
Mechanisms of Resistance and Strategies to Combat Resistance in PD-(L)1 Blockade
Abstract
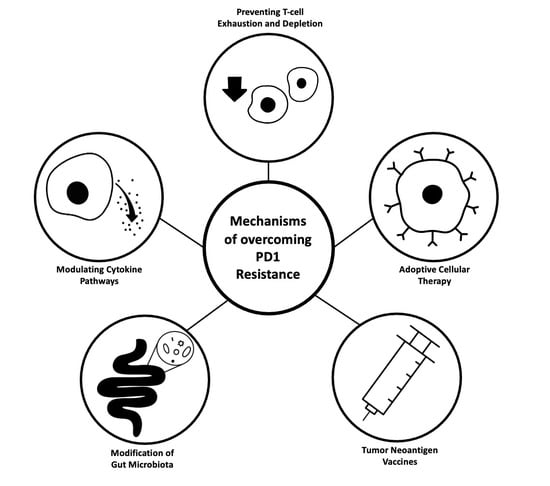
:
1. Introduction
2. PD-1/PD-L1 Expression and Function
3. PD-L1 Overexpression in Tumors and PD-1/PD-L1 Immune Checkpoint Therapy
4. Mechanisms of Resistance to PD-1/PD-L1 Blockade and Corresponding Therapeutic Strategies
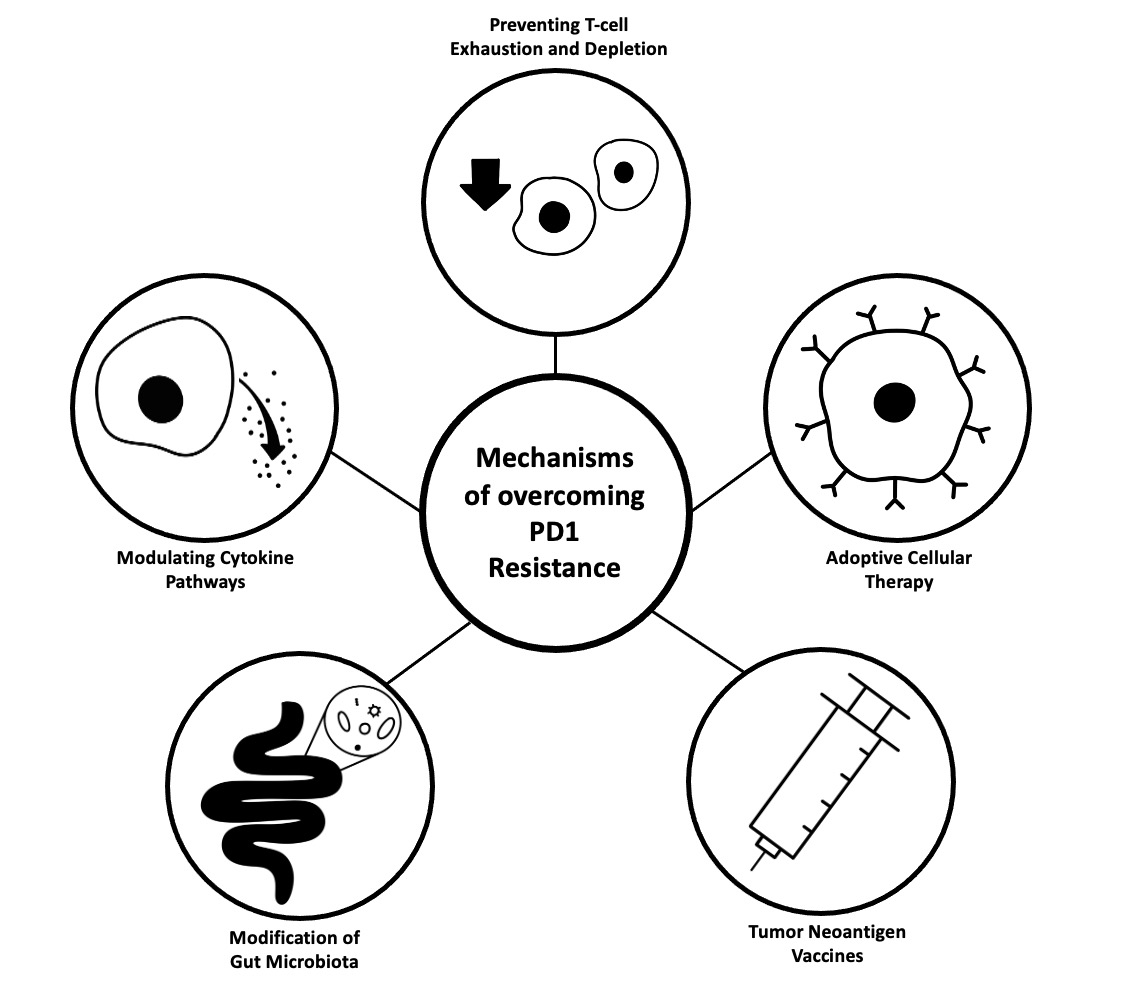
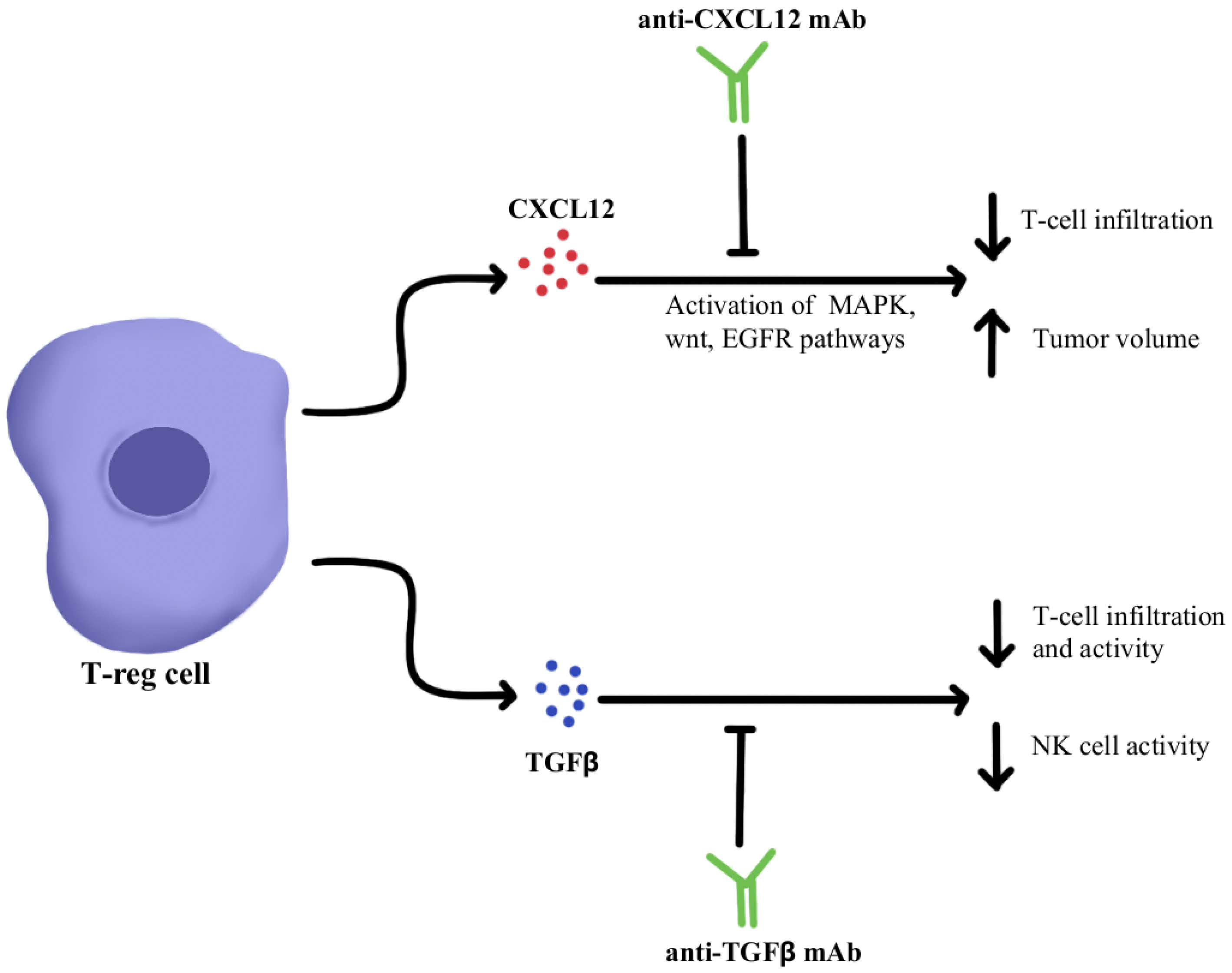
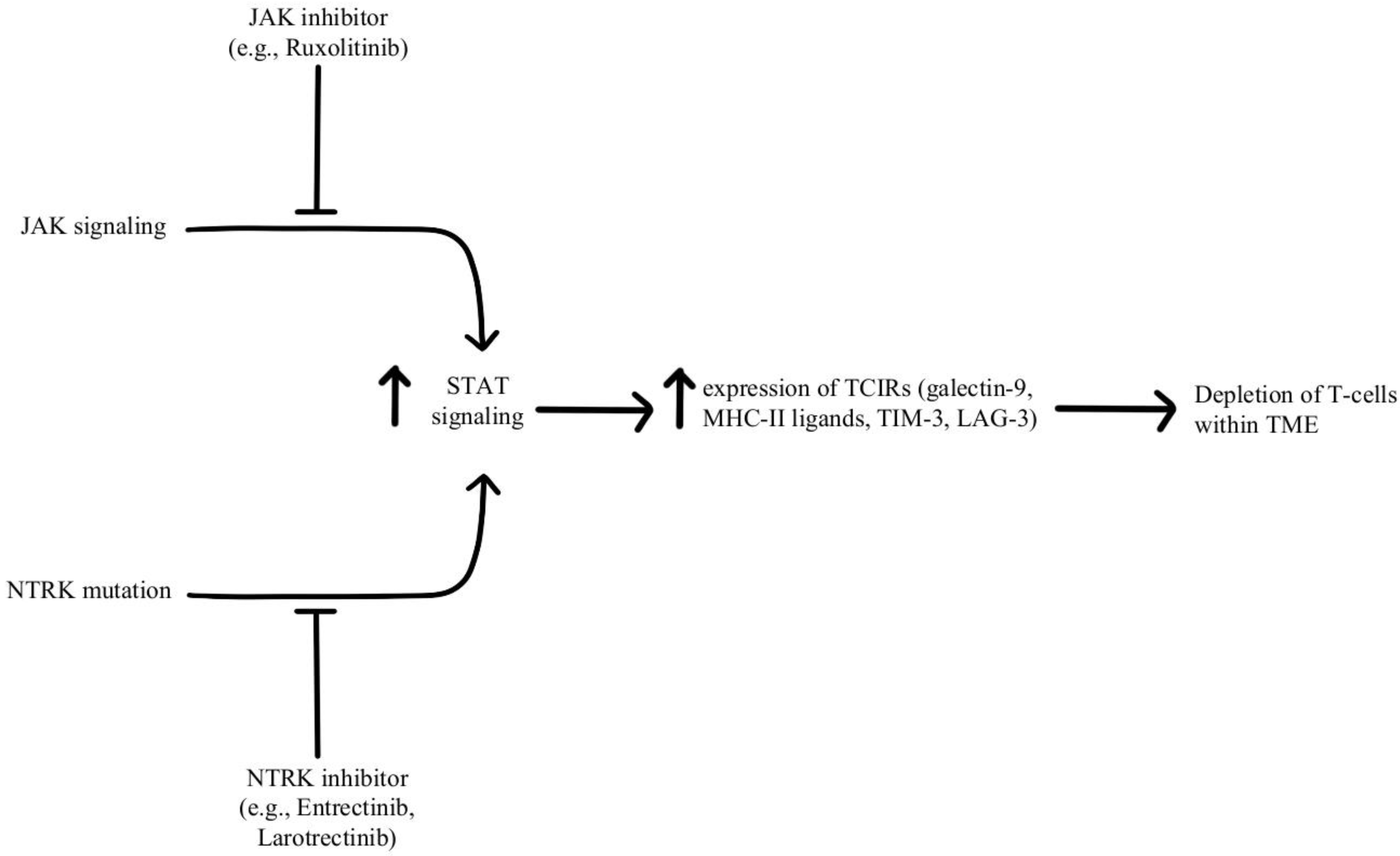
4.1. Immunosuppressive Cytokine Pathways
4.2. T-Cell Exhaustion and Depletion
4.3. Adoptive Cellular Therapy
4.4. Tumor Neoantigen Vaccines
4.5. Gut Microbiota
5. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishijima, T.F.; Shachar, S.S.; Nyrop, K.A.; Muss, H.B. Safety and Tolerability of PD-1/PD-L1 Inhibitors Compared with Chemotherapy in Patients with Advanced Cancer: A Meta-Analysis. Oncologist 2017, 22, 470–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, X.; Zhao, B. Efficacy of PD-1 or PD-L1 Inhibitors and PD-L1 Expression Status in Cancer: Meta-Analysis. BMJ 2018, 362, k3529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- US Food & Drug. Administration Drugs@FDA: FDA-Approved Drugs. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/ (accessed on 19 April 2022).
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced Expression of PD-1, a Novel Member of the Immunoglobulin Gene Superfamily, upon Programmed Cell Death. EMBO J. 1992, 11, 3887–3895. [Google Scholar] [CrossRef]
- Nishimura, H.; Nose, M.; Hiai, H.; Minato, N.; Honjo, T. Development of Lupus-like Autoimmune Diseases by Disruption of the PD-1 Gene Encoding an ITIM Motif-Carrying Immunoreceptor. Immunity 1999, 11, 141–151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dong, H.; Zhu, G.; Tamada, K.; Chen, L. B7-H1, a Third Member of the B7 Family, Co-Stimulates T-Cell Proliferation and Interleukin-10 Secretion. Nat. Med. 1999, 5, 1365–1369. [Google Scholar] [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 Immunoinhibitory Receptor by a Novel B7 Family Member Leads to Negative Regulation of Lymphocyte Activation. J. Exp. Med. 2000, 192, 1027–1034. [Google Scholar] [CrossRef] [Green Version]
- Iwai, Y.; Ishida, M.; Tanaka, Y.; Okazaki, T.; Honjo, T.; Minato, N. Involvement of PD-L1 on Tumor Cells in the Escape from Host Immune System and Tumor Immunotherapy by PD-L1 Blockade. Proc. Natl. Acad. Sci. USA 2002, 99, 12293–12297. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-J.; Jang, B.-C.; Lee, S.-W.; Yang, Y.-I.; Suh, S.-I.; Park, Y.-M.; Oh, S.; Shin, J.-G.; Yao, S.; Chen, L.; et al. Interferon Regulatory Factor-1 Is Prerequisite to the Constitutive Expression and IFN-Gamma-Induced Upregulation of B7-H1 (CD274). FEBS Lett. 2006, 580, 755–762. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Hamrouni, A.; Wolowiec, D.; Coiteux, V.; Kuliczkowski, K.; Hetuin, D.; Saudemont, A.; Quesnel, B. Plasma Cells from Multiple Myeloma Patients Express B7-H1 (PD-L1) and Increase Expression after Stimulation with IFN-{gamma} and TLR Ligands via a MyD88-, TRAF6-, and MEK-Dependent Pathway. Blood 2007, 110, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Gajewski, T.F.; Kline, J. PD-1/PD-L1 Interactions Inhibit Antitumor Immune Responses in a Murine Acute Myeloid Leukemia Model. Blood 2009, 114, 1545–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Taube, J.M.; Anders, R.A.; Pardoll, D.M. Mechanism-Driven Biomarkers to Guide Immune Checkpoint Blockade in Cancer Therapy. Nat. Rev. Cancer 2016, 16, 275–287. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhao, X.; Fu, J.; Wang, H. Progress and Challenges in Precise Treatment of Tumors With PD-1/PD-L1 Blockade. Front. Immunol. 2020, 11, 339. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.-Y.; Zhang, D.; Wu, S.; Xu, M.; Zhou, X.; Lu, X.-J.; Ji, J. Resistance to PD-1/PD-L1 Blockade Cancer Immunotherapy: Mechanisms, Predictive Factors, and Future Perspectives. Biomark. Res. 2020, 8, 35. [Google Scholar] [CrossRef] [PubMed]
- Alsaab, H.O.; Sau, S.; Alzhrani, R.; Tatiparti, K.; Bhise, K.; Kashaw, S.K.; Iyer, A.K. PD-1 and PD-L1 Checkpoint Signaling Inhibition for Cancer Immunotherapy: Mechanism, Combinations, and Clinical Outcome. Front. Pharmacol. 2017, 8, 561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bardhan, K.; Anagnostou, T.; Boussiotis, V.A. The PD1:PD-L1/2 Pathway from Discovery to Clinical Implementation. Front. Immunol. 2016, 7, 550. [Google Scholar] [CrossRef] [Green Version]
- Nurieva, R.; Thomas, S.; Nguyen, T.; Martin-Orozco, N.; Wang, Y.; Kaja, M.-K.; Yu, X.-Z.; Dong, C. T-Cell Tolerance or Function Is Determined by Combinatorial Costimulatory Signals. EMBO J. 2006, 25, 2623–2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Liang, X.-L.; Liu, X.-G.; Chen, N.-P. The Landscape of PD-L1 Expression and Somatic Mutations in Hepatocellular Carcinoma. J. Gastrointest. Oncol. 2021, 12, 1132–1140. [Google Scholar] [CrossRef] [PubMed]
- Keir, M.E.; Liang, S.C.; Guleria, I.; Latchman, Y.E.; Qipo, A.; Albacker, L.A.; Koulmanda, M.; Freeman, G.J.; Sayegh, M.H.; Sharpe, A.H. Tissue Expression of PD-L1 Mediates Peripheral T Cell Tolerance. J. Exp. Med. 2006, 203, 883–895. [Google Scholar] [CrossRef] [PubMed]
- Eppihimer, M.J.; Gunn, J.; Freeman, G.J.; Greenfield, E.A.; Chernova, T.; Erickson, J.; Leonard, J.P. Expression and Regulation of the PD-L1 Immunoinhibitory Molecule on Microvascular Endothelial Cells. Microcirculation 2002, 9, 133–145. [Google Scholar] [CrossRef]
- Ju, X.; Zhang, H.; Zhou, Z.; Wang, Q. Regulation of PD-L1 Expression in Cancer and Clinical Implications in Immunotherapy. Am. J. Cancer Res. 2020, 10, 1–11. [Google Scholar] [PubMed]
- Butte, M.J.; Keir, M.E.; Phamduy, T.B.; Sharpe, A.H.; Freeman, G.J. Programmed Death-1 Ligand 1 Interacts Specifically with the B7-1 Costimulatory Molecule to Inhibit T Cell Responses. Immunity 2007, 27, 111–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francisco, L.M.; Sage, P.T.; Sharpe, A.H. The PD-1 Pathway in Tolerance and Autoimmunity. Immunol. Rev. 2010, 236, 219–242. [Google Scholar] [CrossRef] [PubMed]
- Thompson, R.H.; Gillett, M.D.; Cheville, J.C.; Lohse, C.M.; Dong, H.; Webster, W.S.; Krejci, K.G.; Lobo, J.R.; Sengupta, S.; Chen, L.; et al. Costimulatory B7-H1 in Renal Cell Carcinoma Patients: Indicator of Tumor Aggressiveness and Potential Therapeutic Target. Proc. Natl. Acad. Sci. USA 2004, 101, 17174–17179. [Google Scholar] [CrossRef] [Green Version]
- Ohigashi, Y.; Sho, M.; Yamada, Y.; Tsurui, Y.; Hamada, K.; Ikeda, N.; Mizuno, T.; Yoriki, R.; Kashizuka, H.; Yane, K.; et al. Clinical Significance of Programmed Death-1 Ligand-1 and Programmed Death-1 Ligand-2 Expression in Human Esophageal Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2005, 11, 2947–2953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, C.; Zhu, Y.; Jiang, J.; Zhao, J.; Zhang, X.-G.; Xu, N. Immunohistochemical Localization of Programmed Death-1 Ligand-1 (PD-L1) in Gastric Carcinoma and Its Clinical Significance. Acta Histochem. 2006, 108, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Hamanishi, J.; Mandai, M.; Iwasaki, M.; Okazaki, T.; Tanaka, Y.; Yamaguchi, K.; Higuchi, T.; Yagi, H.; Takakura, K.; Minato, N.; et al. Programmed Cell Death 1 Ligand 1 and Tumor-Infiltrating CD8+ T Lymphocytes Are Prognostic Factors of Human Ovarian Cancer. Proc. Natl. Acad. Sci. USA 2007, 104, 3360–3365. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, J.; Wada, Y.; Matsumoto, K.; Azuma, M.; Kikuchi, K.; Ueda, S. Overexpression of B7-H1 (PD-L1) Significantly Associates with Tumor Grade and Postoperative Prognosis in Human Urothelial Cancers. Cancer Immunol. Immunother. CII 2007, 56, 1173–1182. [Google Scholar] [CrossRef]
- Nomi, T.; Sho, M.; Akahori, T.; Hamada, K.; Kubo, A.; Kanehiro, H.; Nakamura, S.; Enomoto, K.; Yagita, H.; Azuma, M.; et al. Clinical Significance and Therapeutic Potential of the Programmed Death-1 Ligand/Programmed Death-1 Pathway in Human Pancreatic Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 2151–2157. [Google Scholar] [CrossRef] [Green Version]
- Hino, R.; Kabashima, K.; Kato, Y.; Yagi, H.; Nakamura, M.; Honjo, T.; Okazaki, T.; Tokura, Y. Tumor Cell Expression of Programmed Cell Death-1 Ligand 1 Is a Prognostic Factor for Malignant Melanoma. Cancer 2010, 116, 1757–1766. [Google Scholar] [CrossRef]
- Abril-Rodriguez, G.; Ribas, A. SnapShot: Immune Checkpoint Inhibitors. Cancer Cell 2017, 31, 848–848.e1. [Google Scholar] [CrossRef]
- Krummel, M.F.; Allison, J.P. CD28 and CTLA-4 Have Opposing Effects on the Response of T Cells to Stimulation. J. Exp. Med. 1995, 182, 459–465. [Google Scholar] [CrossRef] [Green Version]
- Topalian, S.L.; Sznol, M.; McDermott, D.F.; Kluger, H.M.; Carvajal, R.D.; Sharfman, W.H.; Brahmer, J.R.; Lawrence, D.P.; Atkins, M.B.; Powderly, J.D.; et al. Survival, Durable Tumor Remission, and Long-Term Safety in Patients with Advanced Melanoma Receiving Nivolumab. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2014, 32, 1020–1030. [Google Scholar] [CrossRef] [PubMed]
- Naidoo, J.; Page, D.B.; Li, B.T.; Connell, L.C.; Schindler, K.; Lacouture, M.E.; Postow, M.A.; Wolchok, J.D. Toxicities of the Anti-PD-1 and Anti-PD-L1 Immune Checkpoint Antibodies. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2015, 26, 2375–2391. [Google Scholar] [CrossRef] [PubMed]
- Vaddepally, R.K.; Kharel, P.; Pandey, R.; Garje, R.; Chandra, A.B. Review of Indications of FDA-Approved Immune Checkpoint Inhibitors per NCCN Guidelines with the Level of Evidence. Cancers 2020, 12, 738. [Google Scholar] [CrossRef] [Green Version]
- Ai, L.; Chen, J.; Yan, H.; He, Q.; Luo, P.; Xu, Z.; Yang, X. Research Status and Outlook of PD-1/PD-L1 Inhibitors for Cancer Therapy. Drug Des. Dev. Ther. 2020, 14, 3625–3649. [Google Scholar] [CrossRef] [PubMed]
- Cercek, A.; Lumish, M.; Sinopoli, J.; Weiss, J.; Shia, J.; Lamendola-Essel, M.; El Dika, I.H.; Segal, N.; Shcherba, M.; Sugarman, R.; et al. PD-1 Blockade in Mismatch Repair-Deficient, Locally Advanced Rectal Cancer. N. Engl. J. Med. 2022, 386, 2363–2376. [Google Scholar] [CrossRef]
- Yearley, J.H.; Gibson, C.; Yu, N.; Moon, C.; Murphy, E.; Juco, J.; Lunceford, J.; Cheng, J.; Chow, L.Q.M.; Seiwert, T.Y.; et al. PD-L2 Expression in Human Tumors: Relevance to Anti-PD-1 Therapy in Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 3158–3167. [Google Scholar] [CrossRef] [Green Version]
- Couillault, C.; Srinivasamani, A.; Hedge, S.; Liu, Q.; Jaiswal, A.; Zha, D.; Curran, M. 291 Dual-Specific Antibodies Blocking Both PD-L1 and PD-L2 Engagement of PD-1 Restore Anti-Tumor Immunity. J. Immunother. Cancer 2021, 9, A315. [Google Scholar] [CrossRef]
- Nowicki, T.S.; Hu-Lieskovan, S.; Ribas, A. Mechanisms of Resistance to PD-1 and PD-L1 Blockade. Cancer J. Sudbury Mass. 2018, 24, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Gianchecchi, E.; Fierabracci, A. Inhibitory Receptors and Pathways of Lymphocytes: The Role of PD-1 in Treg Development and Their Involvement in Autoimmunity Onset and Cancer Progression. Front. Immunol. 2018, 9, 2374. [Google Scholar] [CrossRef] [Green Version]
- Cai, J.; Wang, D.; Zhang, G.; Guo, X. The Role Of PD-1/PD-L1 Axis In Treg Development And Function: Implications For Cancer Immunotherapy. OncoTargets Ther. 2019, 12, 8437–8445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; Wang, Y.; Liu, J.; Mok, S.C.; Xue, F.; Zhang, W. CXCL12/CXCR4: A Symbiotic Bridge Linking Cancer Cells and Their Stromal Neighbors in Oncogenic Communication Networks. Oncogene 2016, 35, 816–826. [Google Scholar] [CrossRef]
- Liu, C.; Liu, R.; Wang, B.; Lian, J.; Yao, Y.; Sun, H.; Zhang, C.; Fang, L.; Guan, X.; Shi, J.; et al. Blocking IL-17A Enhances Tumor Response to Anti-PD-1 Immunotherapy in Microsatellite Stable Colorectal Cancer. J. Immunother. Cancer 2021, 9, e001895. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ Attenuates Tumour Response to PD-L1 Blockade by Contributing to Exclusion of T Cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Zboralski, D.; Hoehlig, K.; Eulberg, D.; Frömming, A.; Vater, A. Increasing Tumor-Infiltrating T Cells through Inhibition of CXCL12 with NOX-A12 Synergizes with PD-1 Blockade. Cancer Immunol. Res. 2017, 5, 950–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feig, C.; Jones, J.O.; Kraman, M.; Wells, R.J.B.; Deonarine, A.; Chan, D.S.; Connell, C.M.; Roberts, E.W.; Zhao, Q.; Caballero, O.L.; et al. Targeting CXCL12 from FAP-Expressing Carcinoma-Associated Fibroblasts Synergizes with Anti-PD-L1 Immunotherapy in Pancreatic Cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 20212–20217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Riese, D.J.; Shen, J. The Role of the CXCL12/CXCR4/CXCR7 Chemokine Axis in Cancer. Front. Pharmacol. 2020, 11, 574667. [Google Scholar] [CrossRef]
- Chen, I.X.; Chauhan, V.P.; Posada, J.; Ng, M.R.; Wu, M.W.; Adstamongkonkul, P.; Huang, P.; Lindeman, N.; Langer, R.; Jain, R.K. Blocking CXCR4 Alleviates Desmoplasia, Increases T-Lymphocyte Infiltration, and Improves Immunotherapy in Metastatic Breast Cancer. Proc. Natl. Acad. Sci. USA 2019, 116, 4558–4566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bockorny, B.; Semenisty, V.; Macarulla, T.; Borazanci, E.; Wolpin, B.M.; Stemmer, S.M.; Golan, T.; Geva, R.; Borad, M.J.; Pedersen, K.S.; et al. BL-8040, a CXCR4 Antagonist, in Combination with Pembrolizumab and Chemotherapy for Pancreatic Cancer: The COMBAT Trial. Nat. Med. 2020, 26, 878–885. [Google Scholar] [CrossRef]
- Suarez-Carmona, M.; Williams, A.; Schreiber, J.; Hohmann, N.; Pruefer, U.; Krauss, J.; Jäger, D.; Frömming, A.; Beyer, D.; Eulberg, D.; et al. Combined Inhibition of CXCL12 and PD-1 in MSS Colorectal and Pancreatic Cancer: Modulation of the Microenvironment and Clinical Effects. J. Immunother. Cancer 2021, 9, e002505. [Google Scholar] [CrossRef] [PubMed]
- Glasgow, E.; Mishra, L. Transforming Growth Factor-Beta Signaling and Ubiquitinators in Cancer. Endocr. Relat. Cancer 2008, 15, 59–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strainic, M.G.; Shevach, E.M.; An, F.; Lin, F.; Medof, M.E. Absence of Signaling into CD4+ Cells via C3aR and C5aR Enables Autoinductive TGF-Β1 Signaling and Induction of Foxp3+ Regulatory T Cells. Nat. Immunol. 2013, 14, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lind, H.; Gameiro, S.R.; Jochems, C.; Donahue, R.N.; Strauss, J.; Gulley, J.L.; Palena, C.; Schlom, J. Dual Targeting of TGF-β and PD-L1 via a Bifunctional Anti-PD-L1/TGF-ΒRII Agent: Status of Preclinical and Clinical Advances. J. Immunother. Cancer 2020, 8, e000433. [Google Scholar] [CrossRef] [Green Version]
- Knudson, K.M.; Hicks, K.C.; Luo, X.; Chen, J.-Q.; Schlom, J.; Gameiro, S.R. M7824, a Novel Bifunctional Anti-PD-L1/TGFβ Trap Fusion Protein, Promotes Anti-Tumor Efficacy as Monotherapy and in Combination with Vaccine. Oncoimmunology 2018, 7, e1426519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lan, Y.; Zhang, D.; Xu, C.; Hance, K.W.; Marelli, B.; Qi, J.; Yu, H.; Qin, G.; Sircar, A.; Hernández, V.M.; et al. Enhanced Preclinical Antitumor Activity of M7824, a Bifunctional Fusion Protein Simultaneously Targeting PD-L1 and TGF-β. Sci. Transl. Med. 2018, 10, eaan5488. [Google Scholar] [CrossRef] [Green Version]
- Strauss, J.; Heery, C.R.; Schlom, J.; Madan, R.A.; Cao, L.; Kang, Z.; Lamping, E.; Marté, J.L.; Donahue, R.N.; Grenga, I.; et al. Phase I Trial of M7824 (MSB0011359C), a Bifunctional Fusion Protein Targeting PD-L1 and TGFβ, in Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 1287–1295. [Google Scholar] [CrossRef] [Green Version]
- Yoo, C.; Oh, D.-Y.; Choi, H.J.; Kudo, M.; Ueno, M.; Kondo, S.; Chen, L.-T.; Osada, M.; Helwig, C.; Dussault, I.; et al. Phase I Study of Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-β and PD-L1, in Patients with Pretreated Biliary Tract Cancer. J. Immunother. Cancer 2020, 8, e000564. [Google Scholar] [CrossRef] [PubMed]
- Iwakura, Y.; Ishigame, H.; Saijo, S.; Nakae, S. Functional Specialization of Interleukin-17 Family Members. Immunity 2011, 34, 149–162. [Google Scholar] [CrossRef] [Green Version]
- Martin-Orozco, N.; Muranski, P.; Chung, Y.; Yang, X.O.; Yamazaki, T.; Lu, S.; Hwu, P.; Restifo, N.P.; Overwijk, W.W.; Dong, C. T Helper 17 Cells Promote Cytotoxic T Cell Activation in Tumor Immunity. Immunity 2009, 31, 787–798. [Google Scholar] [CrossRef] [PubMed]
- Kryczek, I.; Banerjee, M.; Cheng, P.; Vatan, L.; Szeliga, W.; Wei, S.; Huang, E.; Finlayson, E.; Simeone, D.; Welling, T.H.; et al. Phenotype, Distribution, Generation, and Functional and Clinical Relevance of Th17 Cells in the Human Tumor Environments. Blood 2009, 114, 1141–1149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benchetrit, F.; Ciree, A.; Vives, V.; Warnier, G.; Gey, A.; Sautès-Fridman, C.; Fossiez, F.; Haicheur, N.; Fridman, W.H.; Tartour, E. Interleukin-17 Inhibits Tumor Cell Growth by Means of a T-Cell-Dependent Mechanism. Blood 2002, 99, 2114–2121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, P.; Wu, D.; Ni, C.; Ye, J.; Chen, W.; Hu, G.; Wang, Z.; Wang, C.; Zhang, Z.; Xia, W.; et al. ΓδT17 Cells Promote the Accumulation and Expansion of Myeloid-Derived Suppressor Cells in Human Colorectal Cancer. Immunity 2014, 40, 785–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Ye, X.; Pitmon, E.; Lu, M.; Wan, J.; Jellison, E.R.; Adler, A.J.; Vella, A.T.; Wang, K. IL-17 Inhibits CXCL9/10-Mediated Recruitment of CD8+ Cytotoxic T Cells and Regulatory T Cells to Colorectal Tumors. J. Immunother. Cancer 2019, 7, 324. [Google Scholar] [CrossRef] [PubMed]
- Llosa, N.J.; Luber, B.; Tam, A.J.; Smith, K.N.; Siegel, N.; Awan, A.H.; Fan, H.; Oke, T.; Zhang, J.; Domingue, J.; et al. Intratumoral Adaptive Immunosuppression and Type 17 Immunity in Mismatch Repair Proficient Colorectal Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5250–5259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brevi, A.; Cogrossi, L.L.; Grazia, G.; Masciovecchio, D.; Impellizzieri, D.; Lacanfora, L.; Grioni, M.; Bellone, M. Much More Than IL-17A: Cytokines of the IL-17 Family Between Microbiota and Cancer. Front. Immunol. 2020, 11, 565470. [Google Scholar] [CrossRef] [PubMed]
- Chauvin, J.-M.; Pagliano, O.; Fourcade, J.; Sun, Z.; Wang, H.; Sander, C.; Kirkwood, J.M.; Chen, T.T.; Maurer, M.; Korman, A.J.; et al. TIGIT and PD-1 Impair Tumor Antigen-Specific CD8+ T Cells in Melanoma Patients. J. Clin. Investig. 2015, 125, 2046–2058. [Google Scholar] [CrossRef]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 Expression on HIV-Specific T Cells Is Associated with T-Cell Exhaustion and Disease Progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Johnston, R.J.; Comps-Agrar, L.; Hackney, J.; Yu, X.; Huseni, M.; Yang, Y.; Park, S.; Javinal, V.; Chiu, H.; Irving, B.; et al. The Immunoreceptor TIGIT Regulates Antitumor and Antiviral CD8(+) T Cell Effector Function. Cancer Cell 2014, 26, 923–937. [Google Scholar] [CrossRef] [Green Version]
- Wherry, E.J.; Ha, S.-J.; Kaech, S.M.; Haining, W.N.; Sarkar, S.; Kalia, V.; Subramaniam, S.; Blattman, J.N.; Barber, D.L.; Ahmed, R. Molecular Signature of CD8+ T Cell Exhaustion during Chronic Viral Infection. Immunity 2007, 27, 670–684. [Google Scholar] [CrossRef]
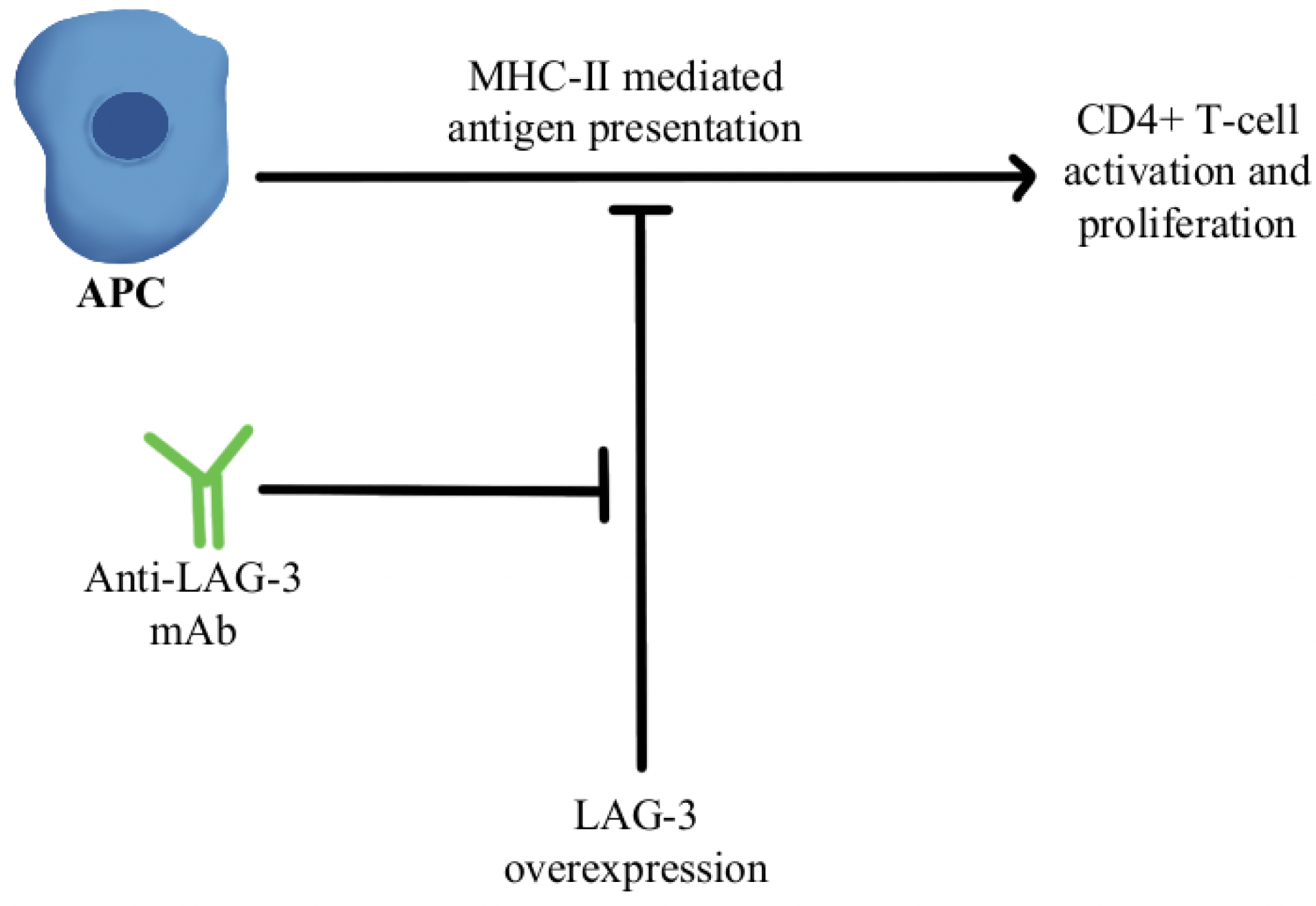
- Woo, S.-R.; Turnis, M.E.; Goldberg, M.V.; Bankoti, J.; Selby, M.; Nirschl, C.J.; Bettini, M.L.; Gravano, D.M.; Vogel, P.; Liu, C.L.; et al. Immune Inhibitory Molecules LAG-3 and PD-1 Synergistically Regulate T-Cell Function to Promote Tumoral Immune Escape. Cancer Res. 2012, 72, 917–927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chemnitz, J.M.; Parry, R.V.; Nichols, K.E.; June, C.H.; Riley, J.L. SHP-1 and SHP-2 Associate with Immunoreceptor Tyrosine-Based Switch Motif of Programmed Death 1 upon Primary Human T Cell Stimulation, but Only Receptor Ligation Prevents T Cell Activation. J. Immunol. 2004, 173, 945–954. [Google Scholar] [CrossRef] [Green Version]
- Hosseinkhani, N.; Shadbad, M.A.; Asghari Jafarabadi, M.; Karim Ahangar, N.; Asadzadeh, Z.; Mohammadi, S.M.; Lotfinejad, P.; Alizadeh, N.; Brunetti, O.; Fasano, R.; et al. A Systematic Review and Meta-Analysis on the Significance of TIGIT in Solid Cancers: Dual TIGIT/PD-1 Blockade to Overcome Immune-Resistance in Solid Cancers. Int. J. Mol. Sci. 2021, 22, 10389. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.S.K.; Sansom, D.M. The Emerging Role of CTLA4 as a Cell-Extrinsic Regulator of T Cell Responses. Nat. Rev. Immunol. 2011, 11, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dai, Z.; Wu, W.; Wang, Z.; Zhang, N.; Zhang, L.; Zeng, W.-J.; Liu, Z.; Cheng, Q. Regulatory Mechanisms of Immune Checkpoints PD-L1 and CTLA-4 in Cancer. J. Exp. Clin. Cancer Res. 2021, 40, 184. [Google Scholar] [CrossRef]
- Goldberg, M.V.; Drake, C.G. LAG-3 in Cancer Immunotherapy. Curr. Top. Microbiol. Immunol. 2011, 344, 269–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ascierto, P.A.; Melero, I.; Bhatia, S.; Bono, P.; Sanborn, R.E.; Lipson, E.J.; Callahan, M.K.; Gajewski, T.; Gomez-Roca, C.A.; Hodi, F.S.; et al. Initial Efficacy of Anti-Lymphocyte Activation Gene-3 (Anti–LAG-3; BMS-986016) in Combination with Nivolumab (Nivo) in Pts with Melanoma (MEL) Previously Treated with Anti–PD-1/PD-L1 Therapy. J. Clin. Oncol. 2017, 35, 9520. [Google Scholar] [CrossRef]
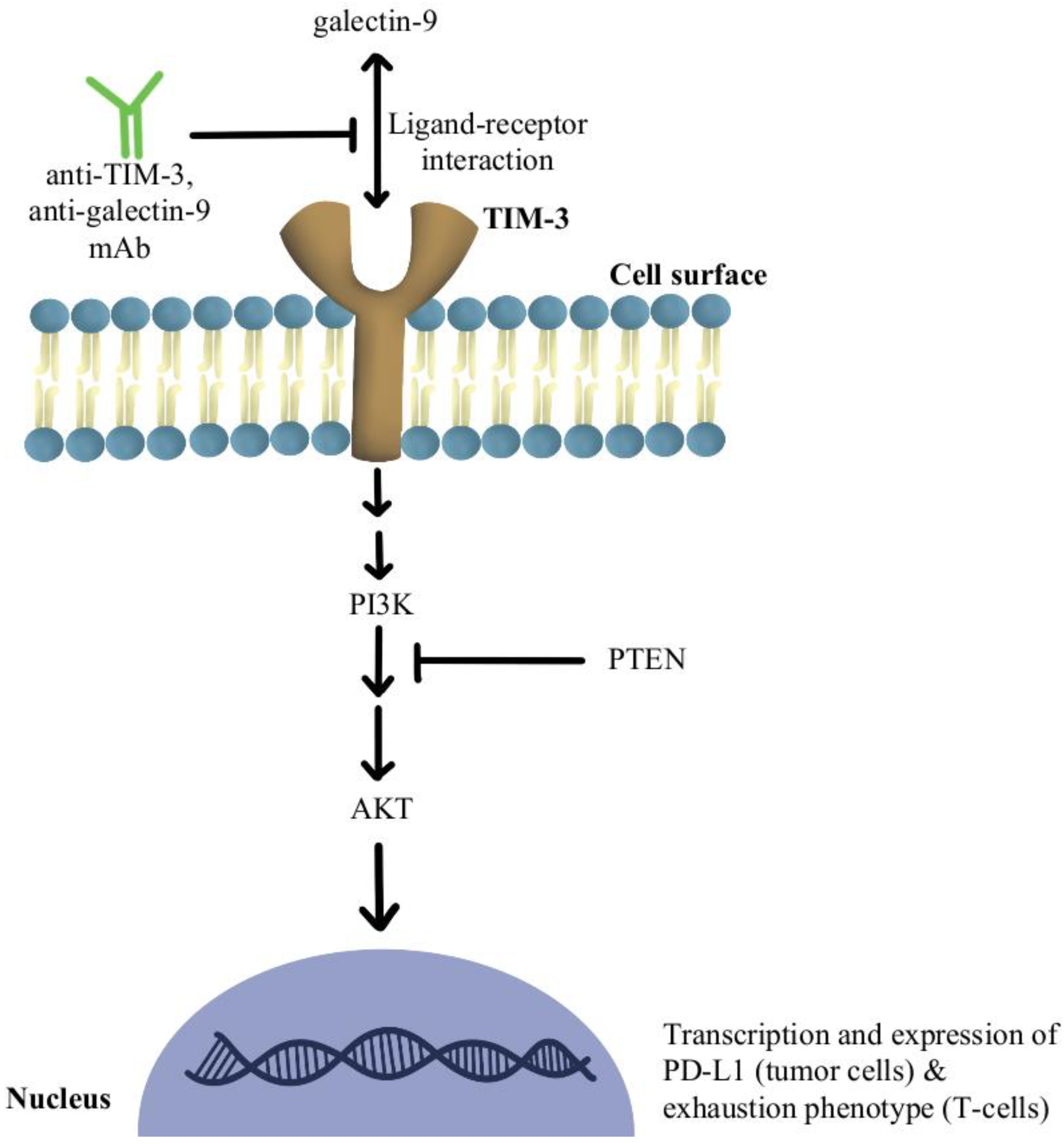
- Fourcade, J.; Sun, Z.; Benallaoua, M.; Guillaume, P.; Luescher, I.F.; Sander, C.; Kirkwood, J.M.; Kuchroo, V.; Zarour, H.M. Upregulation of Tim-3 and PD-1 Expression Is Associated with Tumor Antigen-Specific CD8+ T Cell Dysfunction in Melanoma Patients. J. Exp. Med. 2010, 207, 2175–2186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, L.; Xu, Y.; Fang, J.; Liu, W.; Chen, J.; Liu, Z.; Xu, Q. Targeting STAT3 Abrogates Tim-3 Upregulation of Adaptive Resistance to PD-1 Blockade on Regulatory T Cells of Melanoma. Front. Immunol. 2021, 12, 654749. [Google Scholar] [CrossRef]
- de Mingo Pulido, Á.; Gardner, A.; Hiebler, S.; Soliman, H.; Rugo, H.S.; Krummel, M.F.; Coussens, L.M.; Ruffell, B. TIM-3 Regulates CD103+ Dendritic Cell Function and Response to Chemotherapy in Breast Cancer. Cancer Cell 2018, 33, 60–74. [Google Scholar] [CrossRef]
- Shayan, G.; Srivastava, R.; Li, J.; Schmitt, N.; Kane, L.P.; Ferris, R.L. Adaptive Resistance to Anti-PD1 Therapy by Tim-3 Upregulation Is Mediated by the PI3K-Akt Pathway in Head and Neck Cancer. Oncoimmunology 2017, 6, e1261779. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016, 6, 202–216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curigliano, G.; Gelderblom, H.; Mach, N.; Doi, T.; Tai, D.; Forde, P.M.; Sarantopoulos, J.; Bedard, P.L.; Lin, C.-C.; Hodi, F.S.; et al. Phase I/Ib Clinical Trial of Sabatolimab, an Anti-TIM-3 Antibody, Alone and in Combination with Spartalizumab, an Anti-PD-1 Antibody, in Advanced Solid Tumors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2021, 27, 3620–3629. [Google Scholar] [CrossRef] [PubMed]
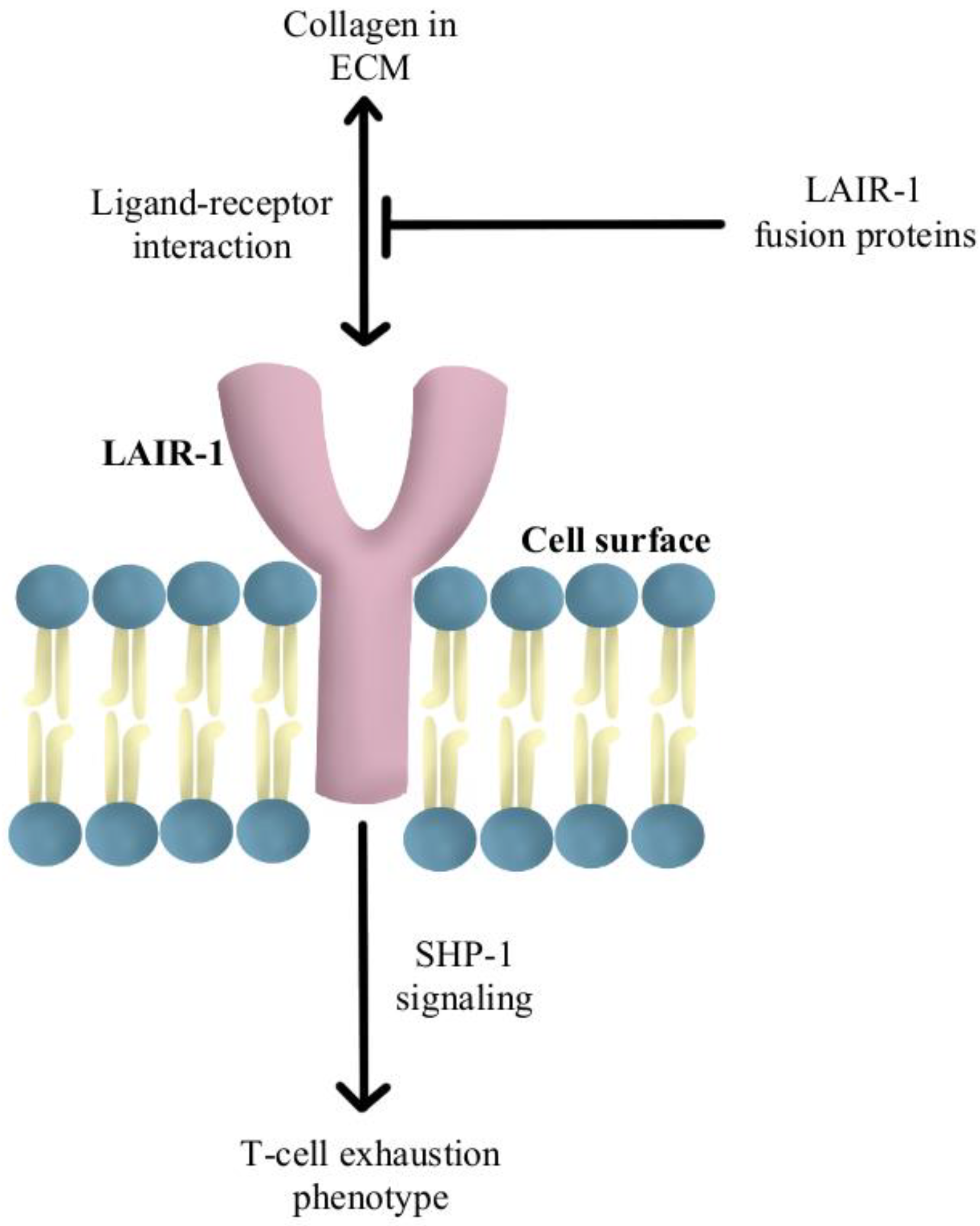
- Peng, D.H.; Rodriguez, B.L.; Diao, L.; Chen, L.; Wang, J.; Byers, L.A.; Wei, Y.; Chapman, H.A.; Yamauchi, M.; Behrens, C.; et al. Collagen Promotes Anti-PD-1/PD-L1 Resistance in Cancer through LAIR1-Dependent CD8+ T Cell Exhaustion. Nat. Commun. 2020, 11, 4520. [Google Scholar] [CrossRef]
- Lebbink, R.J.; van den Berg, M.C.W.; de Ruiter, T.; Raynal, N.; van Roon, J.A.G.; Lenting, P.J.; Jin, B.; Meyaard, L. The Soluble Leukocyte-Associated Ig-like Receptor (LAIR)-2 Antagonizes the Collagen/LAIR-1 Inhibitory Immune Interaction. J. Immunol. 2008, 180, 1662–1669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyaard, L.; Adema, G.J.; Chang, C.; Woollatt, E.; Sutherland, G.R.; Lanier, L.L.; Phillips, J.H. LAIR-1, a Novel Inhibitory Receptor Expressed on Human Mononuclear Leukocytes. Immunity 1997, 7, 283–290. [Google Scholar] [CrossRef]
- Gettinger, S.; Choi, J.; Hastings, K.; Truini, A.; Datar, I.; Sowell, R.; Wurtz, A.; Dong, W.; Cai, G.; Melnick, M.A.; et al. Impaired HLA Class I Antigen Processing and Presentation as a Mechanism of Acquired Resistance to Immune Checkpoint Inhibitors in Lung Cancer. Cancer Discov. 2017, 7, 1420–1435. [Google Scholar] [CrossRef] [Green Version]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.-P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to Checkpoint Blockade Therapy through Inactivation of Antigen Presentation. Nat. Commun. 2017, 8, 1136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zaretsky, J.M.; Garcia-Diaz, A.; Shin, D.S.; Escuin-Ordinas, H.; Hugo, W.; Hu-Lieskovan, S.; Torrejon, D.Y.; Abril-Rodriguez, G.; Sandoval, S.; Barthly, L.; et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N. Engl. J. Med. 2016, 375, 819–829. [Google Scholar] [CrossRef]
- Benci, J.L.; Johnson, L.R.; Choa, R.; Xu, Y.; Qiu, J.; Zhou, Z.; Xu, B.; Ye, D.; Nathanson, K.L.; June, C.H.; et al. Opposing Functions of Interferon Coordinate Adaptive and Innate Immune Responses to Cancer Immune Checkpoint Blockade. Cell 2019, 178, 933–948.e14. [Google Scholar] [CrossRef]
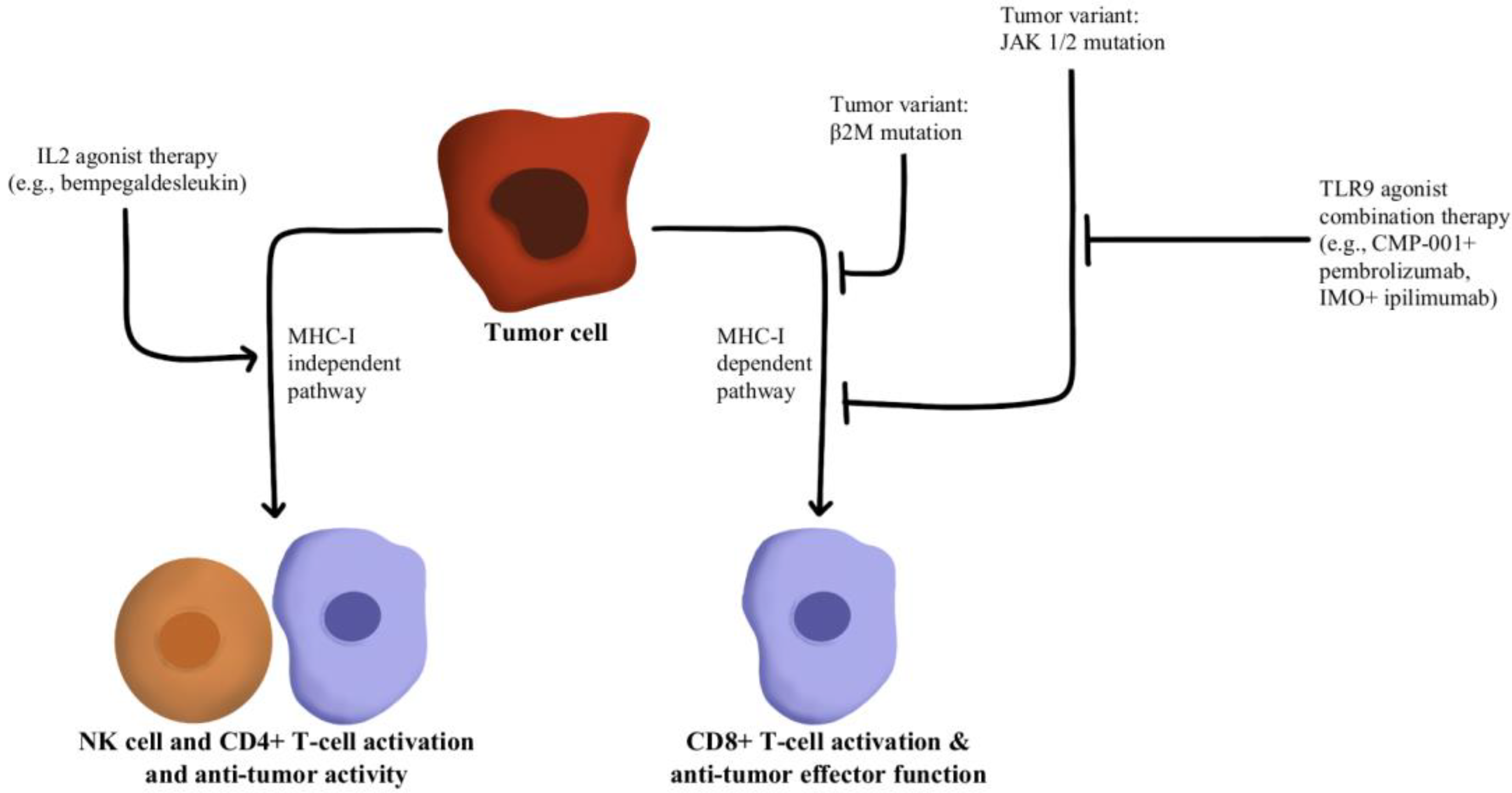
- Hurwitz, M.E.; Cho, D.C.; Balar, A.V.; Curti, B.D.; Siefker-Radtke, A.O.; Sznol, M.; Kluger, H.M.; Bernatchez, C.; Fanton, C.; Iacucci, E.; et al. Baseline Tumor-Immune Signatures Associated with Response to Bempegaldesleukin (NKTR-214) and Nivolumab. J. Clin. Oncol. 2019, 37, 2623. [Google Scholar] [CrossRef]
- Sharma, M.; Khong, H.; Fa’ak, F.; Bentebibel, S.-E.; Janssen, L.M.E.; Chesson, B.C.; Creasy, C.A.; Forget, M.-A.; Kahn, L.M.S.; Pazdrak, B.; et al. Bempegaldesleukin Selectively Depletes Intratumoral Tregs and Potentiates T Cell-Mediated Cancer Therapy. Nat. Commun. 2020, 11, 661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torrejon, D.Y.; Abril-Rodriguez, G.; Champhekar, A.S.; Tsoi, J.; Campbell, K.M.; Kalbasi, A.; Parisi, G.; Zaretsky, J.M.; Garcia-Diaz, A.; Puig-Saus, C.; et al. Overcoming Genetically Based Resistance Mechanisms to PD-1 Blockade. Cancer Discov. 2020, 10, 1140–1157. [Google Scholar] [CrossRef] [PubMed]
- Diab, A.; Rahimian, S.; Haymaker, C.L.; Bernatchez, C.; Andtbacka, R.H.I.; James, M.; Johnson, D.B.; Markowitz, J.; Murthy, R.; Puzanov, I.; et al. A Phase 2 Study to Evaluate the Safety and Efficacy of Intratumoral (IT) Injection of the TLR9 Agonist IMO-2125 (IMO) in Combination with Ipilimumab (Ipi) in PD-1 Inhibitor Refractory Melanoma. J. Clin. Oncol. 2018, 36, 9515. [Google Scholar] [CrossRef]
- Ren, D.; Hua, Y.; Yu, B.; Ye, X.; He, Z.; Li, C.; Wang, J.; Mo, Y.; Wei, X.; Chen, Y.; et al. Predictive Biomarkers and Mechanisms Underlying Resistance to PD1/PD-L1 Blockade Cancer Immunotherapy. Mol. Cancer 2020, 19, 19. [Google Scholar] [CrossRef] [Green Version]
- Benci, J.L.; Xu, B.; Qiu, Y.; Wu, T.J.; Dada, H.; Twyman-Saint Victor, C.; Cucolo, L.; Lee, D.S.M.; Pauken, K.E.; Huang, A.C.; et al. Tumor Interferon Signaling Regulates a Multigenic Resistance Program to Immune Checkpoint Blockade. Cell 2016, 167, 1540–1554.e12. [Google Scholar] [CrossRef] [Green Version]
- Konen, J.M.; Rodriguez, B.L.; Fradette, J.J.; Gibson, L.; Davis, D.; Minelli, R.; Peoples, M.D.; Kovacs, J.; Carugo, A.; Bristow, C.; et al. Ntrk1 Promotes Resistance to PD-1 Checkpoint Blockade in Mesenchymal Kras/P53 Mutant Lung Cancer. Cancers 2019, 11, 462. [Google Scholar] [CrossRef] [Green Version]
- Mian, A.; Hill, B.T. Brexucabtagene Autoleucel for the Treatment of Relapsed/Refractory Mantle Cell Lymphoma. Expert Opin. Biol. Ther. 2021, 21, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Morgan, R.A.; Dudley, M.E.; Wunderlich, J.R.; Hughes, M.S.; Yang, J.C.; Sherry, R.M.; Royal, R.E.; Topalian, S.L.; Kammula, U.S.; Restifo, N.P.; et al. Cancer Regression in Patients after Transfer of Genetically Engineered Lymphocytes. Science 2006, 314, 126–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Bishop, M.R.; Tam, C.S.; Waller, E.K.; Borchmann, P.; McGuirk, J.P.; Jäger, U.; Jaglowski, S.; Andreadis, C.; Westin, J.R.; et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N. Engl. J. Med. 2019, 380, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Deng, Q.; Jiang, Y.-Y.; Zhang, R.; Zhu, H.-B.; Meng, J.-X.; Li, Y.-M. CAR-T 19 Combined with Reduced-Dose PD-1 Blockade Therapy for Treatment of Refractory Follicular Lymphoma: A Case Report. Oncol. Lett. 2019, 18, 4415–4420. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Lu, W.; Sun, R.; Jin, X.; Cheng, L.; He, X.; Wang, L.; Yuan, T.; Lyu, C.; Zhao, M. Anti-CD19 Chimeric Antigen Receptor T Cells in Combination With Nivolumab Are Safe and Effective Against Relapsed/Refractory B-Cell Non-Hodgkin Lymphoma. Front. Oncol. 2019, 9, 767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- John, L.B.; Kershaw, M.H.; Darcy, P.K. Blockade of PD-1 Immunosuppression Boosts CAR T-Cell Therapy. Oncoimmunology 2013, 2, e26286. [Google Scholar] [CrossRef]
- Gargett, T.; Yu, W.; Dotti, G.; Yvon, E.S.; Christo, S.N.; Hayball, J.D.; Lewis, I.D.; Brenner, M.K.; Brown, M.P. GD2-Specific CAR T Cells Undergo Potent Activation and Deletion Following Antigen Encounter but Can Be Protected From Activation-Induced Cell Death by PD-1 Blockade. Mol. Ther. J. Am. Soc. Gene Ther. 2016, 24, 1135–1149. [Google Scholar] [CrossRef] [Green Version]
- Marotte, L.; Simon, S.; Vignard, V.; Dupre, E.; Gantier, M.; Cruard, J.; Alberge, J.-B.; Hussong, M.; Deleine, C.; Heslan, J.-M.; et al. Increased Antitumor Efficacy of PD-1-Deficient Melanoma-Specific Human Lymphocytes. J. Immunother. Cancer 2020, 8, e000311. [Google Scholar] [CrossRef] [Green Version]
- Marotte, L.; Capitao, M.; Deleine, C.; Beauvais, T.; Cadiou, G.; Perrin, J.; Chérel, M.; Scotet, E.; Guilloux, Y.; Bruchertseifer, F.; et al. Anti-Tumor Efficacy of a Combination Therapy with PD-L1 Targeted Alpha Therapy and Adoptive Cell Transfer of PD-1 Deficient Melanoma-Specific Human T-Lymphocytes. Oncoimmunology 2021, 10, 1940676. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Jiang, H.; Shi, B.; Zhou, M.; Zhang, H.; Shi, Z.; Du, G.; Luo, H.; Wu, X.; Wang, Y.; et al. Disruption of PD-1 Enhanced the Anti-Tumor Activity of Chimeric Antigen Receptor T Cells Against Hepatocellular Carcinoma. Front. Pharmacol. 2018, 9, 1118. [Google Scholar] [CrossRef] [PubMed]
- Rupp, L.J.; Schumann, K.; Roybal, K.T.; Gate, R.E.; Ye, C.J.; Lim, W.A.; Marson, A. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Anti-Tumor Efficacy of Human Chimeric Antigen Receptor T Cells. Sci. Rep. 2017, 7, 737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Zi, Z.; Jin, Y.; Li, G.; Shao, K.; Cai, Q.; Ma, X.; Wei, F. CRISPR/Cas9-Mediated PD-1 Disruption Enhances Human Mesothelin-Targeted CAR T Cell Effector Functions. Cancer Immunol. Immunother. CII 2019, 68, 365–377. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Siriwon, N.; Zhang, X.; Yang, S.; Jin, T.; He, F.; Kim, Y.J.; Mac, J.; Lu, Z.; Wang, S.; et al. Enhanced Cancer Immunotherapy by Chimeric Antigen Receptor-Modified T Cells Engineered to Secrete Checkpoint Inhibitors. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 6982–6992. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, M.; Sakoda, Y.; Adachi, K.; Nagano, H.; Tamada, K. Improved Survival of Chimeric Antigen Receptor-Engineered T (CAR-T) and Tumor-Specific T Cells Caused by Anti-Programmed Cell Death Protein 1 Single-Chain Variable Fragment-Producing CAR-T Cells. Cancer Sci. 2019, 110, 3079–3088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suarez, E.R.; Chang, D.K.; Sun, J.; Sui, J.; Freeman, G.J.; Signoretti, S.; Zhu, Q.; Marasco, W.A. Chimeric Antigen Receptor T Cells Secreting Anti-PD-L1 Antibodies More Effectively Regress Renal Cell Carcinoma in a Humanized Mouse Model. Oncotarget 2016, 7, 34341–34355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, J.S.; Long, G.V.; Scolyer, R.A.; Teng, M.W.L.; Smyth, M.J. Resistance to PD1/PDL1 Checkpoint Inhibition. Cancer Treat. Rev. 2017, 52, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Pennock, G.K.; Chow, L.Q.M. The Evolving Role of Immune Checkpoint Inhibitors in Cancer Treatment. The Oncologist 2015, 20, 812–822. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Mo, Y.; Wang, Y.; Wu, P.; Zhang, Y.; Xiong, F.; Guo, C.; Wu, X.; Li, Y.; Li, X.; et al. Neoantigen Vaccine: An Emerging Tumor Immunotherapy. Mol. Cancer 2019, 18, 128. [Google Scholar] [CrossRef] [Green Version]
- Bassani-Sternberg, M.; Digklia, A.; Huber, F.; Wagner, D.; Sempoux, C.; Stevenson, B.J.; Thierry, A.-C.; Michaux, J.; Pak, H.; Racle, J.; et al. A Phase Ib Study of the Combination of Personalized Autologous Dendritic Cell Vaccine, Aspirin, and Standard of Care Adjuvant Chemotherapy Followed by Nivolumab for Resected Pancreatic Adenocarcinoma-A Proof of Antigen Discovery Feasibility in Three Patients. Front. Immunol. 2019, 10, 1832. [Google Scholar] [CrossRef] [Green Version]
- Nesselhut, J.; Marx, D.; Lange, H.; Regalo, G.; Cillien, N.; Chang, R.Y.; Nesselhut, T. Systemic Treatment with Anti-PD-1 Antibody Nivolumab in Combination with Vaccine Therapy in Advanced Pancreatic Cancer. J. Clin. Oncol. 2016, 34, 3092. [Google Scholar] [CrossRef]
- Zhang, B.; Wu, Q.; Li, B.; Wang, D.; Wang, L.; Zhou, Y.L. M6A Regulator-Mediated Methylation Modification Patterns and Tumor Microenvironment Infiltration Characterization in Gastric Cancer. Mol. Cancer 2020, 19, 53. [Google Scholar] [CrossRef] [Green Version]
- Nemunaitis, J. Vaccines in Cancer: GVAX, a GM-CSF Gene Vaccine. Expert Rev. Vaccines 2005, 4, 259–274. [Google Scholar] [CrossRef]
- Becher, B.; Tugues, S.; Greter, M. GM-CSF: From Growth Factor to Central Mediator of Tissue Inflammation. Immunity 2016, 45, 963–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duraiswamy, J.; Kaluza, K.M.; Freeman, G.J.; Coukos, G. Dual Blockade of PD-1 and CTLA-4 Combined with Tumor Vaccine Effectively Restores T-Cell Rejection Function in Tumors. Cancer Res. 2013, 73, 3591–3603. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-Y.; Hutzen, B.; Wedekind, M.F.; Cripe, T.P. Oncolytic Virus and PD-1/PD-L1 Blockade Combination Therapy. Oncolytic Virotherapy 2018, 7, 65–77. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Zheng, Z.; Barman, A.K.; Wang, Z.; Wang, L.; Zeng, W.; Wang, L.; Qin, Y.; Pandey, A.; Zhang, C.; et al. Optimal Combination Treatment Regimens of Vaccine and Radiotherapy Augment Tumor-Bearing Host Immunity. Commun. Biol. 2021, 4, 78. [Google Scholar] [CrossRef]
- Carabotti, M.; Scirocco, A.; Maselli, M.A.; Severi, C. The Gut-Brain Axis: Interactions between Enteric Microbiota, Central and Enteric Nervous Systems. Ann. Gastroenterol. 2015, 28, 203–209. [Google Scholar] [PubMed]
- Holmes, E.; Li, J.V.; Athanasiou, T.; Ashrafian, H.; Nicholson, J.K. Understanding the Role of Gut Microbiome-Host Metabolic Signal Disruption in Health and Disease. Trends Microbiol. 2011, 19, 349–359. [Google Scholar] [CrossRef]
- Martin, A.M.; Sun, E.W.; Rogers, G.B.; Keating, D.J. The Influence of the Gut Microbiome on Host Metabolism Through the Regulation of Gut Hormone Release. Front. Physiol. 2019, 10, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The Gut Microbiome in Health and in Disease. Curr. Opin. Gastroenterol. 2015, 31, 69–75. [Google Scholar] [CrossRef]
- Sun, L.; Zhang, X.; Zhang, Y.; Zheng, K.; Xiang, Q.; Chen, N.; Chen, Z.; Zhang, N.; Zhu, J.; He, Q. Antibiotic-Induced Disruption of Gut Microbiota Alters Local Metabolomes and Immune Responses. Front. Cell. Infect. Microbiol. 2019, 9, 99. [Google Scholar] [CrossRef] [Green Version]
- Weersma, R.K.; Zhernakova, A.; Fu, J. Interaction between Drugs and the Gut Microbiome. Gut 2020, 69, 1510–1519. [Google Scholar] [CrossRef] [PubMed]
- Vétizou, M.; Pitt, J.M.; Daillère, R.; Lepage, P.; Waldschmitt, N.; Flament, C.; Rusakiewicz, S.; Routy, B.; Roberti, M.P.; Duong, C.P.M.; et al. Anticancer Immunotherapy by CTLA-4 Blockade Relies on the Gut Microbiota. Science 2015, 350, 1079–1084. [Google Scholar] [CrossRef] [Green Version]
- Sivan, A.; Corrales, L.; Hubert, N.; Williams, J.B.; Aquino-Michaels, K.; Earley, Z.M.; Benyamin, F.W.; Lei, Y.M.; Jabri, B.; Alegre, M.-L.; et al. Commensal Bifidobacterium Promotes Antitumor Immunity and Facilitates Anti-PD-L1 Efficacy. Science 2015, 350, 1084–1089. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishnan, V.; Spencer, C.N.; Nezi, L.; Reuben, A.; Andrews, M.C.; Karpinets, T.V.; Prieto, P.A.; Vicente, D.; Hoffman, K.; Wei, S.C.; et al. Gut Microbiome Modulates Response to Anti-PD-1 Immunotherapy in Melanoma Patients. Science 2018, 359, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Dong, H.; Xia, L.; Yang, Y.; Zhu, Y.; Shen, Y.; Zheng, H.; Yao, C.; Wang, Y.; Lu, S. The Diversity of Gut Microbiome Is Associated With Favorable Responses to Anti-Programmed Death 1 Immunotherapy in Chinese Patients With NSCLC. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2019, 14, 1378–1389. [Google Scholar] [CrossRef]
- Mao, J.; Wang, D.; Long, J.; Yang, X.; Lin, J.; Song, Y.; Xie, F.; Xun, Z.; Wang, Y.; Wang, Y.; et al. Gut Microbiome Is Associated with the Clinical Response to Anti-PD-1 Based Immunotherapy in Hepatobiliary Cancers. J. Immunother. Cancer 2021, 9, e003334. [Google Scholar] [CrossRef] [PubMed]
- Peng, Z.; Cheng, S.; Kou, Y.; Wang, Z.; Jin, R.; Hu, H.; Zhang, X.; Gong, J.-F.; Li, J.; Lu, M.; et al. The Gut Microbiome Is Associated with Clinical Response to Anti-PD-1/PD-L1 Immunotherapy in Gastrointestinal Cancer. Cancer Immunol. Res. 2020, 8, 1251–1261. [Google Scholar] [CrossRef]
- Routy, B.; Le Chatelier, E.; Derosa, L.; Duong, C.P.M.; Alou, M.T.; Daillère, R.; Fluckiger, A.; Messaoudene, M.; Rauber, C.; Roberti, M.P.; et al. Gut Microbiome Influences Efficacy of PD-1-Based Immunotherapy against Epithelial Tumors. Science 2018, 359, 91–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Wang, T.; Tu, X.; Huang, Y.; Zhang, H.; Tan, D.; Jiang, W.; Cai, S.; Zhao, P.; Song, R.; et al. Gut Microbiome Affects the Response to Anti-PD-1 Immunotherapy in Patients with Hepatocellular Carcinoma. J. Immunother. Cancer 2019, 7, 193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, M.C.; Duong, C.P.M.; Gopalakrishnan, V.; Iebba, V.; Chen, W.-S.; Derosa, L.; Khan, M.A.W.; Cogdill, A.P.; White, M.G.; Wong, M.C.; et al. Gut Microbiota Signatures Are Associated with Toxicity to Combined CTLA-4 and PD-1 Blockade. Nat. Med. 2021, 27, 1432–1441. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Hodi, F.S.; Robert, C.; Weber, J.S.; Margolin, K.; Hamid, O.; Patt, D.; Chen, T.-T.; Berman, D.M.; Wolchok, J.D. Pooled Analysis of Long-Term Survival Data From Phase II and Phase III Trials of Ipilimumab in Unresectable or Metastatic Melanoma. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 1889–1894. [Google Scholar] [CrossRef]
- Ribas, A.; Hamid, O.; Daud, A.; Hodi, F.S.; Wolchok, J.D.; Kefford, R.; Joshua, A.M.; Patnaik, A.; Hwu, W.-J.; Weber, J.S.; et al. Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA 2016, 315, 1600–1609. [Google Scholar] [CrossRef]
- Bai, R.; Chen, N.; Li, L.; Du, N.; Bai, L.; Lv, Z.; Tian, H.; Cui, J. Mechanisms of Cancer Resistance to Immunotherapy. Front. Oncol. 2020, 10, 1290. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Kasic, T.; Gri, G.; Gallana, K.; Borsellino, G.; Marigo, I.; Battistini, L.; Iafrate, M.; Prayer-Galetti, T.; Pagano, F.; et al. Boosting Antitumor Responses of T Lymphocytes Infiltrating Human Prostate Cancers. J. Exp. Med. 2005, 201, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell 2017, 168, 707–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pitt, J.M.; Vétizou, M.; Daillère, R.; Roberti, M.P.; Yamazaki, T.; Routy, B.; Lepage, P.; Boneca, I.G.; Chamaillard, M.; Kroemer, G.; et al. Resistance Mechanisms to Immune-Checkpoint Blockade in Cancer: Tumor-Intrinsic and -Extrinsic Factors. Immunity 2016, 44, 1255–1269. [Google Scholar] [CrossRef] [Green Version]
- Joyce, J.A.; Fearon, D.T. T Cell Exclusion, Immune Privilege, and the Tumor Microenvironment. Science 2015, 348, 74–80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in Cancer Immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational Heterogeneity in Cancer and the Search for New Cancer-Associated Genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zitvogel, L.; Pitt, J.M.; Daillère, R.; Smyth, M.J.; Kroemer, G. Mouse Models in Oncoimmunology. Nat. Rev. Cancer 2016, 16, 759–773. [Google Scholar] [CrossRef]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-Intrinsic β-Catenin Signalling Prevents Anti-Tumour Immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Hugo, W.; Zaretsky, J.M.; Sun, L.; Song, C.; Moreno, B.H.; Hu-Lieskovan, S.; Berent-Maoz, B.; Pang, J.; Chmielowski, B.; Cherry, G.; et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016, 165, 35–44. [Google Scholar] [CrossRef] [Green Version]
- Goodman, A.M.; Kato, S.; Bazhenova, L.; Patel, S.P.; Frampton, G.M.; Miller, V.; Stephens, P.J.; Daniels, G.A.; Kurzrock, R. Tumor Mutational Burden as an Independent Predictor of Response to Immunotherapy in Diverse Cancers. Mol. Cancer Ther. 2017, 16, 2598–2608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Havel, J.J.; Chowell, D.; Chan, T.A. The Evolving Landscape of Biomarkers for Checkpoint Inhibitor Immunotherapy. Nat. Rev. Cancer 2019, 19, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paley, M.A.; Kroy, D.C.; Odorizzi, P.M.; Johnnidis, J.B.; Dolfi, D.V.; Barnett, B.E.; Bikoff, E.K.; Robertson, E.J.; Lauer, G.M.; Reiner, S.L.; et al. Progenitor and Terminal Subsets of CD8+ T Cells Cooperate to Contain Chronic Viral Infection. Science 2012, 338, 1220–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, D.R.; Kaminski, J.; Barnitz, R.A.; Kurachi, M.; Gerdemann, U.; Yates, K.B.; Tsao, H.-W.; Godec, J.; LaFleur, M.W.; Brown, F.D.; et al. The Epigenetic Landscape of T Cell Exhaustion. Science 2016, 354, 1165–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Im, S.J.; Hashimoto, M.; Gerner, M.Y.; Lee, J.; Kissick, H.T.; Burger, M.C.; Shan, Q.; Hale, J.S.; Lee, J.; Nasti, T.H.; et al. Defining CD8+ T Cells That Provide the Proliferative Burst after PD-1 Therapy. Nature 2016, 537, 417–421. [Google Scholar] [CrossRef] [Green Version]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of Exhausted CD8+ T Cells Differentially Mediate Tumor Control and Respond to Checkpoint Blockade. Nat. Immunol. 2019, 20, 326–336. [Google Scholar] [CrossRef]
- Siddiqui, I.; Schaeuble, K.; Chennupati, V.; Fuertes Marraco, S.A.; Calderon-Copete, S.; Pais Ferreira, D.; Carmona, S.J.; Scarpellino, L.; Gfeller, D.; Pradervand, S.; et al. Intratumoral Tcf1+PD-1+CD8+ T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity 2019, 50, 195–211. [Google Scholar] [CrossRef] [Green Version]
- Sakuishi, K.; Apetoh, L.; Sullivan, J.M.; Blazar, B.R.; Kuchroo, V.K.; Anderson, A.C. Targeting Tim-3 and PD-1 Pathways to Reverse T Cell Exhaustion and Restore Anti-Tumor Immunity. J. Exp. Med. 2010, 207, 2187–2194. [Google Scholar] [CrossRef] [PubMed]
- Arlauckas, S.P.; Garris, C.S.; Kohler, R.H.; Kitaoka, M.; Cuccarese, M.F.; Yang, K.S.; Miller, M.A.; Carlson, J.C.; Freeman, G.J.; Anthony, R.M.; et al. In Vivo Imaging Reveals a Tumor-Associated Macrophage-Mediated Resistance Pathway in Anti-PD-1 Therapy. Sci. Transl. Med. 2017, 9, eaal3604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikucki, M.E.; Fisher, D.T.; Matsuzaki, J.; Skitzki, J.J.; Gaulin, N.B.; Muhitch, J.B.; Ku, A.W.; Frelinger, J.G.; Odunsi, K.; Gajewski, T.F.; et al. Non-Redundant Requirement for CXCR3 Signalling during Tumoricidal T-Cell Trafficking across Tumour Vascular Checkpoints. Nat. Commun. 2015, 6, 7458. [Google Scholar] [CrossRef] [Green Version]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; McSkane, M.; Baba, H.; Lenz, H.-J. CXCL9, CXCL10, CXCL11/CXCR3 Axis for Immune Activation—A Target for Novel Cancer Therapy. Cancer Treat. Rev. 2018, 63, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-T.; Sun, Z.-J. Turning Cold Tumors into Hot Tumors by Improving T-Cell Infiltration. Theranostics 2021, 11, 5365–5386. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moise, J.; Murthy, J.; Dabir, D.; Yu, S.; Kisto, F.; Herron, E.; Aulakh, S. Mechanisms of Resistance and Strategies to Combat Resistance in PD-(L)1 Blockade. Immuno 2022, 2, 671-691. https://doi.org/10.3390/immuno2040041
Moise J, Murthy J, Dabir D, Yu S, Kisto F, Herron E, Aulakh S. Mechanisms of Resistance and Strategies to Combat Resistance in PD-(L)1 Blockade. Immuno. 2022; 2(4):671-691. https://doi.org/10.3390/immuno2040041
Chicago/Turabian StyleMoise, John, Jeevan Murthy, Dolma Dabir, Stephen Yu, Farah Kisto, Emily Herron, and Sonikpreet Aulakh. 2022. "Mechanisms of Resistance and Strategies to Combat Resistance in PD-(L)1 Blockade" Immuno 2, no. 4: 671-691. https://doi.org/10.3390/immuno2040041