Scholar] [CrossRef] [PubMed]
- Gossen, M.; Bujard, H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA 1992, 89, 5547–5551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gossen, M.; Freundlieb, S.; Bender, G.; Muller, G.; Hillen, W.; Bujard, H. Transcriptional activation by tetracyclines in mammalian cells. Science 1995, 268, 1766–1769. [Google Scholar] [CrossRef]
- Sipo, I.; HurtadoPicó, A.; Wang, X.; Eberle, J.; Petersen, I.; Weger, S.; Poller, W.; Fechner, H. An improved Tet-On regulatable FasL-adenovirus vector system for lung cancer therapy. J. Mol. Med. 2006, 84, 215–225. [Google Scholar] [CrossRef]
- Malaver-Ortega, L.F.; Rosenbluh, J. Immortalised Cas9-expressing Cell lines for Gene interrogation. Methods Mol. Biol. 2022, 2495, 91–97. [Google Scholar]
- Maqsood, M.I.; Matin, M.M.; Bahrami, A.R.; Ghasroldasht, M.M. Immortality of celllines: Challenges and advantages of establishment. Cell Biol. Int. 2013, 37, 1038–1045. [Google Scholar] [CrossRef]
- Inoue, Y.; Hasegawa, S.; Hasebe, Y.; Kawagishi-Hotta, M.; Okuno, R.; Yamada, T.; Adachi, H.; Miyachi, K.; Ishii, Y.; Sugiura, K. Establishment of Three Types of Immortalized Human SkinStem Cell Lines Derived from the Single Donor. Biol. Pharm. Bull. 2021, 44, 1403–1412. [Google Scholar] [CrossRef]
- Nakamura, S.; Sugimoto, N.; Eto, K. Ex vivo generation of plateletproducts from humaniPScells. Inflamm. Regen. 2020, 40, 30. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mechanisms Enrolled | |
|---|---|
| ALT pathways | SLX4/RAD52 |
| BML-POLD3/4 |
| Strategy | Method |
|---|---|
| Expression of the catalytic subunit of telomerase | TERT |
| Induction of viral oncogenes that inactivate cell cycle proteins (p14, p16, p21, p53, Rb) | -SV40 T antigen -Small dsDNA virus HPV -myc’s oncogenes |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Bardet, J.C.; Cardentey, C.R.; González, B.L.; Patrone, D.; Mulet, I.L.; Siniscalco, D.; Robinson-Agramonte, M.d.l.A. Cell Immortalization: In Vivo Molecular Bases and In Vitro Techniques for Obtention. BioTech 2023, 12, 14. https://doi.org/10.3390/biotech12010014
de Bardet JC, Cardentey CR, González BL, Patrone D, Mulet IL, Siniscalco D, Robinson-Agramonte MdlA. Cell Immortalization: In Vivo Molecular Bases and In Vitro Techniques for Obtention. BioTech. 2023; 12(1):14. https://doi.org/10.3390/biotech12010014
Chicago/Turabian Stylede Bardet, Javier Curi, Celeste Ramírez Cardentey, Belkis López González, Deanira Patrone, Idania Lores Mulet, Dario Siniscalco, and María de los Angeles Robinson-Agramonte. 2023. "Cell Immortalization: In Vivo Molecular Bases and In Vitro Techniques for Obtention" BioTech 12, no. 1: 14. https://doi.org/10.3390/biotech12010014