
Congenital Atrophic Dermatofibrosarcoma Protuberans: A Case Report and Review of the Literature
 and
and
Abstract
:1. Introduction
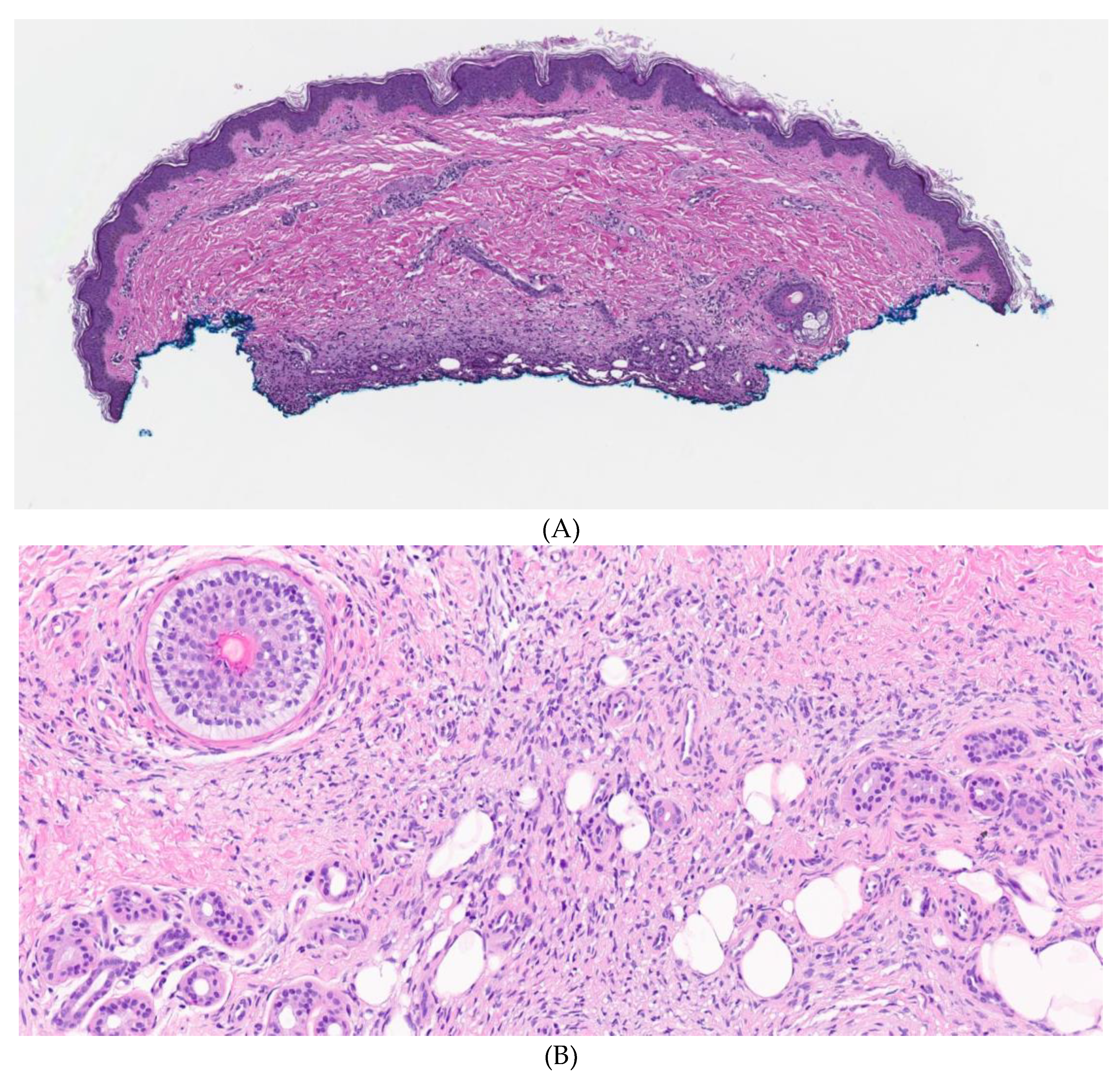
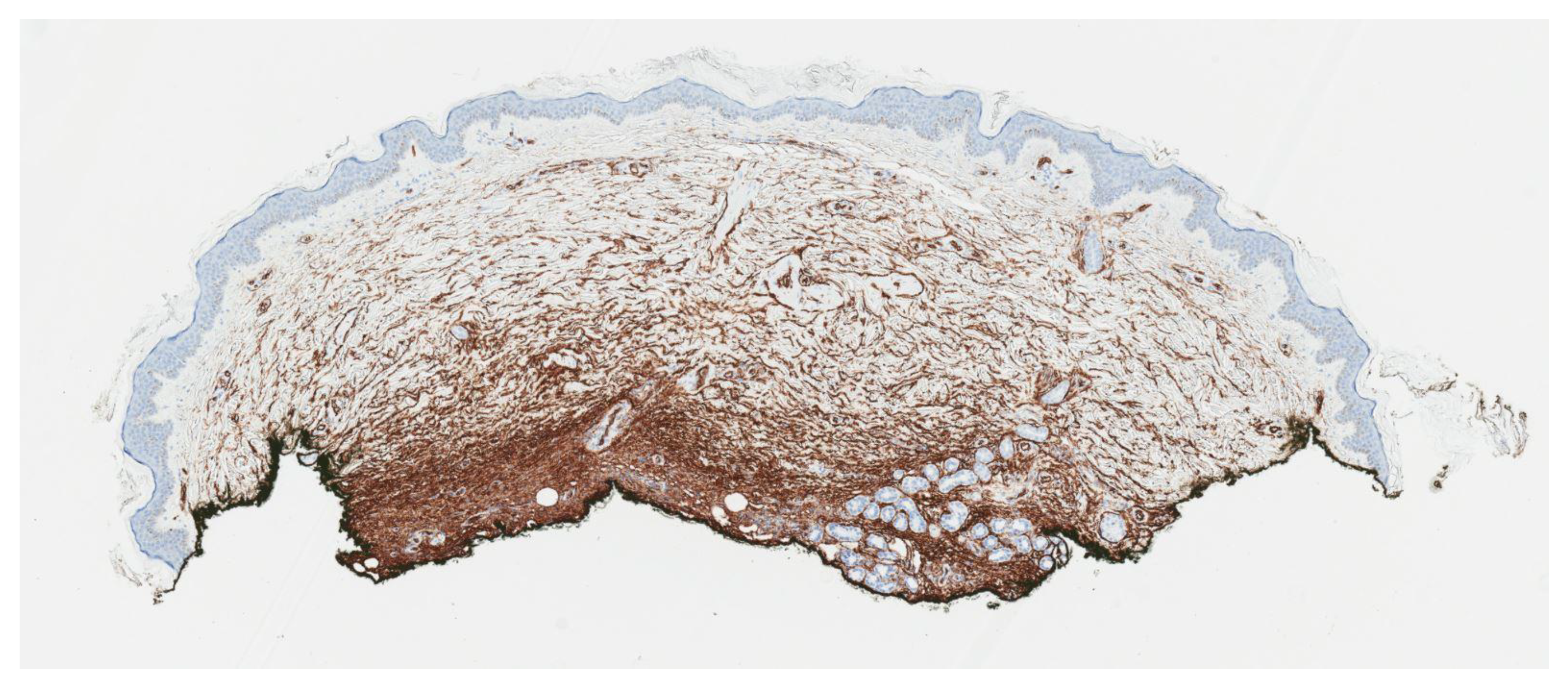
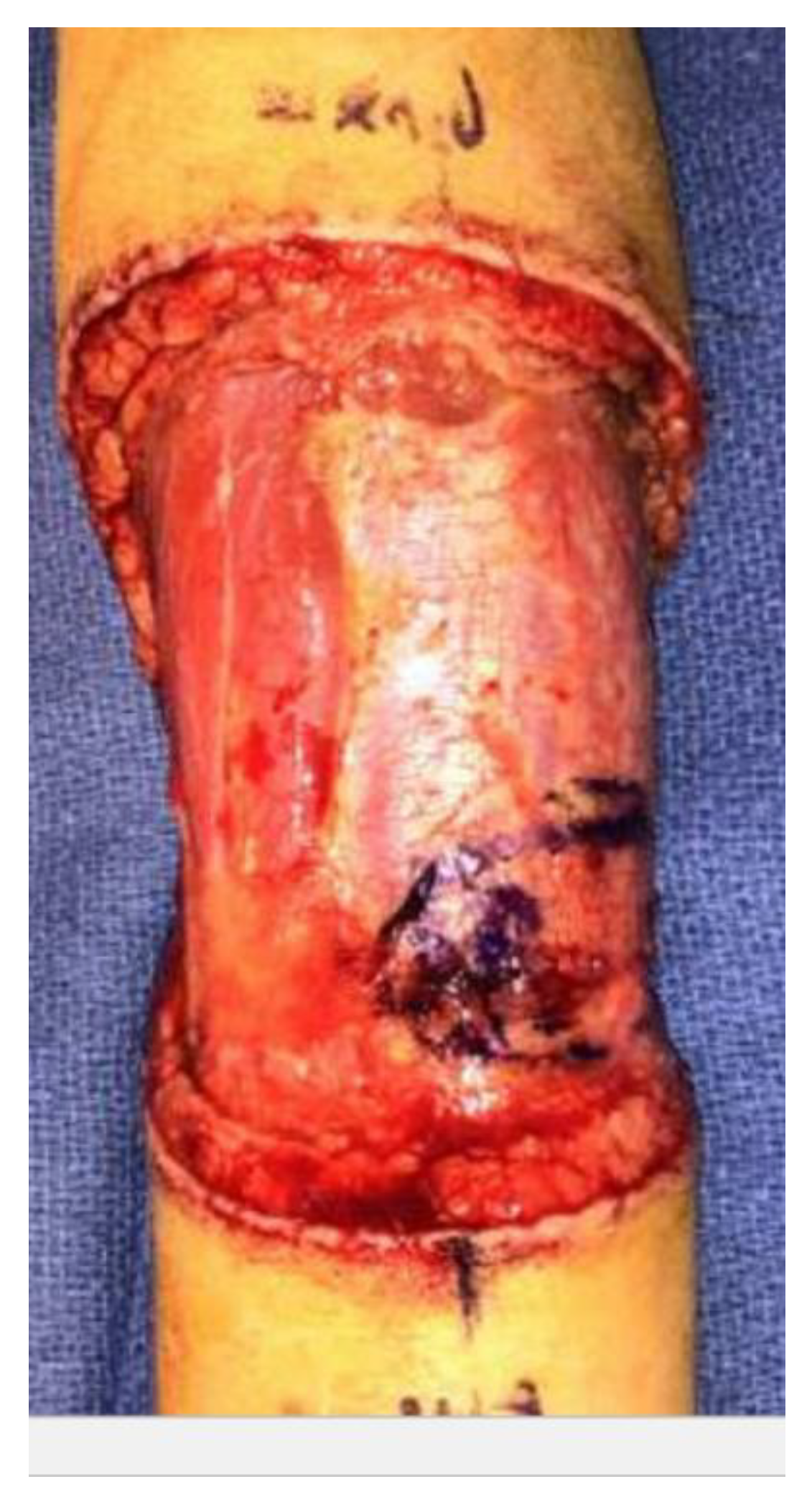
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brooks, J.; Ramsey, M.L. Dermatofibrosarcoma Protuberans. 2018. Available online: https://europepmc.org/article/NBK/nbk513305 (accessed on 19 July 2018).
- Van Lee, C.; Kan, W.C.; Gran, S.; Mooyaart, A.; Mureau, M.; Williams, H.; Matin, R.; Van Den Bos, R.; Hollestein, L. Dermatofibrosarcoma protuberans re-excision and recurrence rates in the Netherlands Between 1989 and 2016. Acta Derm.-Venereol. 2019, 99, 1160–1165. [Google Scholar] [CrossRef] [PubMed]
- Badhey, A.K.; Tikhtman, R.; Tang, A.L. Management of dermatofibrosarcoma protuberans. Curr. Opin. Otolaryngol. Head Neck Surg. 2021, 29, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Shi, Y.Z.; Feng, X.H.; Wang, X.T.; He, X.L.; Zhao, M. Novel TNC-PDGFD fusion in fibrosarcomatous dermatofibrosarcoma protuberans: A case report. Diagn. Pathol. 2021, 16, 1–6. [Google Scholar] [CrossRef]
- Reddy, C.; Hayward, P.; Thompson, P.; Kan, A. Dermatofibrosarcoma protuberans in children. J. Plast. Reconstr. Aesthetic Surg. 2009, 62, 819–823. [Google Scholar] [CrossRef]
- Posso-De Los Rios, C.J.; Lara-Corrales, I.; Ho, N. Dermatofibrosarcoma protuberans in pediatric patients: A report of 17 cases. J. Cutan. Med. Surg. 2014, 18, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Lam, S.W.; Cleton-Jansen, A.M.; Cleven, A.H.; Ruano, D.; van Wezel, T.; Szuhai, K.; Bovée, J.V. Molecular analysis of gene fusions in bone and soft tissue tumors by anchored multiplex PCR–based targeted next-generation sequencing. J. Mol. Diagn. 2018, 20, 653–663. [Google Scholar] [CrossRef]
- Zhou, Y.; Chin, J.; Strutin, M.D.; Lomiguen, C.M. Unmasking dermatofibrosarcoma protuberans: Case report of an atypical presentation complicated by post-surgical excision. Int. J. Surg. Case Rep. 2020, 69, 101–104. [Google Scholar] [CrossRef]
- Souiki, T.; Belhaj, A.; Ait Abderrhim, A.; Alami, B.; Tahiri, L.; Chbani, L.; Ibn Majdoub, K.; Toughrai, I.; Mazaz, K. Dermatofibrosarcoma protuberans of the anterior abdominal wall: Case report and literature review. J. Surg. Case Rep. 2022, 2022, rjac272. [Google Scholar] [CrossRef]
- Bakry, O.; Attia, A. Atrophic dermatofibrosarcoma protuberans. J. Dermatol. Case Rep. 2012, 6, 14. [Google Scholar] [CrossRef]
- Feramisco, J.; Larsen, F.; Weitzul, S.; Cockerell, C.; Ghali, F. Congenital atrophic dermatofibrosarcoma protuberans in a 7-month-old boy treated with Mohs micrographic surgery. Pediatr. Dermatol. 2008, 25, 455–459. [Google Scholar] [CrossRef]
- Llombart, B.; Serra, C.; Requena, C.; Alsina, M.; Morgado-Carrasco, D.; Través, V.; Sanmartín, O. Guidelines for diagnosis and treatment of cutaneous sarcomas: Dermatofibrosarcoma protuberans. Actas Dermo-Sifiliográficas 2018, 109, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Rao, C.; Shetty K, J.; Prasad, H.L.K. An uncommon malignancy of the breast; dermatofibrosarcoma protruberans—A case report. Indian J. Surg. Oncol. 2012, 3, 242–244. [Google Scholar] [CrossRef]
- Reilly, D.J.; Loo, Y.L.; Alexander, W.M.; Wilks, D.J.; MacGregor, D.; Coombs, C.J. Diagnostic and Management Considerations in Pediatric Dermatofibrosarcoma Protuberans. Ann. Plast. Surg. 2022, 88, 319–322. [Google Scholar] [CrossRef] [PubMed]
- Valdivielso-Ramos, M.; Hernanz, J.M. Dermatofibrosarcoma protuberans en la infancia. Actas Dermo-Sifiliogr. 2012, 103, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhang, B.; Han, X.; Ma, L. Pediatric atrophic dermatofibrosarcoma protuberans. Pediatr. Investig. 2017, 1, 50. [Google Scholar] [CrossRef] [PubMed]
- Akay, B.N.; Unlu, E.; Erdem, C.; Heper, A.O. Dermatoscopic findings of atrophic dermatofibrosarcoma protuberans. Dermatol. Pract. Concept. 2015, 5, 71. [Google Scholar] [CrossRef] [PubMed]
- Makino, M.; Sasaoka, S.; Nakanishi, G.; Makino, E.; Fujimoto, W. Congenital atrophic dermatofibrosarcoma protuberans detected by COL1A1-PDGFB rearrangement. Diagn. Pathol. 2016, 11, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Hanabusa, M.; Kamo, R.; Harada, T.; Ishii, M. Dermatofibrosarcoma protuberans with atrophic appearance at early stage of the tumor. J. Dermatol. 2007, 34, 336–339. [Google Scholar] [CrossRef] [PubMed]
- Marini, M.; Saponaro, A.; Magariños, G.; De Baldrich, A.; Lynch, P.; Remorino, L. Congenital atrophic dermatofibrosarcoma protuberans. Int. J. Dermatol. 2001, 40, 448–450. [Google Scholar] [CrossRef]
- Han, H.H.; Lim, S.Y.; Park, Y.M.; Rhie, J.W. Congenital dermatofibrosarcoma protuberans: A case report and literature review. Ann. Dermatol. 2015, 27, 597–600. [Google Scholar] [CrossRef]
- Weinstein, J.M.; Drolet, B.A.; Esterly, N.B.; Rogers, M.; Bauer, B.S.; Wagner, A.M.; Mancini, A.J. Congenital dermatofibrosarcoma protuberans: Variability in presentation. Arch. Dermatol. 2003, 139, 207–211. [Google Scholar] [CrossRef]
- Martin, L.; Combemale, P.; Dupin, M.; Chouvet, B.; Kanitakis, J.; Bouyssou-Gauthier, M.L.; Dubreuil, G.; Claudy, A.; Grimand, P.G. The atrophic variant of dermatofibrosarcoma protuberans in childhood: A report of six cases. Br. J. Dermatol. 1998, 139, 719–725. [Google Scholar] [PubMed]
- Martin, L.; Piette, F.; Blanc, P.; Mortier, L.; Avril, M.F.; Delaunay, M.M.; Dréno, B.; Granel, F.; Mantoux, F.; Aubin, F.; et al. Clinical variants of the preprotuberant stage of dermatofibrosarcoma protuberans. Br. J. Dermatol. 2005, 153, 932–936. [Google Scholar] [CrossRef] [PubMed]
- Maire, G.; Fraitag, S.; Galmiche, L.; Keslair, F.; Ebran, N.; Terrier-Lacombe, M.J.; de Prost, Y.; Pedeutour, F. A clinical, histologic, and molecular study of 9 cases of congenital dermatofibrosarcoma protuberans. Arch. Dermatol. 2007, 143, 203–210. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Zhao, L.; Wang, J. Atrophic dermatofibrosarcoma protuberans: A clinicopathological study of 16 cases. Pathology 2019, 51, 615–620. [Google Scholar] [CrossRef]
- Ouzzani, M.; Hammady, H.; Fedorowicz, Z.; Elmagarmid, A. Rayyan — a web and mobile app for systematic reviews. Syst. Rev. 2016, 5, 210. [Google Scholar] [CrossRef]
- Hao, X.; Billings, S.D.; Wu, F.; Stultz, T.W.; Procop, G.W.; Mirkin, G.; Vidimos, A.T. Dermatofibrosarcoma protuberans: Update on the diagnosis and treatment. J. Clin. Med. 2020, 9, 1752. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.K.; Bandyopadhyay, D.; Ghosh, A.; Biswas, S.K.; Barma, K.D. Acquired multifocal tufted angiomas in an immunocompetent young adult. Indian J. Dermatol. 2011, 56, 412. [Google Scholar] [CrossRef]
- Jandali, S.; Kirschner, R.E. Congenital leukemia cutis presenting as multiple violaceous lesions in a newborn. Ann. Plast. Surg. 2011, 66, 310–312. [Google Scholar] [CrossRef]
- Tantcheva-Poor, I.; Marathovouniotis, N.; Kutzner, H.; Mentzel, T. Vascular congenital dermatofibrosarcoma protuberans: A new histological variant of dermatofibrosarcoma protuberans. Am. J. Dermatopathol. 2012, 34, e46–e49. [Google Scholar] [CrossRef]
- Marque, M.; Bessis, D.; Pedeutour, F.; Viseux, V.; Guillot, B.; Fraitag-Spinner, S. Medallion-like dermal dendrocyte hamartoma: The main diagnostic pitfall is congenital atrophic dermatofibrosarcoma. Br. J. Dermatol. 2008, 160, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Bai, J.; Liu, B.; Liu, T.; Qiao, J.; Fang, H. Atrophic Pigmented Dermatofibrosarcoma Protuberans: A Case Report and Literature Review. Front. Oncol. 2021, 11, 669754. [Google Scholar] [CrossRef]
- Rodríguez-Jurado, R.; Palacios, C.; Durán-McKinster, C.; Mercadillo, P.; Orozco-Covarrubias, L.; del Mar Saez-de-Ocariz, M.; Ruiz-Maldonado, R. Medallion-like dermal dendrocyte hamartoma: A new clinically and histopathologically distinct lesion. J. Am. Acad. Dermatol. 2004, 51, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Shah, K.N.; Anderson, E.; Junkins-Hopkins, J.; James, W.D. Medallion-like dermal dendrocyte hamartoma. Pediatr. Dermatol. 2007, 24, 632–636. [Google Scholar] [CrossRef] [PubMed]
- Sirvent, N.; Maire, G.; Pedeutour, F. Genetics of dermatofibrosarcoma protuberans family of tumors: From ring chromosomes to tyrosine kinase inhibitor treatment. Genes Chromosomes Cancer 2003, 37, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kadam, S.S.; Kadam, T. Recurrent Dermatofibrosarcoma Protruberance over Shoulder: An Unresolved Problem. Int. Res. J. Oncol. 2021, 4, 11–20. [Google Scholar]
- Sleiwah, A.; Wright, T.C.; Chapman, T.; Dangoor, A.; Maggiani, F.; Clancy, R. Dermatofibrosarcoma protuberans in children. Curr. Treat. Options Oncol. 2022, 23, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Chicaud, M.; Frassati-Biaggi, A.; Kaltenbach, S.; Karanian, M.; Orbach, D.; Fraitag, S. Dermatofibrosarcoma protuberans, fibrosarcomatous variant: A rare tumor in children. Pediatr. Dermatol. 2021, 38, 217–222. [Google Scholar] [CrossRef] [PubMed]
- Macarenco, R.S.; Zamolyi, R.; Franco, M.F.; Nascimento, A.G.; Abott, J.J.; Wang, X.; Erickson-Johnson, M.R.; Oliveira, A.M. Genomic gains of COL1A1-PDFGB occur in the histologic evolution of giant cell fibroblastoma into dermatofibrosarcoma protuberans. Genes Chromosomes Cancer 2008, 47, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.P.; Gupta, A.K.; Ansari, M.; Mehra, S.K.; Barolia, D.K. Very rare childhood tumor: Giant cell fibroblastoma. Med. J. Dr. DY Patil Univ. 2018, 11, 452. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Age of Presentation | Age of Diagnosis | Quiescent Period to Protuberant Phase | Location | Misdiagnosis | Translocation T (17; 22)/ COL1A1-PDGFB Fusion Gene | Treatment |
|---|---|---|---|---|---|---|---|
| Salem et al. | One year * | 10 years | Remained atrophic | Forearm | Bruise, morphea | Positive | Wide excision |
| Makino et al. [18] | Birth | 19 years | 10 years | Anterior chest | N/A | Positive | Wide excision |
| Marini et al. [20] | Birth | 16 years | 7 years | Anterior leg | Congenital fibroma | N/A | Mohs surgery |
| Han et al. [21] | Birth | 6 years | 4 years | Posterior neck | Dermatitis | Negative | Wide excision |
| Feramisco et al. [11] | 7 m * | 7 m | Remained atrophic | Left inguinal | N/A | Positive | Mohs surgery |
| Weinstein et al. [22] | 6 years * | 14 years | 6 years | Back | Vacular malformation, Fibrous histocytoma | N/A | Wide excision |
| Martin et al. [23,24] | Birth | 13 years | 7 years | Calf | Hematoma | Positive | Wide excision |
| Weinstein et al. [22] | 6 m * | 1 year | 6 m | Right thigh | Aplasia cutis | N/A | Wide excision |
| Martin et al. [23,24] | Birth | 3 years | 2 years | Periumbilical | Morphea | N/A | Wide excision |
| Maire et al. [25] | N/A | 3 years | N/A | Lumbar | “Difficult to Characterize” | Positive | Wide excision |
| Maire et al. [25] | N/A | 11 years | N/A | Lumbar | N/A | Positive | Wide excision |
| Maire et al. [25] | 2 years * | 7 years | N/A | Occipital | Aplasia cutis, Fibrous hamartoma, Infantile fibromatosis | Positive | Wide excision |
| Maire et al. [25] | N/A | 10 years | N/A | Foot | Difficult to characterize | Positive | Wide excision |
| Maire et al. [25] | 15 years * | 17 years | N/A | Trunk | Fibromatosis, Xanthomatous hamartoma, Aplasia cutis, Infantile fibrosarcoma | Positive | Wide excision |
| Maire et al. [25] | 2 years * | 3 years | N/A | Thorax | Fibrosarcoma, Angioma, Mastocytoma | N/A | Wide excision |
| Wide Local Excision | Mohs Excision | Staged Excision “Slow Mohs ” | |
|---|---|---|---|
| Description | Excision with 2–4 cm margins followed by closure of the defect |
|
|
| Advantage |
|
|
|
| Disadvantage |
|
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Salem, I.; Bradley, K.; Mann, J.A.; Shin, J.H.; LeBoeuf, M.; Sriharan, A. Congenital Atrophic Dermatofibrosarcoma Protuberans: A Case Report and Review of the Literature. Dermato 2023, 3, 97-108. https://doi.org/10.3390/dermato3020008
Salem I, Bradley K, Mann JA, Shin JH, LeBoeuf M, Sriharan A. Congenital Atrophic Dermatofibrosarcoma Protuberans: A Case Report and Review of the Literature. Dermato. 2023; 3(2):97-108. https://doi.org/10.3390/dermato3020008
Chicago/Turabian StyleSalem, Iman, Katherine Bradley, Julianne A. Mann, Joseph H. Shin, Matthew LeBoeuf, and Aravindhan Sriharan. 2023. "Congenital Atrophic Dermatofibrosarcoma Protuberans: A Case Report and Review of the Literature" Dermato 3, no. 2: 97-108. https://doi.org/10.3390/dermato3020008