
Chitin Derived Small Molecule AVR-48 Reprograms the Resting Macrophages to an Intermediate Phenotype and Decrease Pseudomonas aeruginosa Mouse Lung Infection
 ,
, 
Abstract
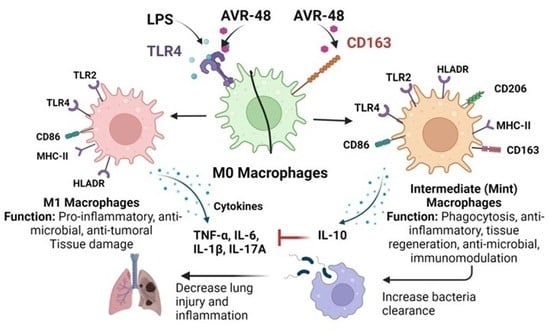
:
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Cells and Cell Lines
2.3. Bacteria
2.4. Chemicals and Reagents
2.5. Cell Viability Assay
2.5.1. Cytokine and CD163 Assay
2.5.2. IL-10 and IL-17A Detection in Lung Homogenates and Serum
2.6. Flow Cytometry Studies
2.6.1. Binding of AVR-48 to Splenic Monocytes/Macrophages
2.6.2. Binding of Biotinylated Conjugated AVR-48 to Splenic Monocytes/Macrophages
2.6.3. Quantification of Macrophages after AVR-48 Treatment to hPBMC Cells
2.7. Phagocytosis and Bacteria CFU Measurement Using THP-1 Cells
2.8. Combination MIC Assay
2.9. Pseudomonas aeruginosa Mouse Lung Infection
2.10. Quantification of Bacterial Load in Mouse Lung and Blood Samples
2.11. Statistical Analysis
3. Results
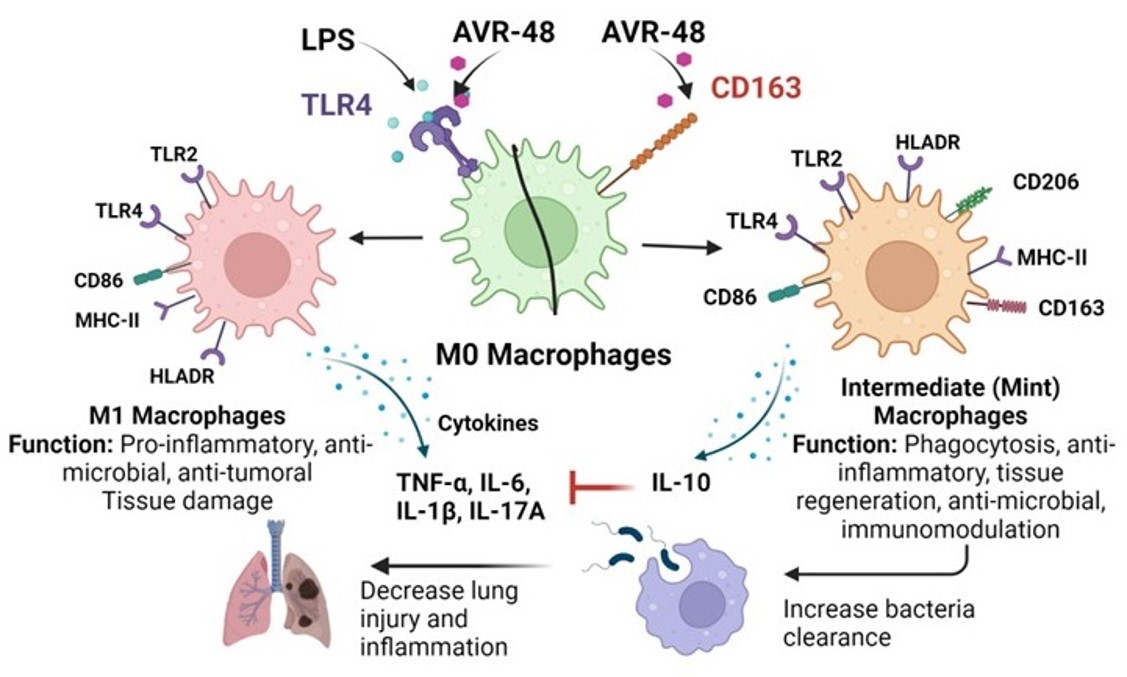
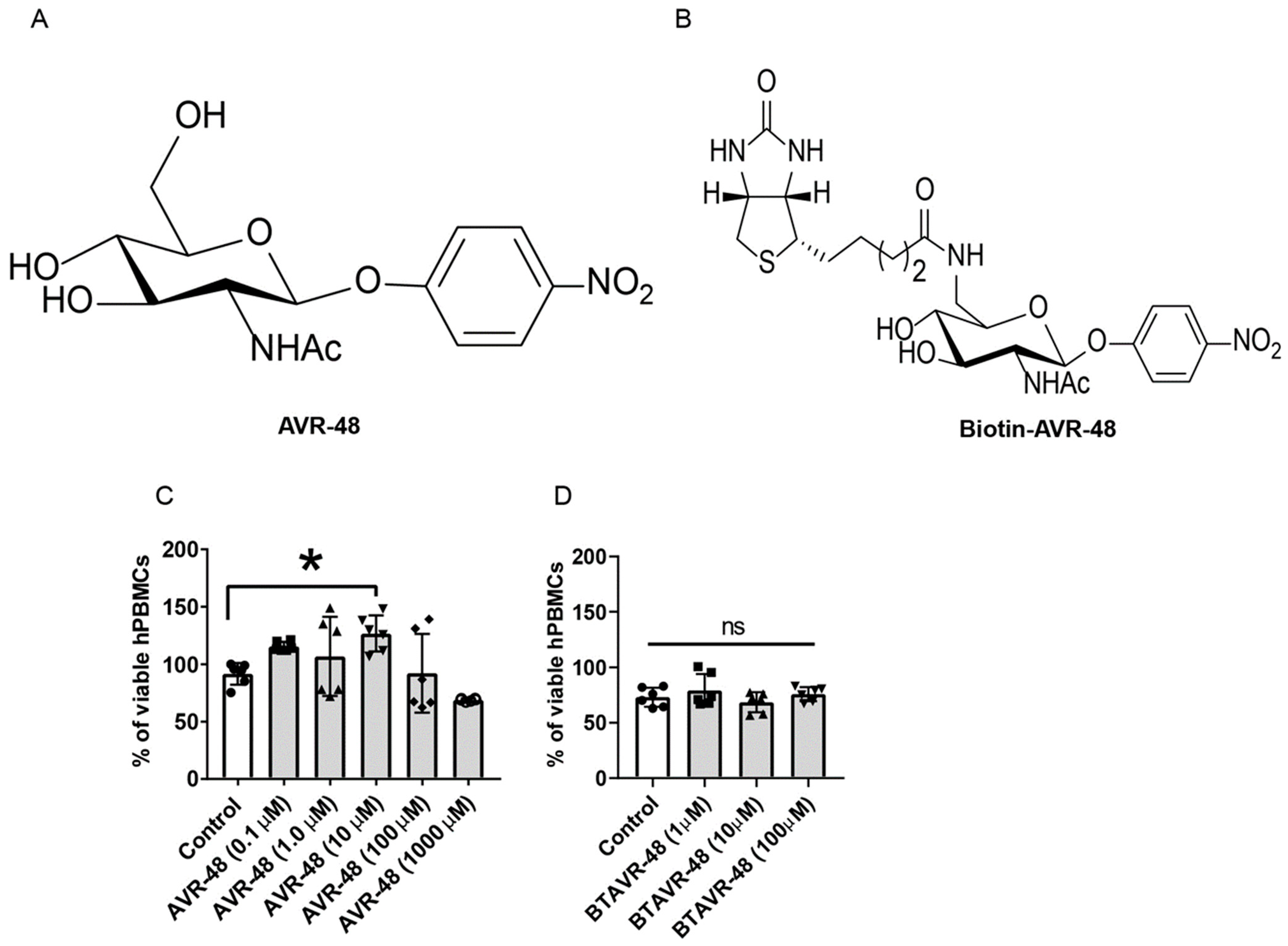
3.1. AVR-48 and Biotin Conjugated AVR-48 Do Not Exhibit Cytotoxicity to hPBMCs In Vitro
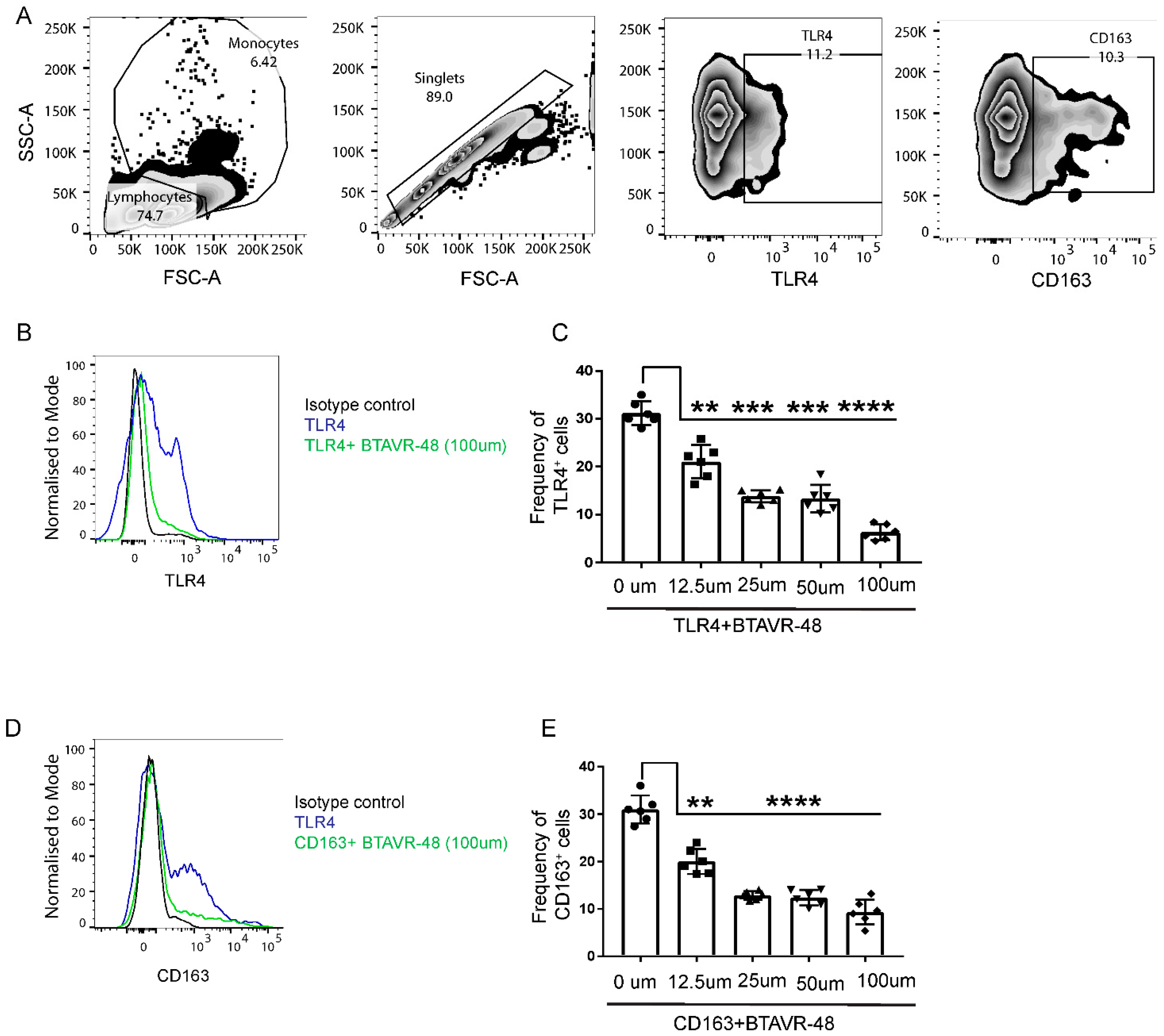
3.2. AVR-48 Binds to Both TLR4 and CD163 Receptors in Primary Monocytes
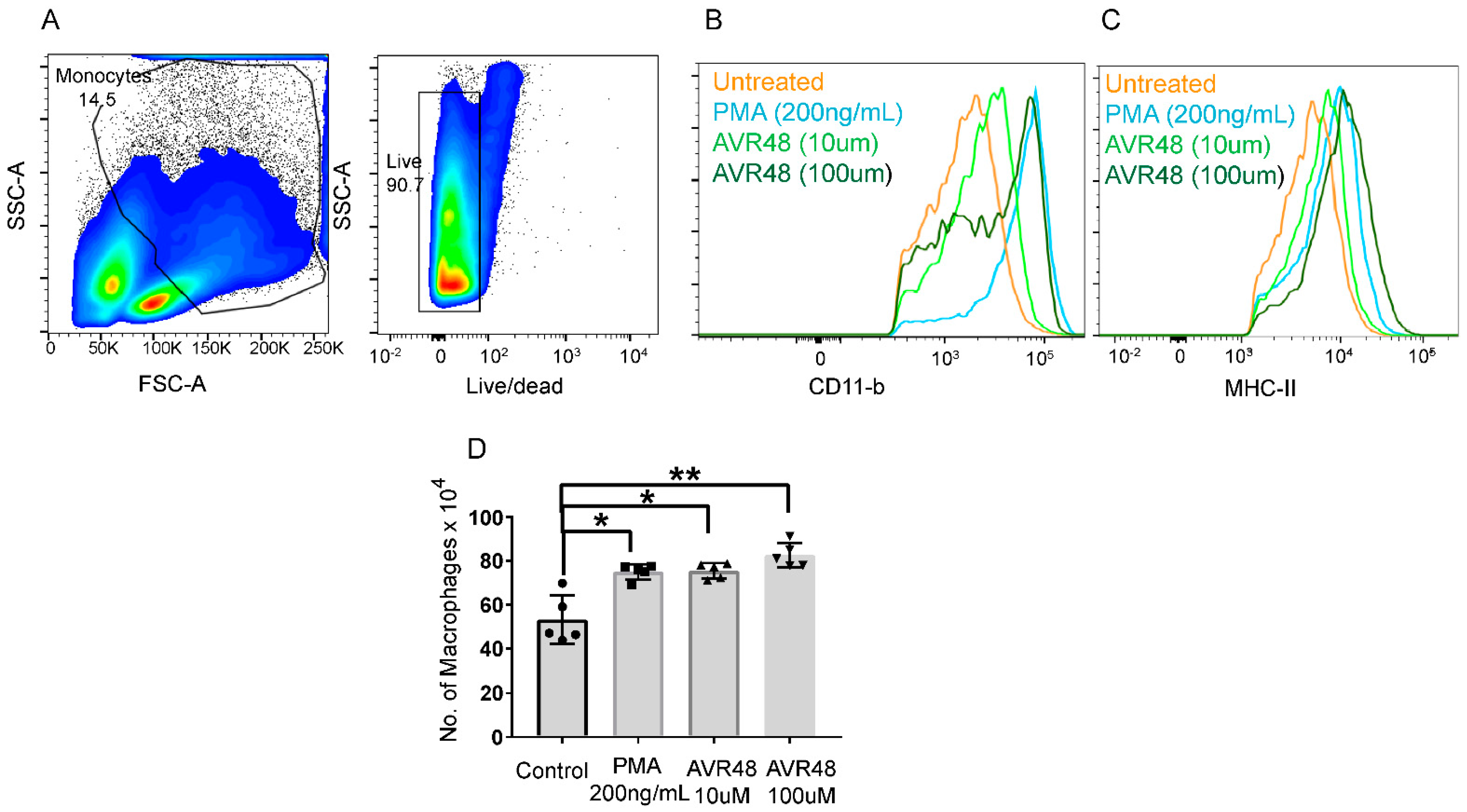
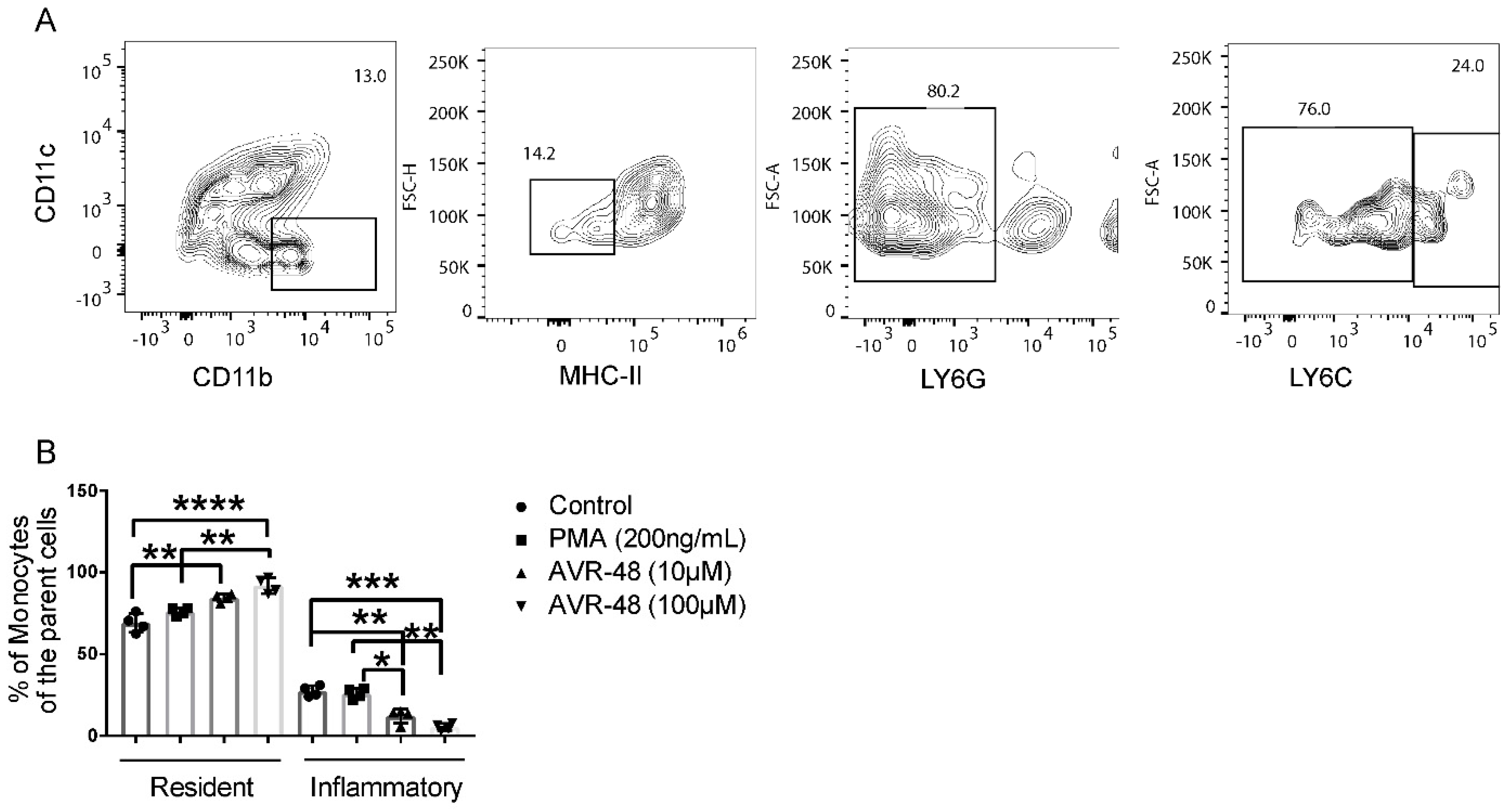
3.3. AVR-48 Treatment Polarizes Mouse Monocytes to Macrophages and Shifts the Monocyte Populations More to a Resident Phenotype
3.4. Treatment of AVR-48 to Human Peripheral Blood Mononuclear Cells Increases the Percentage of Intermediate (Mint) Macrophages
3.5. Effect of LPS and AVR-48 Treatment on Percentage of Macrophages in hPBMCs
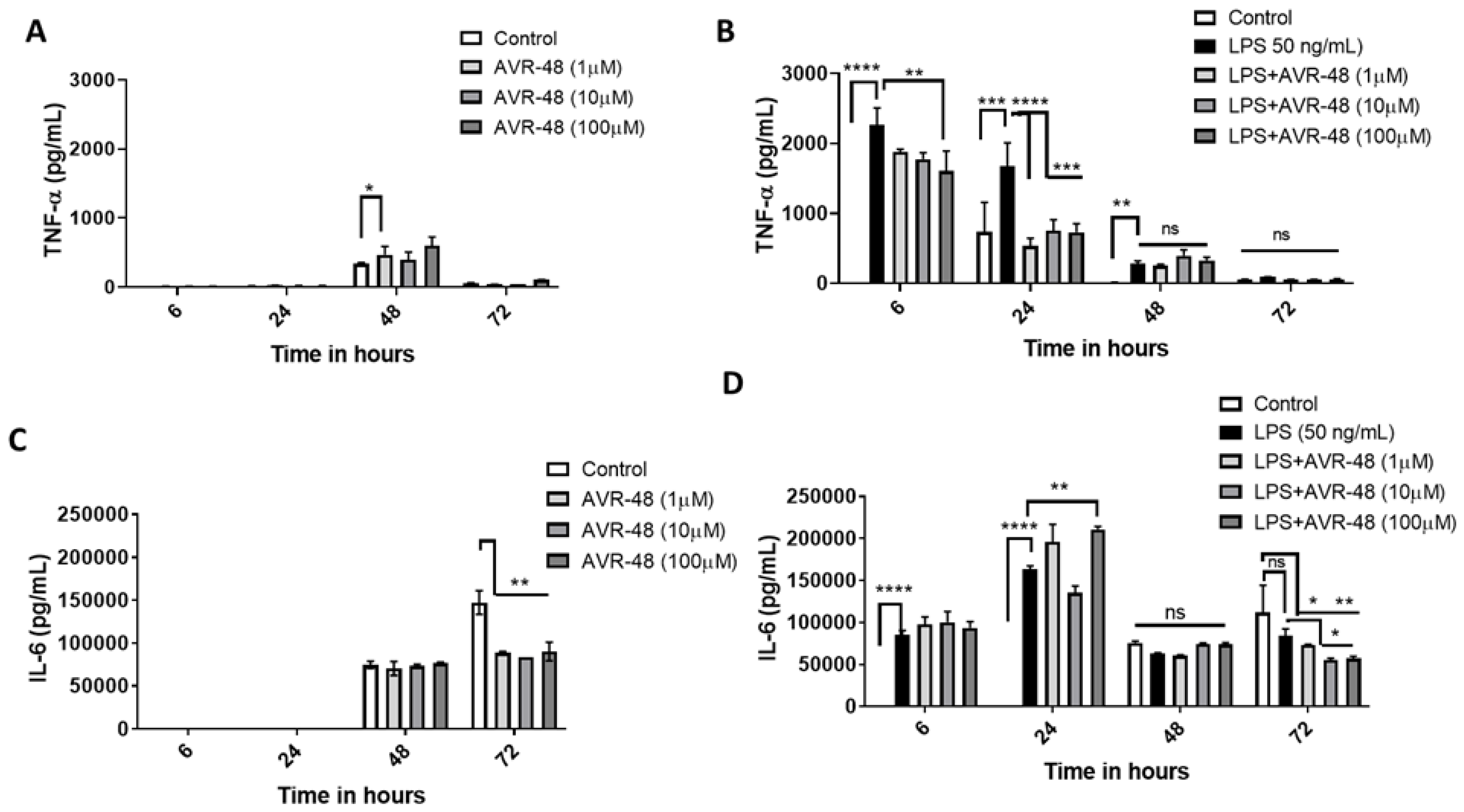
3.6. Effect of AVR-48 and LPS on Concentration of TNF-α and IL-6 in hPBMCs
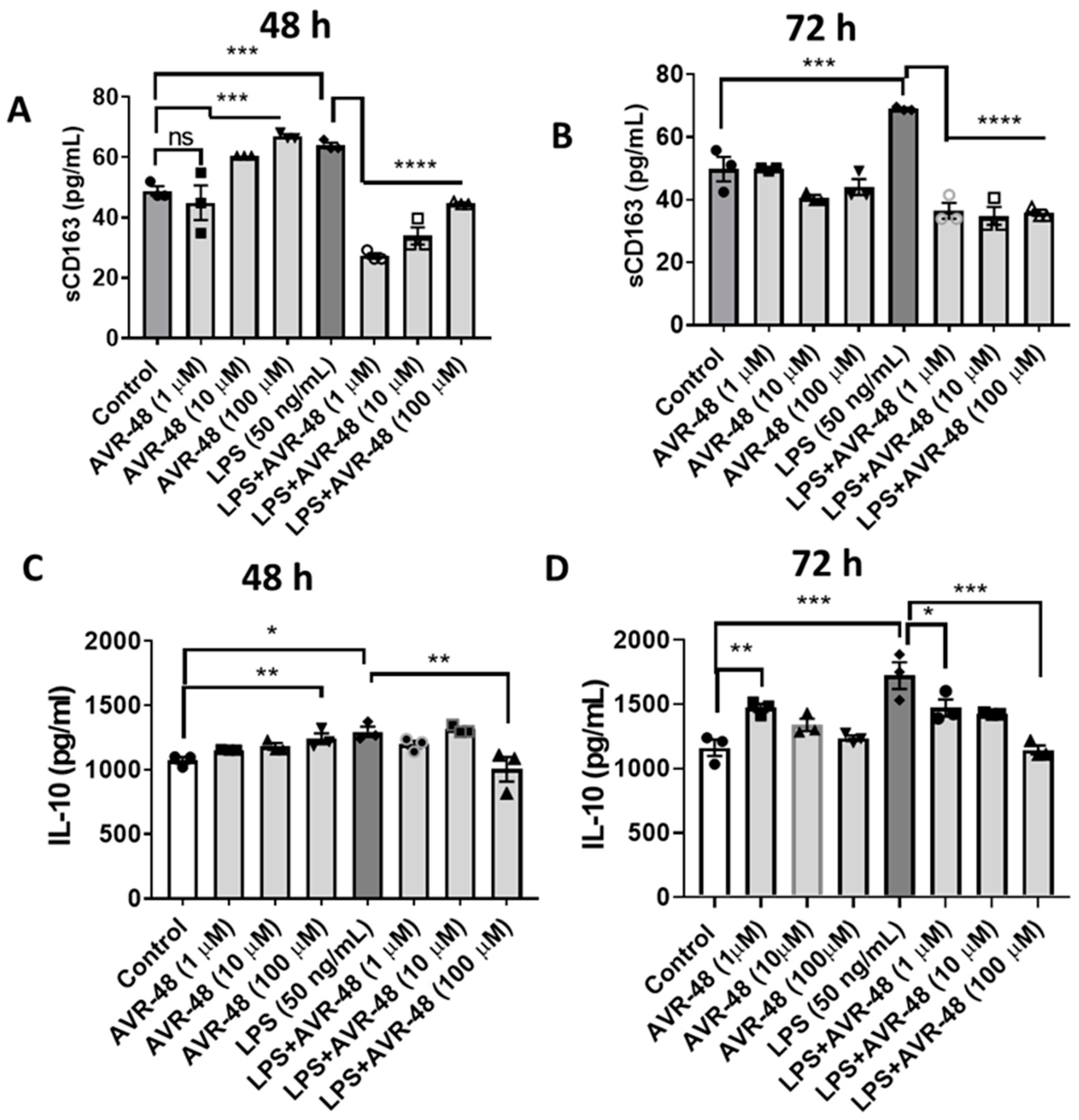
3.7. Effect of AVR-48 and LPS on Concentration of IL-10 and sCD163 in hPBMCs
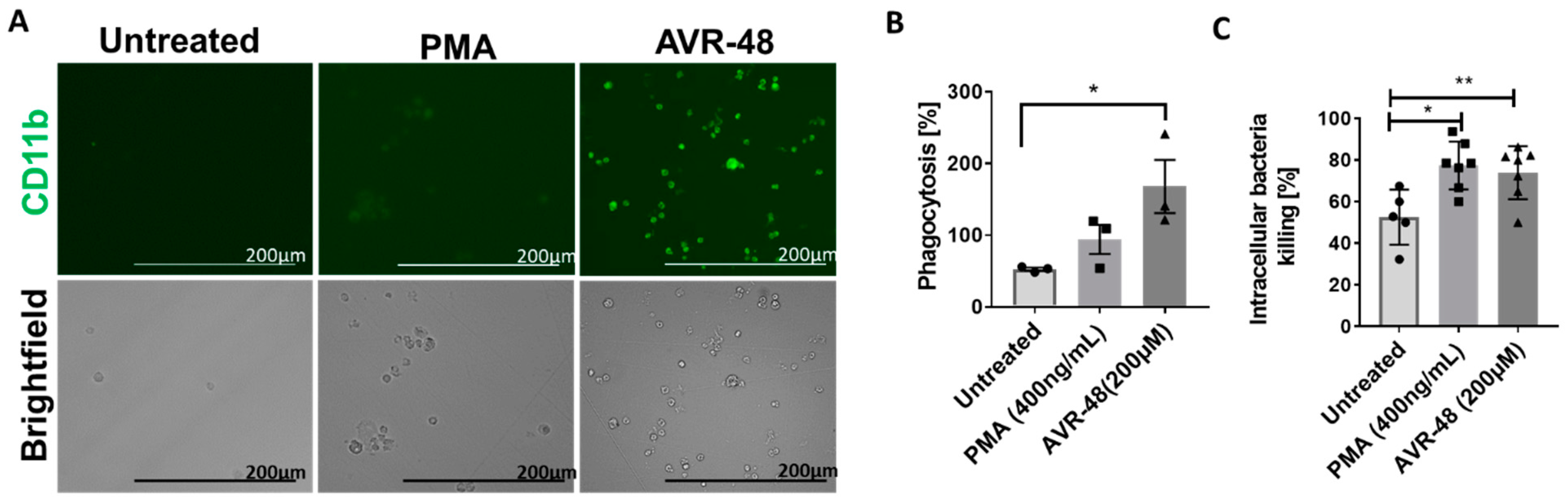
3.8. AVR-48 Induce Differentiation of THP-1 Human Monocytic Cells into Macrophages with Enhanced Phagocytosis of the Bacteria and Promote Intracellular Killing
3.9. AVR-48 Demonstrates Synergy with Standard of Care Antibiotics
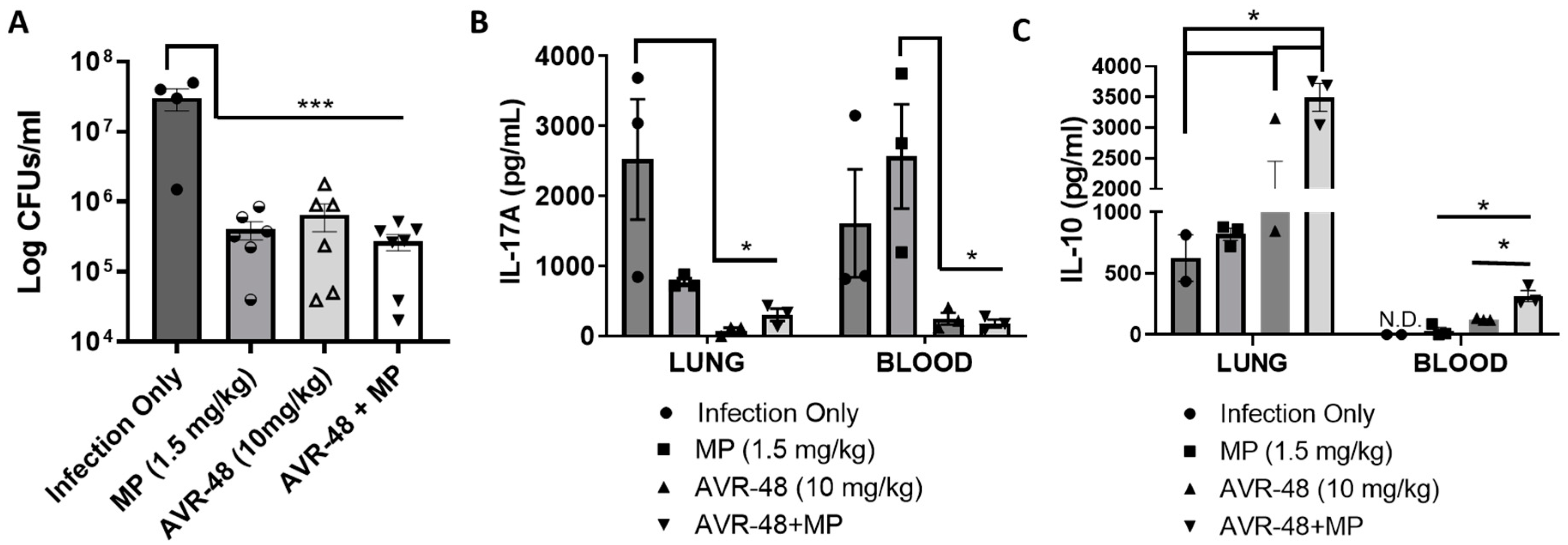
3.10. AVR-48 Decreases Bacteria Load in a Mouse Lung Infection Model and Increases Anti-Inflammatory Cytokine IL-10
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Xia, W.; Liu, P.; Zhang, J.; Chen, J. Biological activities of chitosan and chitooligosaccharides. Food Hydrocoll. 2011, 25, 170–179. [Google Scholar] [CrossRef]
- Panda, S.K.; Kumar, S.; Tupperwar, N.C.; Vaidya, T.; George, A.; Rath, S.; Bal, V.; Ravindran, B. Chitohexaose activates macrophages by alternate pathway through TLR4 and blocks endotoxemia. PLoS Pathog. 2012, 8, e1002717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, D.; Das, P.; Acharya, S.; Agarwal, B.; Christensen, D.J.; Robertson, S.M.; Bhandari, V. Small Immunomodulatory Molecules as Potential Therapeutics in Experimental Murine Models of Acute Lung Injury (ALI)/Acute Respiratory Distress Syndrome (ARDS). Int. J. Mol. Sci. 2021, 22, 2573. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Acharya, S.; Shah, D.; Agarwal, B.; Prahaladan, V.; Bhandari, V. Chitin Analog AVR-25 Prevents Experimental Bronchopulmonary Dysplasia. J. Pediatr. Intensive Care 2020, 9, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Panda, S.K.; Agarwal, B.; Behera, S.; Ali, S.M.; Pulse, M.E.; Solomkin, J.S.; Opal, S.M.; Bhandari, V.; Acharya, S. Novel Chitohexaose Analog Protects Young and Aged mice from CLP Induced Polymicrobial Sepsis. Sci. Rep. 2019, 9, 2904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acharya, S.; Das, P.; Agarwal, B. Novel Immunodulating Small Molecules. U.S. Patent US20200022995A1, 21 June 2022. [Google Scholar]
- Das, P.; Acharya, S.; Prahaladan, V.M.; Kumova, O.K.; Malaeb, S.; Behera, S.; Agarwal, B.; Christensen, D.J.; Carey, A.J.; Bhandari, V. Chitin-Derived AVR-48 Prevents Experimental Bronchopulmonary Dysplasia (BPD) and BPD-Associated Pulmonary Hypertension in Newborn Mice. Int. J. Mol. Sci. 2021, 22, 8547. [Google Scholar] [CrossRef]
- Wu, Y.; Li, D.; Wang, Y.; Liu, X.; Zhang, Y.; Qu, W.; Chen, K.; Francisco, N.M.; Feng, L.; Huang, X.; et al. Beta-Defensin 2 and 3 Promote Bacterial Clearance of Pseudomonas aeruginosa by Inhibiting Macrophage Autophagy through Downregulation of Early Growth Response Gene-1 and c-FOS. Front. Immunol. 2018, 9, 211. [Google Scholar] [CrossRef]
- Lechartier, B.; Hartkoorn, R.C.; Cole, S.T. In vitro combination studies of benzothiazinone lead compound BTZ043 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 5790–5793. [Google Scholar] [CrossRef] [Green Version]
- Rand, K.H.; Houck, H.J.; Brown, P.; Bennett, D. Reproducibility of the microdilution checkerboard method for antibiotic synergy. Antimicrob. Agents Chemother. 1993, 37, 613–615. [Google Scholar] [CrossRef] [Green Version]
- Odds, F.C. Synergy, antagonism, and what the chequerboard puts between them. J. Antimicrob. Chemother. 2003, 52, 1. [Google Scholar] [CrossRef]
- Oishi, Y.; Manabe, I. Macrophages in inflammation, repair and regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef] [Green Version]
- Swirski, F.K.; Nahrendorf, M.; Etzrodt, M.; Wildgruber, M.; Cortez-Retamozo, V.; Panizzi, P.; Figueiredo, J.L.; Kohler, R.H.; Chudnovskiy, A.; Waterman, P.; et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009, 325, 612–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheadle, W.G.; Hershman, M.J.; Wellhausen, S.R.; Polk, H.C. HLA-DR antigen expression on peripheral blood monocytes correlates with surgical infection. Am. J. Surg. 1991, 161, 639–645. [Google Scholar] [CrossRef]
- Palojärvi, A.; Petäjä, J.; Siitonen, S.; Janér, C.; Andersson, S. Low monocyte HLA-DR expression as an indicator of immunodepression in very low birth weight infants. Pediatr. Res. 2013, 73, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Schulz, D.; Severin, Y.; Zanotelli, V.R.T.; Bodenmiller, B. In-Depth Characterization of Monocyte-Derived Macrophages using a Mass Cytometry-Based Phagocytosis Assay. Sci. Rep. 2019, 9, 1925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindner, B.; Burkard, T.; Schuler, M. Phagocytosis assays with different pH-sensitive fluorescent particles and various readouts. BioTechniques 2020, 68, 245–250. [Google Scholar] [CrossRef] [Green Version]
- Bosshart, H.; Heinzelmann, M. THP-1 cells as a model for human monocytes. Ann. Transl. Med. 2016, 4, 22. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Wynn, T.A. Protective and pathogenic functions of macrophage subsets. Nat. Rev. Immunol. 2011, 11, 723–737. [Google Scholar] [CrossRef] [Green Version]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5, 514. [Google Scholar] [CrossRef] [Green Version]
- Murray, P.J.; Wynn, T.A. Obstacles and opportunities for understanding macrophage polarization. J. Leukoc. Biol. 2011, 89, 557–563. [Google Scholar] [CrossRef] [Green Version]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 816460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Porcheray, F.; Viaud, S.; Rimaniol, A.C.; Léone, C.; Samah, B.; Dereuddre-Bosquet, N.; Dormont, D.; Gras, G. Macrophage activation switching: An asset for the resolution of inflammation. Clin. Exp. Immunol. 2005, 142, 481–489. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Yao, X.; Gordon, E.M.; Barochia, A.; Cuento, R.A.; Kaler, M.; Meyer, K.S.; Keeran, K.J.; Nugent, G.Z.; Jeffries, K.R.; et al. A CCL24-dependent pathway augments eosinophilic airway inflammation in house dust mite-challenged Cd163−/−mice. Mucosal Immunol. 2016, 9, 702–717. [Google Scholar] [CrossRef] [Green Version]
- Kwon, J.H.; Kim, M.; Bae, Y.K.; Kim, G.H.; Choi, S.J.; Oh, W.; Um, S.; Jin, H.J. Decorin Secreted by Human Umbilical Cord Blood-Derived Mesenchymal Stem Cells Induces Macrophage Polarization via CD44 to Repair Hyperoxic Lung Injury. Int. J. Mol. Sci. 2019, 20, 4815. [Google Scholar] [CrossRef] [Green Version]
- Timmermann, M.; Högger, P. Oxidative stress and 8-iso-prostaglandin F(2alpha) induce ectodomain shedding of CD163 and release of tumor necrosis factor-alpha from human monocytes. Free Radic. Biol. Med. 2005, 39, 98–107. [Google Scholar] [CrossRef]
- Zhi, Y.; Gao, P.; Xin, X.; Li, W.; Ji, L.; Zhang, L.; Zhang, X.; Zhang, J. Clinical significance of sCD163 and its possible role in asthma (Review). Mol. Med. Rep. 2017, 15, 2931–2939. [Google Scholar] [CrossRef] [Green Version]
- Weaver, L.K.; Pioli, P.A.; Wardwell, K.; Vogel, S.N.; Guyre, P.M. Up-regulation of human monocyte CD163 upon activation of cell-surface Toll-like receptors. J. Leukoc. Biol. 2007, 81, 663–671. [Google Scholar] [CrossRef]
- Kneidl, J.; Löffler, B.; Erat, M.C.; Kalinka, J.; Peters, G.; Roth, J.; Barczyk, K. Soluble CD163 promotes recognition, phagocytosis and killing of Staphylococcus aureus via binding of specific fibronectin peptides. Cell. Microbiol. 2012, 14, 914–936. [Google Scholar] [CrossRef]
- Sun, Y.; Ma, J.; Li, D.; Li, P.; Zhou, X.; Li, Y.; He, Z.; Qin, L.; Liang, L.; Luo, X. Interleukin-10 inhibits interleukin-1β production and inflammasome activation of microglia in epileptic seizures. J. Neuroinflamm. 2019, 16, 66. [Google Scholar] [CrossRef] [Green Version]
- Garingo, A.; Tesoriero, L.; Cayabyab, R.; Durand, M.; Blahnik, M.; Sardesai, S.; Ramanathan, R.; Jones, C.; Kwong, K.; Li, C.; et al. Constitutive IL-10 expression by lung inflammatory cells and risk for bronchopulmonary dysplasia. Pediatr. Res. 2007, 61, 197–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McColm, J.R.; Stenson, B.J.; Biermasz, N.; McIntosh, N. Measurement of interleukin 10 in bronchoalveolar lavage from preterm ventilated infants. Arch. Dis. Childhood. Fetal Neonatal Ed. 2000, 82, F156–F159. [Google Scholar] [CrossRef] [PubMed]
- Oei, J.; Lui, K.; Wang, H.; Henry, R. Decreased interleukin-10 in tracheal aspirates from preterm infants developing chronic lung disease. Acta Paediatr. 2002, 91, 1194–1199. [Google Scholar] [CrossRef] [PubMed]
- Vento, G.; Capoluongo, E.; Matassa, P.G.; Concolino, P.; Vendettuoli, V.; Vaccarella, C.; Frezza, S.; Zuppi, C.; Romagnoli, C.; Ameglio, F. Serum levels of seven cytokines in premature ventilated newborns: Correlations with old and new forms of bronchopulmonary dysplasia. Intensive Care Med. 2006, 32, 723–730. [Google Scholar] [CrossRef]
- Thompson, A.; Bhandari, V. Pulmonary Biomarkers of Bronchopulmonary Dysplasia. Biomark. Insights 2008, 3, 361–373. [Google Scholar] [CrossRef] [Green Version]
- McGowan, E.C.; Kostadinov, S.; McLean, K.; Gotsch, F.; Venturini, D.; Romero, R.; Laptook, A.R.; Sharma, S. Placental IL-10 dysregulation and association with bronchopulmonary dysplasia risk. Pediatr. Res. 2009, 66, 455–460. [Google Scholar] [CrossRef] [Green Version]
- Rossato, M.; Curtale, G.; Tamassia, N.; Castellucci, M.; Mori, L.; Gasperini, S.; Mariotti, B.; Luca, M.D.; Mirolo, M.; Cassatella, M.A.; et al. IL-10 induced microRNA-187 negatively regulates TNF, IL-6, and IL-12p40 production in TLR4-stimulated monocytes. Proc. Natl. Acad. Sci. USA 2012, 109, E3101–E3110. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MIC90 (µg/mL) | |||||||
|---|---|---|---|---|---|---|---|
| Bacteria | Mero | Cipro | Colistin | AVR-48 | Meropenem (AVR-48) | Ciprofloxacin (AVR-48) | Colistin (AVR-48) |
| P. aeruginosa (10145) | 4.0 | 2.0–4.0 | 8.0 | >200 | 1.5 ± 0.3 (4.6 ± 3.0) | 2.0 (3.6 ± 2.3) | 2.0 (3.6 ± 2.3) |
| A. baumannii (19606) | 0.5–1.0 | 2.0–4.0 | ND | >200 | ND | 1.0 (4.6 ± 3.0) | ND |
| MRSA* (BAA 1760) | ND | ND | >200 | >200 | ND | ND | 14.5 ± 9.5 (4.6 ± 3.0) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Behera, S.; Panda, S.K.; Donkor, M.; Acharya, E.; Jones, H.; Acharya, S. Chitin Derived Small Molecule AVR-48 Reprograms the Resting Macrophages to an Intermediate Phenotype and Decrease Pseudomonas aeruginosa Mouse Lung Infection. Immuno 2022, 2, 651-670. https://doi.org/10.3390/immuno2040040
Behera S, Panda SK, Donkor M, Acharya E, Jones H, Acharya S. Chitin Derived Small Molecule AVR-48 Reprograms the Resting Macrophages to an Intermediate Phenotype and Decrease Pseudomonas aeruginosa Mouse Lung Infection. Immuno. 2022; 2(4):651-670. https://doi.org/10.3390/immuno2040040
Chicago/Turabian StyleBehera, Sumita, Santosh K. Panda, Michael Donkor, Eesha Acharya, Harlan Jones, and Suchismita Acharya. 2022. "Chitin Derived Small Molecule AVR-48 Reprograms the Resting Macrophages to an Intermediate Phenotype and Decrease Pseudomonas aeruginosa Mouse Lung Infection" Immuno 2, no. 4: 651-670. https://doi.org/10.3390/immuno2040040