
Influence of Concentration of Thiol-Substituted Poly(dimethylsiloxane)s on the Properties, Phases, and Swelling Behaviors of Their Crosslinked Disulfides

Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Methods
3. Results and Discussion
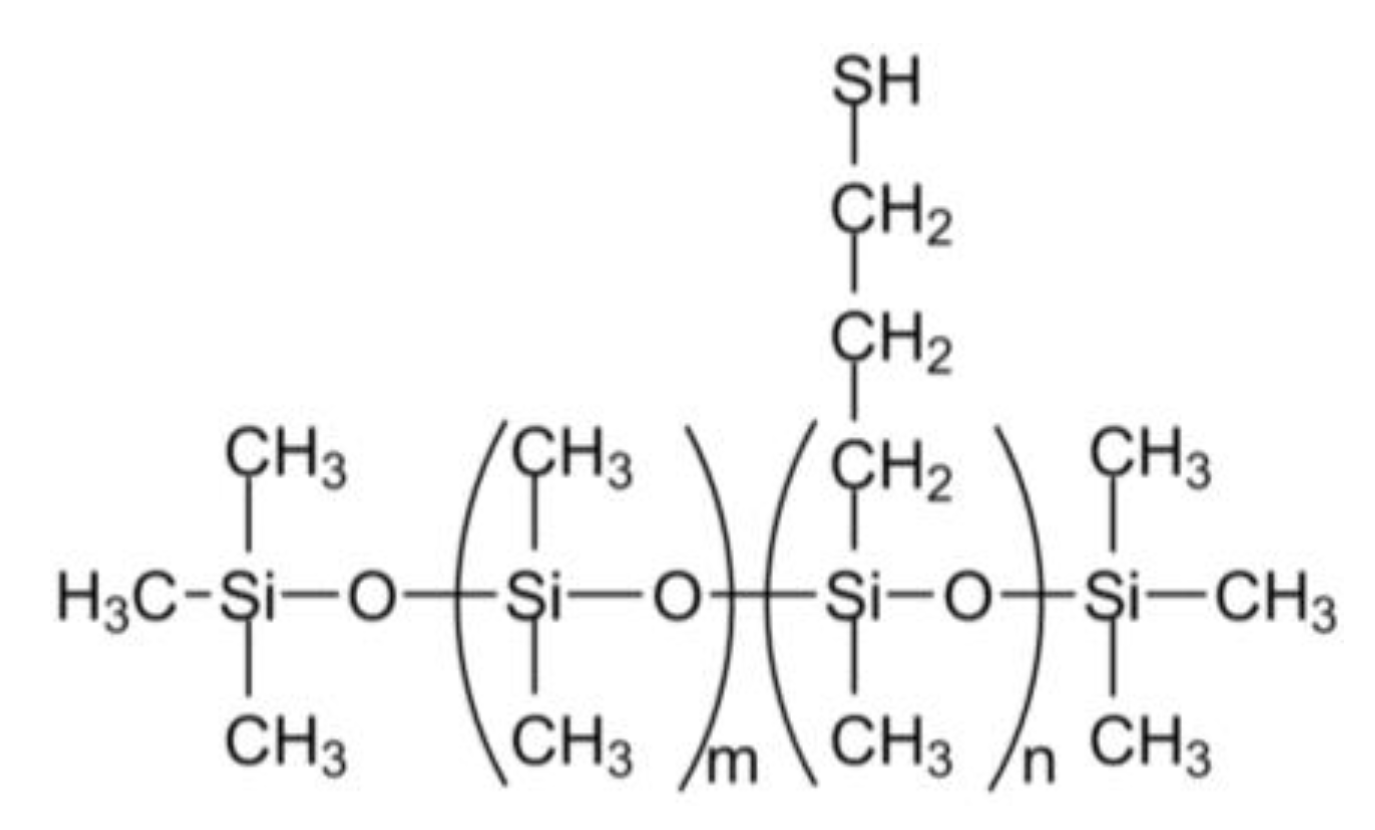
3.1. Starting Materials
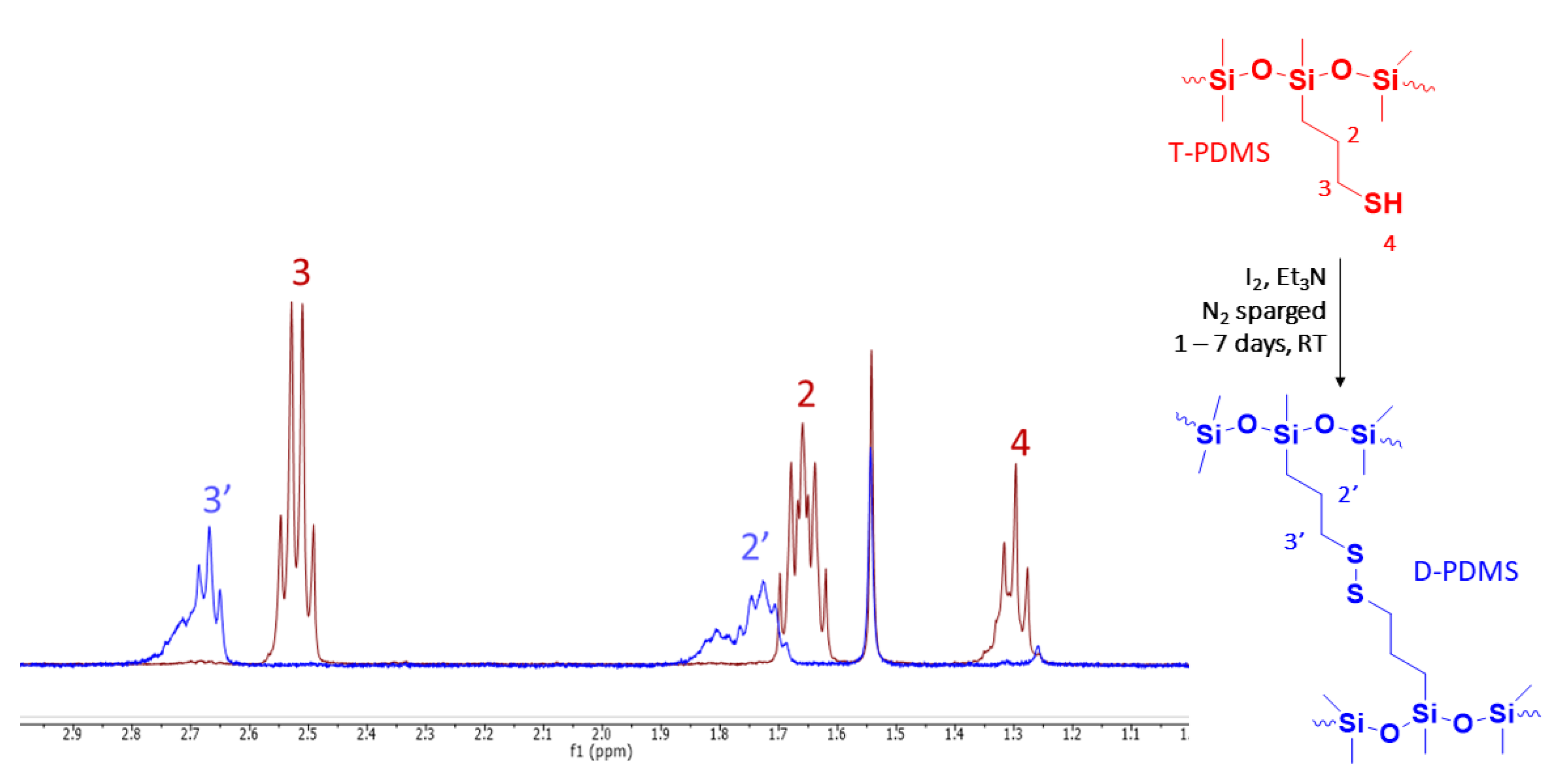
3.2. Synthesis of D-PDMS Materials
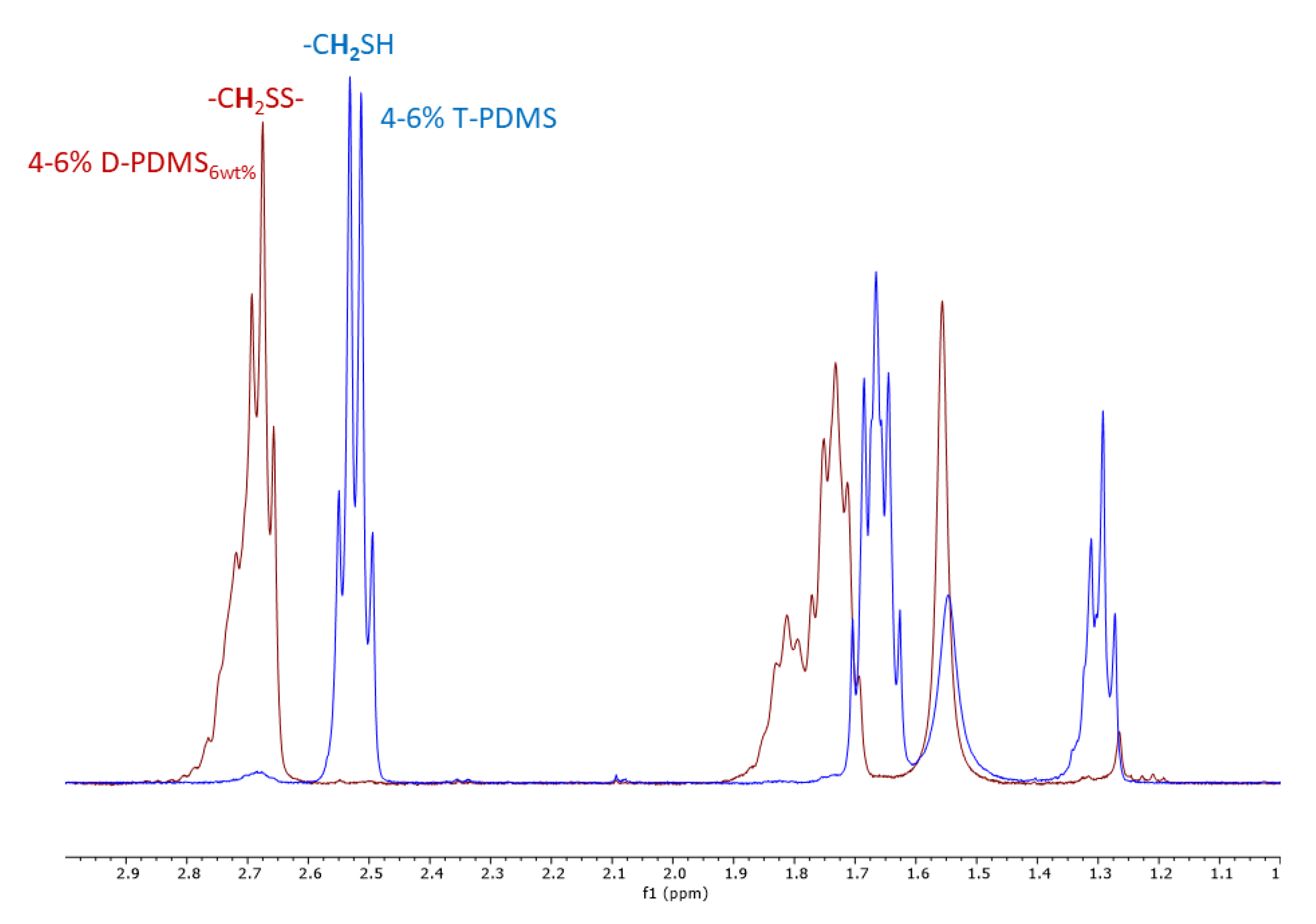
3.3. Characterization of Residual Thiol Content in Liquid Products
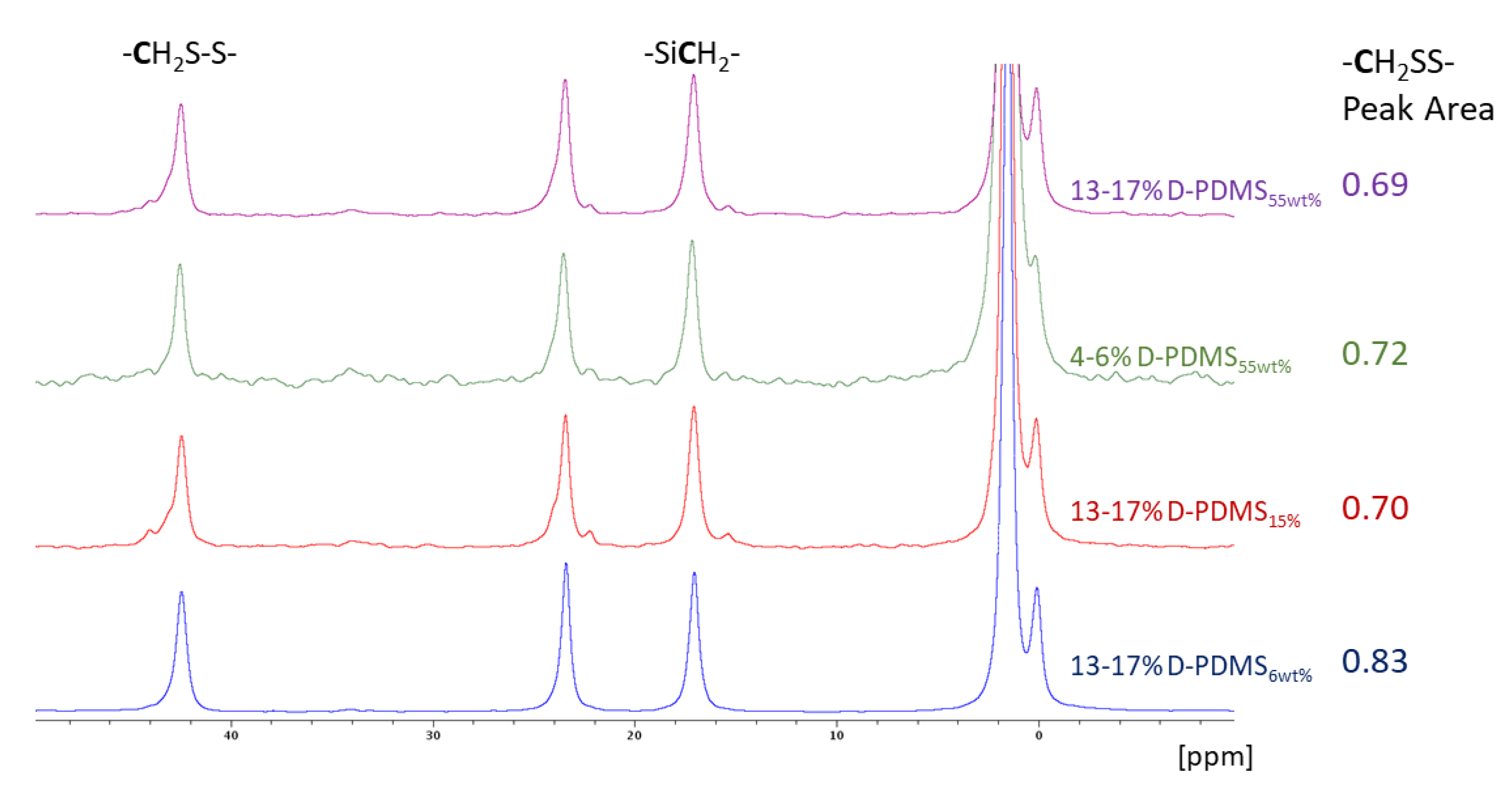
3.4. Characterization of Residual Thiol Content in Solid Products
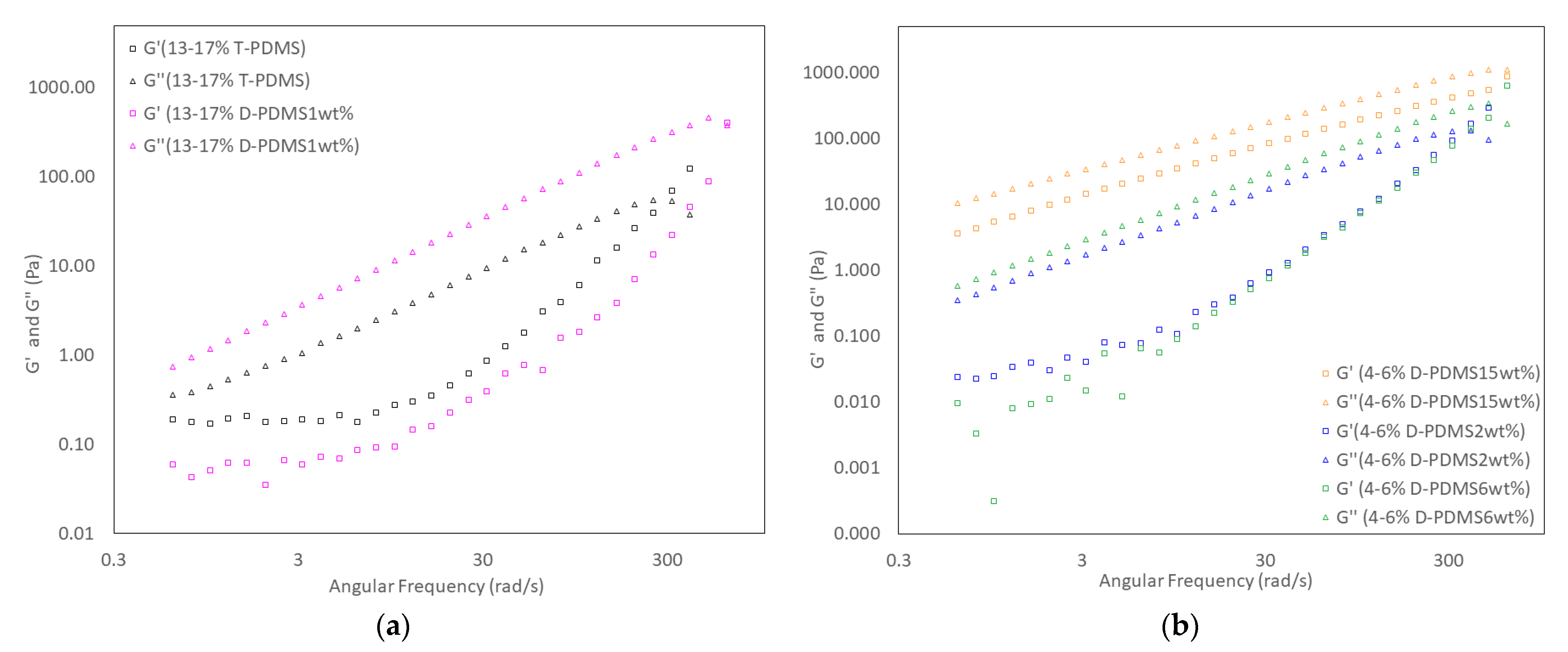
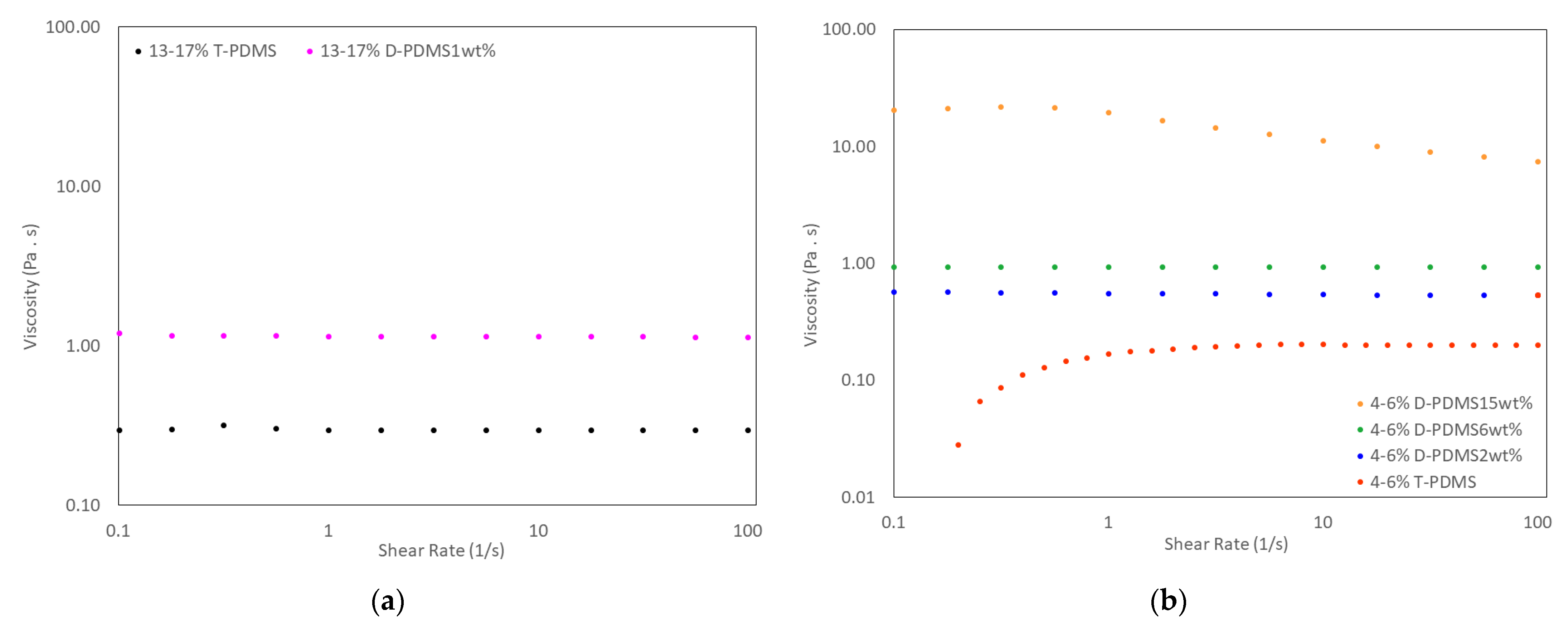
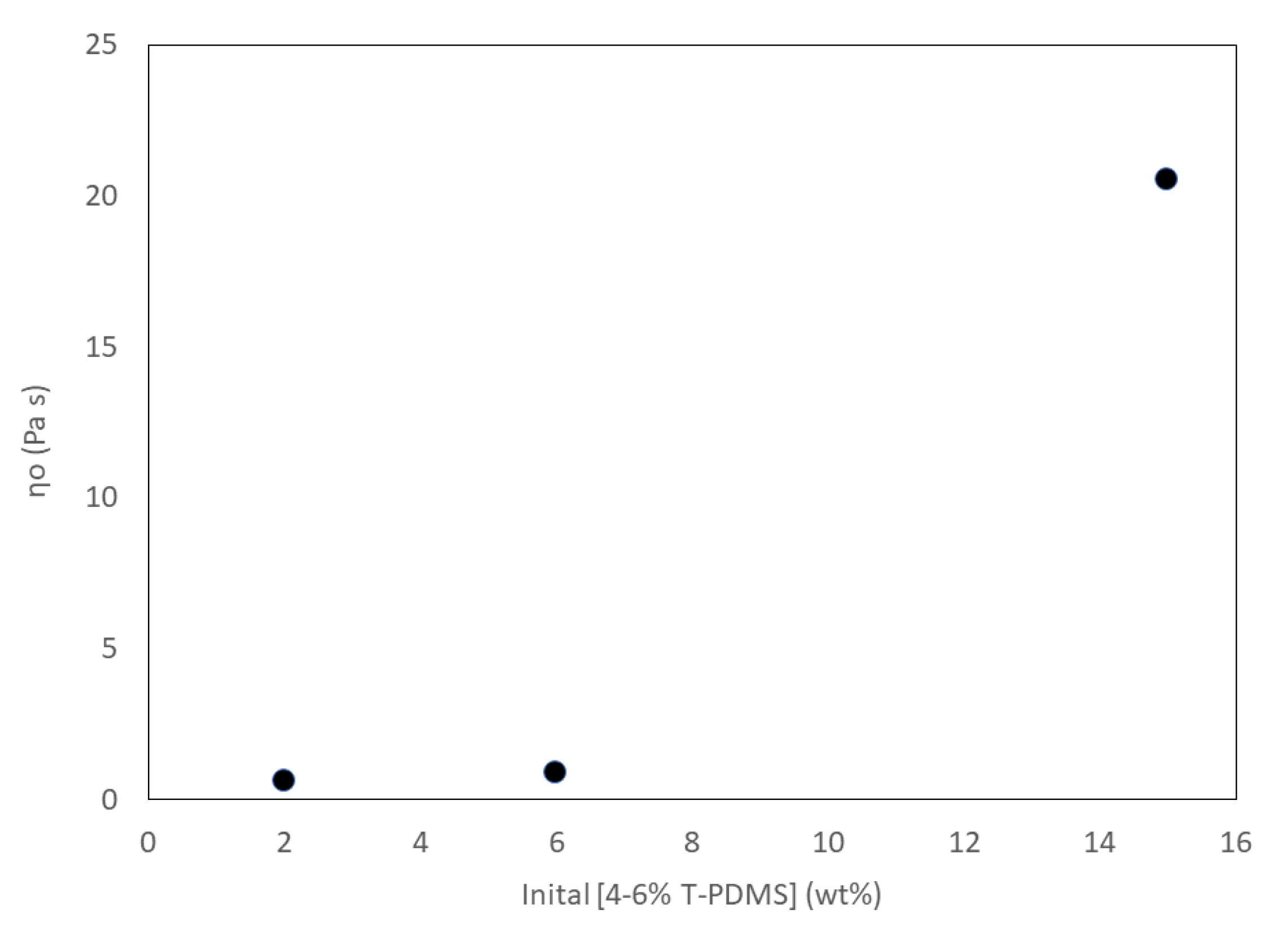
3.5. Physical Properties of Liquid Crosslinked Materials
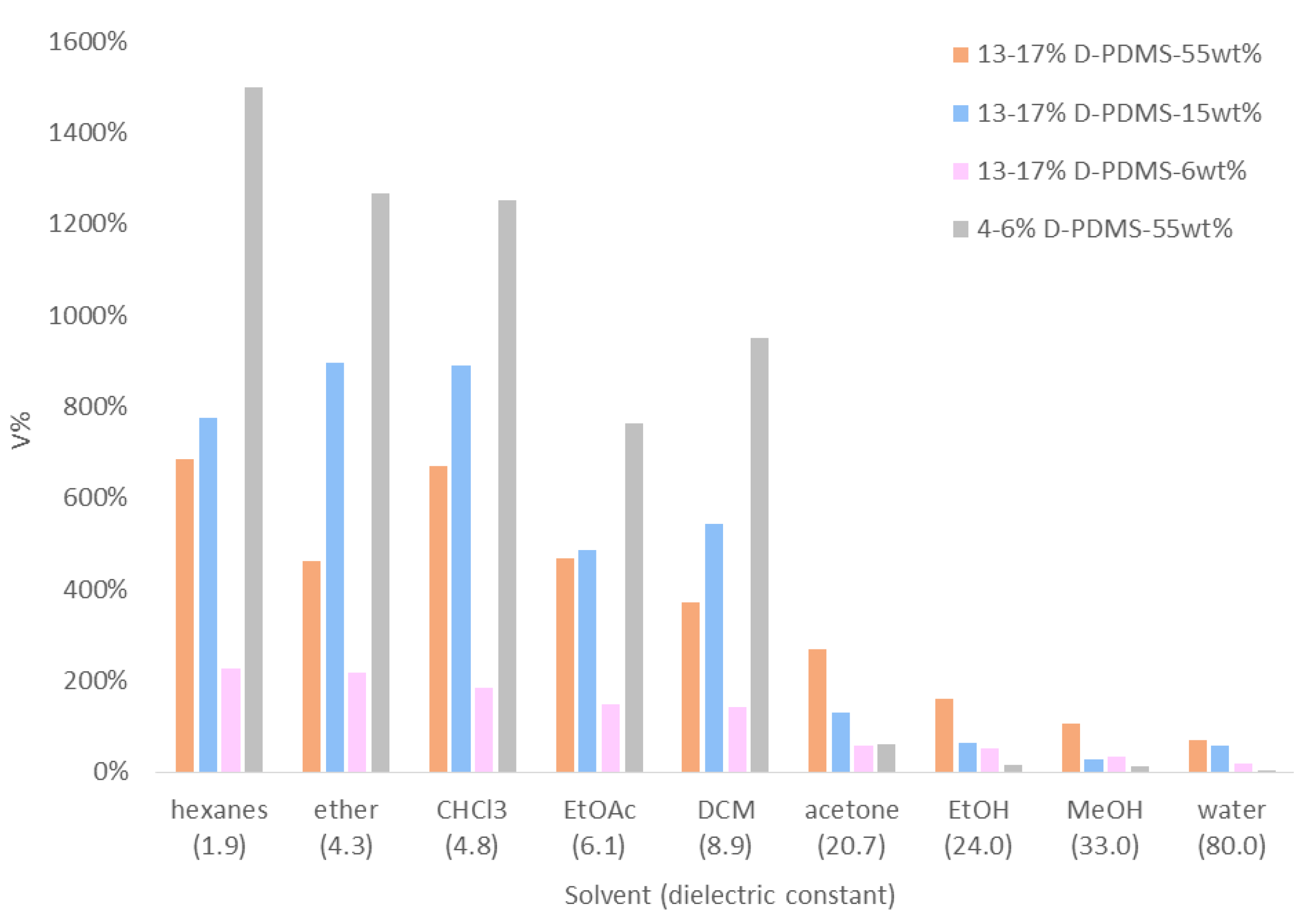
3.6. Swelling Studies of Solid Crosslinked Materials
3.7. TGA and TGA/MS Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beaupre, D.M.; Weiss, R.G. Thiol- and Disulfide-Based Stimulus-Responsive Soft Materials and Self-Assembling Systems. Molecules 2021, 26, 3332. [Google Scholar] [CrossRef]
- Jin, Y.; Yu, C.; Denman, R.J.; Zhang, W. Recent Advances in Dynamic Covalent Chemistry. Chem. Soc. Rev. 2013, 42, 6634–6654. [Google Scholar] [CrossRef]
- Dutta, K.; Das, R.; Medeiros, J.; Thayumanavan, S. Disulfide Bridging Strategies in Viral and Nonviral Platforms for Nucleic Acid Delivery. Biochemistry 2021, 60, 966–990. [Google Scholar] [CrossRef]
- Wang, Q.; Guan, J.; Wan, J.; Li, Z. Disulfide Based Prodrugs for Cancer Therapy. RSC Adv. 2020, 10, 24397–24409. [Google Scholar] [CrossRef]
- Saito, G.; Swanson, J.A.; Lee, K.D. Drug Delivery Strategy Utilizing Conjugation via Reversible Disulfide Linkages: Role and Site of Cellular Reducing Activities. Adv. Drug Deliv. Rev. 2003, 55, 199–215. [Google Scholar] [CrossRef]
- Leichner, C.; Jelkmann, M.; Bernkop-Schnürch, A. Thiolated Polymers: Bioinspired Polymers Utilizing One of the Most Important Bridging Structures in Nature. Adv. Drug Deliv. Rev. 2019, 151–152, 191–221. [Google Scholar] [CrossRef]
- Yang, Y.; Urban, M.W. Self-Healing Polymeric Materials. Chem. Soc. Rev. 2013, 42, 7446–7467. [Google Scholar] [CrossRef]
- Kim, S.M.; Jeon, H.; Shin, S.H.; Park, S.A.; Jegal, J.; Hwang, S.Y.; Oh, D.X.; Park, J. Superior Toughness and Fast Self-Healing at Room Temperature Engineered by Transparent Elastomers. Adv. Mater. 2018, 30, 1–8. [Google Scholar] [CrossRef]
- Ling, L.; Li, J.; Zhang, G.; Sun, R.; Wong, C.P. Self-Healing and Shape Memory Linear Polyurethane Based on Disulfide Linkages with Excellent Mechanical Property. Macromol. Res. 2018, 26, 365–373. [Google Scholar] [CrossRef]
- Zhang, X.; Han, L.; Liu, M.; Wang, K.; Tao, L.; Wan, Q.; Wei, Y. Recent Progress and Advances in Redox-Responsive Polymers as Controlled Delivery Nanoplatforms. Mater. Chem. Front. 2017, 1, 807–822. [Google Scholar] [CrossRef]
- Zhuang, J.; Gordon, M.R.; Ventura, J.; Li, L.; Thayumanavan, S. Multi-Stimuli Responsive Macromolecules and Their Assemblies. Chem. Soc. Rev. 2013, 42, 7421–7435. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.P.; Rong, M.Z.; Zhang, M.Q. Polymer Engineering Based on Reversible Covalent Chemistry: A Promising Innovative Pathway towards New Materials and New Functionalities. Prog. Polym. Sci. 2018, 80, 39–93. [Google Scholar] [CrossRef]
- Lv, C.; Zhao, K.; Zheng, J. A Highly Stretchable Self-Healing Poly(Dimethylsiloxane) Elastomer with Reprocessability and Degradability. Macromol. Rapid Commun. 2018, 39, 1–6. [Google Scholar] [CrossRef]
- Tomei, M.R.; Cinti, S.; Interino, N.; Manovella, V.; Moscone, D.; Arduini, F. Paper-Based Electroanalytical Strip for User-Friendly Blood Glutathione Detection. Sens. Actuator. B Chem. 2019, 294, 291–297. [Google Scholar] [CrossRef]
- Yin, W.; Ke, W.; Lu, N.; Wang, Y.Y.; Japir, A.A.W.M.M.; Mohammed, F.; Wang, Y.Y.; Pan, Y.; Ge, Z. Glutathione and Reactive Oxygen Species Dual-Responsive Block Copolymer Prodrugs for Boosting Tumor Site-Specific Drug Release and Enhanced Antitumor Efficacy. Biomacromolecules 2020, 21, 921–929. [Google Scholar] [CrossRef]
- Gao, Y.; Dong, C.M. Reduction- and Thermo-Sensitive Core-Cross-Linked Polypeptide Hybrid Micelles for Triggered and Intracellular Drug Release. Polym. Chem. 2017, 8, 1223–1232. [Google Scholar] [CrossRef]
- Utrera-Barrios, S.; Verdejo, R.; López-Manchado, M.A.; Hernández Santana, M. Evolution of Self-Healing Elastomers, from Extrinsic to Combined Intrinsic Mechanisms: A Review. Mater. Horizons 2020, 7, 2882–2902. [Google Scholar] [CrossRef]
- Kolšek, K.; Aponte-Santamaría, C.; Gräter, F. Accessibility Explains Preferred Thiol-Disulfide Isomerization in a Protein Domain. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Patai, S. The Chemistry of the Thiol Group. In The Chemistry of the Thiol Group: Patai’s Chemistry of Functional Groups Vol. 2; Wiley: London, UK, 1974; pp. 163–269. [Google Scholar]
- Amamoto, Y.; Otsuka, H.; Takahara, A.; Matyjaszewski, K. Self-Healing of Covalently Cross-Linked Polymers by Reshuffling Thiuram Disulfide Moieties in Air under Visible Light. Adv. Mater. 2012, 24, 3975–3980. [Google Scholar] [CrossRef]
- Parker, A.J.; Kharasch, N. The Scission Of The Sulfur-Sulfur Bond. Chem. Rev. 1959, 59, 583–628. [Google Scholar] [CrossRef]
- Cremlyn, R.J. Thiols, Sulfides, and Sulfenic Acids. In An Introduction to Organosulfur Chemistry; Wiley: Chichester, UK, 1996; pp. 40–61. [Google Scholar]
- Witt, D. Recent Developments in Disulfide Bond Formation. Synthesis. 2008, 16, 2491–2509. [Google Scholar] [CrossRef]
- Mandal, B.; Basu, B. Recent Advances in S-S Bond Formation. RSC Adv. 2014, 4, 13854–13881. [Google Scholar] [CrossRef]
- Mekaru, H.; Yoshigoe, A.; Nakamura, M.; Doura, T.; Tamanoi, F. Biodegradability of Disulfide-Organosilica Nanoparticles Evaluated by Soft X-ray Photoelectron Spectroscopy: Cancer Therapy Implications. ACS Appl. Nano Mater. 2019, 2, 479–488. [Google Scholar] [CrossRef]
- Doura, T.; Nishio, T.; Tamanoi, F.; Nakamura, M. Relationship between the Glutathione-Responsive Degradability of Thiol-Organosilica Nanoparticles and the Chemical Structures. J. Mater. Res. 2019, 34, 1266–1278. [Google Scholar] [CrossRef]
- Yu, T.; Wakuda, K.; Blair, D.L.; Weiss, R.G. Reversibly Cross-Linking Amino-Polysiloxanes by Simple Triatomic Molecules. Facile Methods for Tuning Thermal, Rheological, and Adhesive Properties. J. Phys. Chem. C 2009, 113, 11546–11553. [Google Scholar] [CrossRef]
- Liu, J.; Yao, Y.; Li, X.; Zhang, Z. Fabrication of Advanced Polydimethylsiloxane-Based Functional Materials: Bulk Modifications and Surface Functionalizations. Chem. Eng. J. 2021, 408, 127262. [Google Scholar] [CrossRef]
- De Keer, L.; Kilic, K.I.; Van Steenberge, P.H.M.; Daelemans, L.; Kodura, D.; Frisch, H.; De Clerck, K.; Reyniers, M.F.; Barner-Kowollik, C.; Dauskardt, R.H.; et al. Computational Prediction of the Molecular Configuration of Three-Dimensional Network Polymers. Nat. Mater. 2021, 20, 1422–1430. [Google Scholar] [CrossRef]
- Quake, S.R.; Scherer, A. From Micro- to Nanofabrication with Soft Materials. Science. 2000, 290, 1536–1540. [Google Scholar] [CrossRef] [Green Version]
- Rus, D.; Tolley, M.T. Design, Fabrication and Control of Soft Robots. Nature 2015, 521, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Eduok, U.; Faye, O.; Szpunar, J. Recent Developments and Applications of Protective Silicone Coatings: A Review of PDMS Functional Materials. Prog. Org. Coatings 2017, 111, 124–163. [Google Scholar] [CrossRef]
- Partenhauser, A.; Laffleur, F.; Rohrer, J.; Bernkop-Schnürch, A. Thiolated Silicone Oil: Synthesis, Gelling and Mucoadhesive Properties. Acta Biomater. 2015, 16, 169–177. [Google Scholar] [CrossRef] [Green Version]
- Partenhauser, A.; Netsomboon, K.; Leonaviciute, G.; Bernkop-Schnürch, A. Evaluation of Thiolated Silicone Oil as Advanced Mucoadhesive Antifoaming Agent. Drug Deliv. 2016, 23, 2711–2719. [Google Scholar] [CrossRef]
- Fürst, A.; Baus, R.A.; Lupo, N.; Bernkop-Schnürch, A. Entirely S-Protected Thiolated Silicone: A Novel Hydrophobic Mucoadhesive and Skin Adhesive. J. Pharm. Sci. 2019, 108, 2887–2894. [Google Scholar] [CrossRef]
- Grießinger, J.A.; Bonengel, S.; Partenhauser, A.; Ijaz, M.; Bernkop-Schnürch, A. Thiolated Polymers: Evaluation of Their Potential as Dermoadhesive Excipients. Drug Dev. Ind. Pharm. 2017, 43, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Kulawik-Pióro, A.; Drabczyk, A.K.; Kruk, J.; Wróblewska, M.; Winnicka, K.; Tchórzewska, J. Thiolated Silicone Oils as New Components of Protective Creams in the Prevention of Skin Diseases. Materials 2021, 14. [Google Scholar] [CrossRef]
- Wei, C.; Chen, M.; Liu, D.; Zhou, W.; Khan, M.; Wu, X.; Huang, N.; Li, L. A Recyclable Disulfide Bond Chemically Cross-Linking, High Toughness, High Conductivity Ion Gel Based on Re-Shaping and Restructuring in the Gel State. Polym. Chem. 2015, 6, 4067–4070. [Google Scholar] [CrossRef]
- Wei, C.; Chen, M.; Liu, D.; Zhou, W.; Khan, M.; Wu, X.; Huang, N.; Li, L. Synthesis of Recyclable, Chemically Cross-Linked, High Toughness, High Conductivity Ion Gels by Sequential Triblock Copolymer Self-Assembly and Disulfide Bond Cross-Linking. RSC Adv. 2015, 5, 22638–22646. [Google Scholar] [CrossRef]
- Naga, N.; Moriyama, K.; Furukawa, H. Synthesis and Properties of Multifunctional Thiol Crosslinked Gels Containing Disulfide Bond in the Network Structure. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 3749–3756. [Google Scholar] [CrossRef]
- Colby, R.H. Structure and Linear Viscoelasticity of Flexible Polymer Solutions: Comparison of Polyelectrolyte and Neutral Polymer Solutions. Rheol. Acta 2010, 49, 425–442. [Google Scholar] [CrossRef]
- Pomposo, J.A.; Perez-Baena, I.; Lo Verso, F.; Moreno, A.J.; Arbe, A.; Colmenero, J. How Far Are Single-Chain Polymer Nanoparticles in Solution from the Globular State? ACS Macro Lett. 2014, 3, 767–772. [Google Scholar] [CrossRef]
- Mavila, S.; Eivgi, O.; Berkovich, I.; Lemcoff, N.G. Intramolecular Cross-Linking Methodologies for the Synthesis of Polymer Nanoparticles. Chem. Rev. 2016, 116, 878–961. [Google Scholar] [CrossRef] [PubMed]
- Prasher, A.; Loynd, C.M.; Tuten, B.T.; Frank, P.G.; Chao, D.; Berda, E.B. Efficient Fabrication of Polymer Nanoparticles via Sonogashira Cross-Linking of Linear Polymers in Dilute Solution. J. Polym. Sci. Part A Polym. Chem. 2016, 54, 209–217. [Google Scholar] [CrossRef]
- Bae, S.; Galant, O.; Diesendruck, C.E.; Silberstein, M.N. The Effect of Intrachain Cross-Linking on the Thermomechanical Behavior of Bulk Polymers Assembled Solely from Single Chain Polymer Nanoparticles. Macromolecules 2018, 51, 7160–7168. [Google Scholar] [CrossRef]
- Gelest, Inc. SMS-042-[4-6% (mercaptopropyl)methylsiloxane]-dimethylsiloxane Copoloymer; SDS No. SMS-042; Gelest, Inc.: Morrisville, PA, USA, 2015. [Google Scholar]
- Gelest, Inc. SMS-142-[13-17% (mercaptopropyl)methylsiloxane]-dimethylsiloxane Copolymer; SDS No. SMS-142; Gelest, Inc.: Morrisville, PA, USA, 2017. [Google Scholar]
- Gottlieb, H.E.; Kotlyar, V.; Nudelman, A. NMR Chemical Shifts of Common Laboratory Solvents as Trace Impurities. J. Org. Chem. 1997, 62, 7512–7515. [Google Scholar] [CrossRef]
- Morcombe, C.R.; Zilm, K.W. Chemical Shift Referencing in MAS Solid State NMR. J. Magn. Reson. 2003, 162, 479–486. [Google Scholar] [CrossRef]
- Nieuwendall, R.C. How to Measure Absolute P3HT Crystallinity via CPMAS NMR. Mag. Reson. Chem. 2016, 54, 740–747. [Google Scholar] [CrossRef]
- Kuhn, W.; Balmer, G. Crosslinking of Single Linear Macromolecules. J. Polym. Sci. 1962, 57, 311–319. [Google Scholar] [CrossRef]
- Bourlès, E.; Alves de Sousa, R.; Galardon, E.; Selkti, M.; Tomas, A.; Artaud, I. Synthesis of Cyclic Mono- and Bis-Disulfides and Their Selective Conversion to Mono- and Bis-Thiosulfinates. Tetrahedron 2007, 63, 2466–2471. [Google Scholar] [CrossRef]
- Lepucki, P.; Dioguardi, A.P.; Karnaushenko, D.; Schmidt, O.G.; Grafe, H.J. The Normalized Limit of Detection in NMR Spectroscopy. J. Magn. Reson. 2021, 332, 107077. [Google Scholar] [CrossRef]
- Samoson, A.; Tuherm, T.; Past, J.; Reinhold, A.; Anupold, T.; Heinmaa, I. New Horizons for Magic-Angle Spinning NMR. In New Techniques in Solid State NMR. Topics in Current Chemistry, vol. 246; Klinowski, J., Ed.; Springer: Berlin/Heidelberg, Germany, 2005; pp. 15–31. [Google Scholar]
- Lee, J.N.; Park, C.; Whitesides, G.M. Solvent Compatibility of Poly(Dimethylsiloxane)-Based Microfluidic Devices. Anal. Chem. 2003, 75, 6544–6554. [Google Scholar] [CrossRef]
- Yilgor, I.; Eynur, T.; Yilgor, E.; Wilkes, G.L. Contribution of Soft Segment Entanglement on the Tensile Properties of Silicone-Urea Copolymers with Low Hard Segment Contents. Polymer 2009, 50, 4432–4437. [Google Scholar] [CrossRef]
- Brennan, D.P.; Dobley, A.; Sideris, P.J.; Oliver, S.R.J. Swollen Poly(Dimethylsiloxane) (PDMS) as a Template for Inorganic Morphologies. Langmuir 2005, 21, 11994–11998. [Google Scholar] [CrossRef] [PubMed]
- Favre, E. Swelling of Crosslinked Polydimethylsiloxane Networks by Pure Solvents: Influence of Temperature. Eur. Polym. J. 1996, 32, 1183–1188. [Google Scholar] [CrossRef]
- Iagatti, A.; Patrizi, B.; Basagni, A.; Marcelli, A.; Alessi, A.; Zanardi, S.; Fusco, R.; Salvalaggio, M.; Bussotti, L.; Foggi, P. Photophysical Properties and Excited State Dynamics of 4,7-Dithien-2-Yl-2,1,3-Benzothiadiazole. Phys. Chem. Chem. Phys. 2017, 19, 13604–13613. [Google Scholar] [CrossRef]
- Mao, H.; Qiu, Z.; Xie, B.; Wang, Z.; Shen, Z.; Hou, W. Development and Application of Ultra-High Temperature Drilling Fluids in Offshore Oilfield around Bohai Sea Bay Basin, China. Offshore Technol. Conf. Asia 2016, OTCA 2016 2016, March, 1201–1222. [Google Scholar] [CrossRef]
- Mallia, V.A.; Blair, D.L.; Weiss, R.G. Oscillatory Rheology and Surface Water Wave Effects on Crude Oil and Corn Oil Gels with (R)-12-Hydroxystearic Acid as Gelator. Ind. Eng. Chem. Res. 2016, 55, 954–960. [Google Scholar] [CrossRef]
- Vandeputte, A.G.; Reyniers, M.F.; Marin, G.B. Theoretical Study of the Thermal Decomposition of Dimethyl Disulfide. J. Phys. Chem. A 2010, 114, 10531–10549. [Google Scholar] [CrossRef]
- Sehon, A.H.; Darwent, B.D. The Thermal Decomposition of Mercaptans. J. Am. Chem. Soc. 1954, 76, 4806–4810. [Google Scholar] [CrossRef]
- Yao, Q.; Wilkie, C.A. How Does Cross-Linking Affect the Thermal Stability of Polyisoprene? Polym. Degrad. Stab. 2000, 69, 287–296. [Google Scholar] [CrossRef]
- Faragher, W.F.; Morrell, J.C.; Comay, S. Thermal Decomposition of Organic Sulfur Compounds. Ind. Eng. Chem. 1928, 20, 527–532. [Google Scholar] [CrossRef]
- Voronkov, M.G.; Klyuchnikov, V.A.; Mironenko, E.V.; Shvets, G.N.; Danilova, T.F.; Khudobin, Y.I. Thermochemistry of Organosilicon Compounds: V. Thermochemical Properties of Perorganyloligocyclosiloxanes. J. Organomet. Chem. 1991, 406, 91–97. [Google Scholar] [CrossRef]
- Advamacs.com Boiling Point Calculator. Available online: http://www.trimen.pl/witek/calculators/wrzenie.html (accessed on 10 March 2022).
- SDBSWeb. Available online: https://sdbs.db.aist.go.jp/sdbs/cgi-bin/direct_frame_top.cgi (accessed on 26 October 2022).
- Kleinberg, J.; Davidson, A.W. The Nature of Iodine Solutions. Chem. Rev. 1948, 42, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Winther, J.R.; Thorpe, C. Quantification of Thiols and Disulfides. Biochim. Biophys. Acta-Gen. Subj. 2014, 1840, 838–846. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, F.; Hoops, P.; Kudzin, Z.; Kudzin, Z.; Witczak, Z. Nuclear Magnetic Resonance Determination of the Site of Acylation of the Tautomeric Nucleophile 4-Thioxopyridine. J. Org. Chem. 1986, 51, 571–573. [Google Scholar] [CrossRef]
- Egwim, I.O.C.; Gruber, H.J. Spectrophotometric Measurement of Mercaptans with 4,4′-Dithiodipyridine. Anal. Biochem. 2001, 288, 188–194. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D-PDMS Material | Thiol Monomer Substitution in T-PDMS | Mw Range (Da) of T-PDMS | Initial Concentration of T-PDMS 1 | Initial Concentration in thiol (M) | Product Phase |
|---|---|---|---|---|---|
| 4–6% D-PDMS2wt% | 4–6% | 6000–8000 | 2 wt% | 0.02 | Liquid |
| 4–6% D-PDMS6wt% | 4–6% | 6000–8000 | 6 wt% | 0.06 | Liquid |
| 4–6% D-PDMS15wt% | 4–6% | 6000–8000 | 15 wt% | 0.18 | Liquid |
| 4–6% D-PDMS55wt% | 4–6% | 6000–8000 | 55 wt% | 0.47 | Solid |
| 13–17% D-PDMS1wt% | 13–17% | 3000–4000 | 1 wt% | 0.02 | Liquid |
| 13–17% D-PDMS6wt% | 13–17% | 3000–4000 | 6 wt% | 0.16 | Solid |
| 13–17% D-PDMS15wt% | 13–17% | 3000–4000 | 15 wt% | 0.32 | Solid |
| 13–17% D-PDMS55wt% | 13–17% | 3000–4000 | 55 wt% | 1.14 | Solid |
| Oxidation Product | Integration Value of the Protons α to the Thiol Group 1 | Calculated Percentage of Thiol Remaining after Oxidation |
|---|---|---|
| 4–6% D-PDMS2wt% | 0.01 | 0.5% |
| 4–6% D-PDMS6wt% | 0.00 | 0% |
| 4–6% D-PDMS15wt% | 0.02 | 1% |
| 13–17% D-PDMS1wt% | 0.07 | 3.5% |
| Solvent | δ [55] | ε [59,60] | Relative Swelling Rank from ref. [55] 1 | 4–6% D-PDMS55wt% | PDMS-Based Materials in the Literature (V%) | |||
|---|---|---|---|---|---|---|---|---|
| Swelling Rank (Based on V%) | V% | Ref. [27] 2 | Ref. [57] 1 | Ref. [58] 3 | ||||
| hexanes | 7.3 | 1.9 4 | 3 | 1 | 1500% | 580% | 190% | |
| ether | 7.5 | 4.3 | 2 | 2 | 1270% | 500% | 200% | |
| CHCl3 | 9.2 | 4.8 | 1 | 3 | 1250% | 700% | ||
| EtOAc | 9.0 | 6.1 | 5 | 5 | 760% | 540% | 80% | |
| DCM | 9.9 | 8.9 | 4 | 4 | 950% | 520% | 140% | |
| acetone | 9.9 | 20.7 | 6 | 6 | 60% | 120% | 40% | |
| EtOH | 12.7 | 24.0 | 7 | 7 | 20% | 30% | 10% | 10% |
| MeOH | 14.5 | 33.0 | 8 | 8 | 10% | |||
| water | 23.5 | 80.0 | 9 | 9 | >10% | 0% | 0% | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beaupre, D.M.; Goroncy, A.K.; Weiss, R.G. Influence of Concentration of Thiol-Substituted Poly(dimethylsiloxane)s on the Properties, Phases, and Swelling Behaviors of Their Crosslinked Disulfides. Macromol 2023, 3, 36-53. https://doi.org/10.3390/macromol3010004
Beaupre DM, Goroncy AK, Weiss RG. Influence of Concentration of Thiol-Substituted Poly(dimethylsiloxane)s on the Properties, Phases, and Swelling Behaviors of Their Crosslinked Disulfides. Macromol. 2023; 3(1):36-53. https://doi.org/10.3390/macromol3010004
Chicago/Turabian StyleBeaupre, Danielle M., Alexander K. Goroncy, and Richard G. Weiss. 2023. "Influence of Concentration of Thiol-Substituted Poly(dimethylsiloxane)s on the Properties, Phases, and Swelling Behaviors of Their Crosslinked Disulfides" Macromol 3, no. 1: 36-53. https://doi.org/10.3390/macromol3010004