

Predisposing Factors, Clinical Picture, and Outcome of B-Cell Non-Hodgkin’s Lymphoma in Sjögren’s Syndrome

Department of Pathophysiology, School of Medicine, National and Kapodistrian University of Athens, Mikras Asias Street 75, 11527 Athens, Greece
*
Author to whom correspondence should be addressed.
Immuno 2022, 2(4), 584-608; https://doi.org/10.3390/immuno2040037
Submission received: 20 September 2022
/
Revised: 14 October 2022
/
Accepted: 17 October 2022
/
Published: 20 October 2022
(This article belongs to the Special Issue Advances in Autoimmune and Rheumatic Diseases: A Theme Issue in Honor of Prof. Dr. Yehuda Shoenfeld)
Abstract
:Among other systemic autoimmune diseases, primary Sjögren syndrome (pSS) bears the highest risk for lymphoma development. In pSS, chronic antigenic stimulation gradually drives the evolution from polyclonal B-cell expansion to oligoclonal/monoclonal B-cell predominance to malignant B-cell transformation. Thus, most pSS-related lymphomas are B-cell non-Hodgkin lymphomas (NHLs), with mucosa-associated lymphoid tissue (MALT) lymphomas predominating, followed by diffuse large B-cell lymphomas (DLBCLs) and nodal marginal zone lymphomas (NMZLs). Since lymphomagenesis is one of the most serious complications of pSS, affecting patients’ survival, a plethora of possible predisposing factors has been studied over the years, ranging from classical clinical, serological, hematological, and histological, to the more recently proposed genetic and molecular, allowing clinicians to timely detect and to closely follow-up the subgroup of pSS patients with increased risk for lymphoma development. Overall predisposing factors for pSS-related lymphomagenesis reflect the status of B-cell hyperactivity. Different clinical features have been described for each of the distinct pSS-related B-cell NHL subtypes. While generally pSS patients developing B-cell NHLs display a fairly good prognosis, outcomes in terms of treatment response and survival rates seem to differ depending on the lymphoma subtype, with MALT lymphomas being characterized by a rather indolent course and DLBCLs gravely affecting patients’ survival.
Keywords:
Sjögren’s syndrome; lymphoma; biomarkers; MALT lymphoma; autoimmunity; lymphoproliferation1. Introduction
Primary Sjögren’s syndrome (pSS) is a systemic autoimmune disease characterized by B-cell hyperactivity [1]. Lymphomagenesis in the setting of pSS is considered to be a multi-step process arising from persistent polyclonal B-cell activation due to chronic antigenic stimulation in the salivary glands (SGs) of pSS patients, which in turn leads to oligoclonal/monoclonal B-cell expansion followed by the selection of premalignant B-cell clones, which will eventually progress to lymphoma [1,2]. Lymphoepithelial sialadenitis, the histologic hallmark of pSS, is characterized by a different lymphocytic composition based on lesion severity, with the B-cell component predominating over T-cells in the more severe lesions [3]. One could argue that this is not merely a histological finding since an overt B-cell hyperactive state defines a phenotypically unique pSS patient subset with features that have been recognized as predisposing factors for lymphomagenesis [1,4]. The stepwise transition to lymphoma is also reflected by distinct serological findings, with polyclonal B-cell proliferation leading to autoantibody positivity and hypergammaglobulinemia, and oligoclonal/monoclonal pre-malignant B-cell expansion resulting in cryoglobulinemia.
B-cell non-Hodgkin lymphomas (NHL) predominate among other hematological malignancies reported in pSS patients [5,6], while not so often, the development of other lymphoid malignancies, such as T-cell lymphomas or multiple myeloma, may occur [6,7]. While in the general population, the three most frequent B-cell NHL subtypes are diffuse large B-cell lymphoma (DLBCL) (40%), followed by follicular lymphoma (FL) (20%) and marginal zone lymphoma (MZL) (7%) [8], the distribution changes in pSS patients, with marginal zone mucosa-associated lymphoid tissue (MALT) lymphomas constituting the most frequent B-cell NHL subtype (48.5–76%), followed by DLBCL (9–17%) and nodal MZL (NMZL) (7–15%) [5,9,10,11,12,13,14,15,16]. There is, though, a subset of studies demonstrating DLBCLs to be the most frequent lymphoma developing in the setting of pSS [17], while others report a similar frequency of MALT lymphomas and DLBCLs [18]. This difference could be attributed to the different nature of the studies, with cohort studies from Rheumatology or other tertiary centers demonstrating MALT lymphomas as the predominant pSS-associated lymphoma subtype, and epidemiological studies rendering similar frequencies of MALT lymphomas and DLBCLs in the setting of pSS.
It has been demonstrated that pSS-related B-cell NHLs are not associated with viruses [i.e., Epstein–Barr virus (EBV), Hepatitis C virus (HCV), human T-cell lymphotropic virus 1(HTLV-1)] known to be present in lymphomas arising in non-pSS patients [16]. Some of the cytogenetic abnormalities [i.e., translocations t(14;18) or trisomies 18, 3, and 12), and mutations of oncogenes (i.e., p53) described in non-pSS-related lymphomas have also been described in lymphomas complicating pSS [16,19,20,21].
Overall, pSS seems to have a slight impact on patients’ survival compared to the general population, with standardized mortality rates (SMR) ranging from 1.02 [95% confidence interval (CI) 0.4–2.0] to 4.66 (95% CI 3.85–5.6) in different studies [15,22,23,24,25,26]. We should notice, though, that pSS patients who develop lymphoma tend to have higher mortality rates compared to those without lymphoma. Namely, Theander et al. reported that for pSS patients developing lymphoproliferative disorders, the SMR was as high as 7.89 (95% CI 2.89–17.18) compared to the general population of pSS patients with an estimated SMR of 1.17 (95% CI 0.81–1.63) [26], while in the study by Voulgarelis et al., the SMR for pSS patients with lymphoma was 3.25 (95% CI 1.32–6.76) compared to 1.08 (95% CI 0.79–1.45) for those without lymphoma [15].
Despite the increase in mortality rates attributed to lymphoma, the prognosis of pSS patients with B-cell NHL is generally favorable, and the overall survival (OS) rates are rather high [5,11,12,15,17]. This could be attributed to the “benign” nature of the most commonly observed MALT lymphomas, but considering the improved OS described in more recent studies compared to those of the past years [5,11,12,14,15,17], we should reflect on two possible contributing factors: (a) the improved therapeutic response after the addition of rituximab in the administered regimens, and (b) the increased awareness over the years of the predisposing factors that render pSS patients at increased risk for lymphomagenesis, leading to more intensive follow-up and earlier lymphoma diagnosis, as well as timely therapeutic intervention.
The latter is vividly reflected by the strenuous focus of the scientific community on the definition and validation of biomarkers that could predict lymphoma development in the setting of pSS. Apart from the classical predisposing factors (demographic, clinical, serological, and histological) extensively studied, novel molecular and genetic biomarkers are more recently reported, while the upcoming role of imaging studies as a tool for lymphoma prediction also gains ground. Thus, in the first part of this review, we aim to analyze the above-mentioned predisposing factors for lymphoma development in the setting of pSS. The following part focuses on the distinct clinical characteristics of the different B-cell NHL subtypes associated with pSS, while in the last part, the outcome of pSS-related lymphomas, both in terms of response to treatment and survival, is reviewed.
2. Predisposing Factors
2.1. Demographic Features
It has been demonstrated that gender and age at pSS diagnosis may act as predisposing factors for lymphoma development. In 1998, Ramos-Casals reported a higher incidence of lymphoma for pSS patients with disease onset at a young age (20 ± 34 years, mean 28 years) [27]. The majority of other published studies, though, do not support the notion that young age at pSS diagnosis is a predisposing feature for lymphoma development [6,18,28,29,30]. A recent multicenter study, including 1997 pSS patients, demonstrated that pSS patients with disease onset at an age of ≤35 years, as well as pSS patients with disease onset at an age of ≥65 years, are at increased risk for lymphoma development compared to patients with disease onset at an age of 35–65 years. Of note, the authors demonstrated two incidence peaks of lymphoma for the early-onset pSS patients (≤35 years old), within 3 years of onset and after 10 years of onset, while for late-onset pSS patients (≥65 years old), lymphoma was diagnosed within the first 6 years [31].
The majority of published research supports an increased incidence of lymphoma for male pSS patients compared to females [10,32,33,34], while a subset of studies reports no such difference [30,35]. It should be noted, though, that in the general population as well, NHL is slightly more common in men [36]. The male gender has been identified as an independent predisposing factor for pSS-related lymphoma development in one study [33], while Gondran et al. described that for male pSS patients who develop lymphoma, the time interval between pSS and lymphoma diagnosis is shorter compared to females [37]. These findings prompt us to consider whether gender itself acts as a lymphoma-predisposing feature or whether male patients simply accumulate other characteristics that have been validated as lymphoma risk factors. However, it has been demonstrated that there are no differences in the frequencies of classical lymphoma predictors between males and females [33]; further research is required.
2.2. Clinical Predisposing Factors
The most common clinical predictor for lymphoma development in the setting of pSS is persistent salivary gland enlargement (SGE). The first report of SGE conferring an increased risk for pSS-associated lymphoma development came in 1978 by Kasan et al. [Relative Risk (RR) 66.7 for pSS patients with SGE vs. 12.5 for pSS patients without] [38]. Thereafter, SGE has been validated as an independent risk factor for lymphoma development in multiple series of pSS-related NHL patients [11,18,24,28,29,32,39,40,41,42,43,44,45]. Since SGE is present in about one-third of pSS patients but only a subset of them develops lymphoma, Quartuccio et al. demonstrated that for patients with SGE, the presence of additional prognostic markers better defines the subgroup with increased risk for lymphoma development (namely, at least two of the following: cryoglobulinemia, low C4, leukopenia, anti-La/SSB positivity) [42]. The duration of parotid gland swelling also affects the risk for lymphoma development, with patients presenting parotid gland swelling for more than 12 months displaying a higher risk for lymphomagenesis compared to those with swelling durations ranging from 2 to 12 months [46].
Cutaneous vasculitic manifestations related to cryoglobulinemia, such as palpable purpura or skin ulcers, comprise another clinical entity independently associated with increased risk for lymphoma development [6,14,18,24,25,39,44]. Of note, it has been demonstrated that skin purpura is a clinical feature that can be used to distinguish patients with active lymphoma [43]. Other immune complex-mediated clinical manifestations, such as peripheral neuropathy [14,43] and glomerulonephritis [25,43,47], apart from their predictive value for future lymphoma development, tend to co-exist with active lymphoma in some pSS patients [43].
Lymphadenopathy, as a clinical manifestation of B-cell overactivity, has also been reported to be a possible predisposing factor for lymphoma development, mainly in earlier published studies [9,14,38,44,45], while only one study published in the last decade documents lymphadenopathy as a clinical predictor for lymphoma [29]. Splenomegaly [9,38] and Raynaud’s phenomenon [29] have been proposed as predisposing factors for pSS-associated lymphoma in fewer published studies.
2.3. Serological Predisposing Factors
The detection of serum IgMκ type II cryoglobulins has been demonstrated in almost 20% of pSS patients [48]. In 1996, Tzioufas et al. first proposed mixed monoclonal cryoglobulinemia as a serological marker predictive of pSS-related lymphoma development [49]. Ever since, cryoglobulinemia has been consistently reported in the published literature as a strong predisposing factor for lymphomagenesis in the setting of pSS [9,10,11,40,42,50,51,52]. In a more recent study by Chatzis et al. cryoglobulinemia has been demonstrated to be an independent prognostic factor, especially for pSS-associated MALT lymphoma [11]. Of note, in a large cohort of 1083 pSS patients, among which 10.6% had cryoglobulinemia, one-third of those who presented cryoglobulinemic vasculitis (CV) developed B-cell NHL within the first 5 years of the CV course [53].
Other cryoglobulin-related serological markers, such as C3/C4 hypocomplementemia, rheumatoid factor (RF) positivity, and the presence of monoclonal immunoglobulins, have also been proposed as predictive factors for lymphoma development. Skopouli et al. demonstrated low serum C4 levels (RR = 7.5, p = 0.0016) and cryoglobulinemia (RR = 7.9, p = 0.0012) to be strong predictors of lymphoma development [25]. C4 hypocomplementemia [9,10,18,24,25,29,32,42,43,50,52,54] is more frequently reported as a predisposing factor for lymphoma compared to C3 hypocomplementemia [6,10,18,32,50]. Brito-Zerón et al. reported low serum C4 levels as a prognostic serological marker for non-MALT B-NHL and low serum C3 levels for MALT lymphoma [10]. RF positivity, especially with concomitant C4 hypocomplementemia, is indicative of type II mixed cryoglobulinemia [49]. It has thus been associated with other predictors for lymphoma development and proposed as a possible predisposing factor [29,41,49,50]. Though a subset of studies supported the presence of monoclonal immunoglobulins as a serological finding conferring increased risk for pSS-associated lymphoma [29,43,45], Gottenberg et al. demonstrated an association only with pSS disease activity but not with lymphoma [55]. In an earlier study by Walters et al., serum and urine immunoglobulin light chains and their kappa/lambda ratio have been correlated with the burden of proliferating B-cells and have thus been proposed as a possible lymphoma biomarker [56]. More recent research, though, supports that higher levels of kappa- and lambda-free light chains associate only with higher pSS disease activity and not lymphoma [55].
Though Fragkioudaki et al. reported the presence of Ro/La antibodies as an independent predictor of pSS-related lymphoma [29], other studies of large pSS patients’ cohorts did not support a statistically significant association between anti-Ro/La positivity and lymphoma development [6,10,28,30]. A more recent study in a Greek cohort of 121 pSS-associated lymphoma patients reported an increased frequency of anti-La positivity at pSS diagnosis for patients subsequently developing MALT lymphoma compared to those that did not (51% vs. 34%, p = 0.0049), though anti-La positivity was not eventually proven to be an independent predisposing factor for MALT lymphoma development [11]. Of note, anti-Ro/La negative pSS patients demonstrate a lower frequency of manifestations associated with lymphoproliferation, as well as a lower risk of lymphoma development [57,58].
Higher baseline β2 microglobulin levels in pSS patients that subsequently developed lymphoma have been reported in a Finnish pSS patients’ cohort [59]. A more recent study, though, by Gottenberg et al., demonstrated that increased β2 microglobulin levels were associated with increased pSS disease activity, but no association with lymphoma was proven [55]. Notably, pSS patients with a history of lymphoma seem to maintain higher β2 microglobulin compared to those without lymphoma history [55].
Finally, Agmon-Levin et al. proposed a possible link between low vitamin-D levels and lymphoma predisposition in pSS patients [60].
2.4. Hematological Predisposing Factors
Fewer data are available in the published literature regarding hematologic aberrations as possible predisposing factors for pSS-related lymphoma. Theander et al. reported that CD4+ T lymphocytopenia and a CD4+/CD8+ T-cell ratio ≤ 0.8 correlated with an increased risk for DLBCL development in the setting of pSS [6]. In accordance, Baimpa et al. documented lymphocytopenia at pSS diagnosis as a predictive factor for DLBCL [9]. Leukopenia and neutropenia have also been identified as hematological parameters that could be used to discriminate pSS patients at increased risk for lymphoma development [10,14,18]. Finally, the presence of anemia has been correlated with DLBCL development in pSS patients [10].
2.5. Histological Predisposing Factors
The degree of inflammatory infiltration in the SG of pSS patients has been correlated with systemic involvement and disease severity [61]. Moreover, B-cells predominate over T-cells in more severe lesions, and as expected, their presence has been correlated with RF positivity, cryoglobulinemia, and low serum C4 levels [3], serologic features associated with increased risk for lymphoma development. High focus score (FS), defined as the number of lymphocytic foci per 4 mm2 of the SG tissue, has been documented in high-risk pSS patients who are more prone to develop systemic disease and have a poor outcome [62]. It also comprises a histological marker that can be used to discriminate the subset of pSS patients at higher risk for lymphomagenesis. Carubbi et al. showed that minor SG FS could be used as a prognostic histopathological feature for pSS-related lymphomagenesis [63]. Riselada et al. demonstrated that a minor SG FS ≥3 is an independent predictor of NHL development [64]. The independent prognostic value of minor SG FS is also supported by a more recent study additionally showing that patients who developed lymphoma with a minor SG FS ≥ 4 at pSS diagnosis were characterized by statistically significant shorter time intervals from pSS to lymphoma diagnosis compared to those with minor SG FS < 4 (4 vs. 9 years, respectively, p = 0.008) [65]. The same group also documented that FS is an independent prognostic risk factor, especially for pSS-related MALT lymphoma [11]. We should note though that the above-mentioned studies assessed the prognostic value of minor SG FS [64,65], while another subset of studies supports that that parotid gland biopsy may be better for predicting early-stage lymphoma, given that pSS-associated lymphoma often primarily occurs in the parotid gland [66,67,68]. Considering that minor SG and parotid gland biopsy has been shown to have similar specificity and sensitivity for pSS diagnosis [68], as well as diagnostic accuracy based on the American College of Rheumatology–European League Against Rheumatism (ACR–EULAR) 2016 criteria [69], further studies are needed to confirm whether they also display the same prognostic value for lymphoma prediction in terms of FS.
One of the pSS hallmarks is antigen-driven B-cell activation and proliferation [70,71]. B-cell clones are detected with high prevalence in the SGs of pSS patients [72]. These B-cell clones use a limited repertoire of immunoglobulin (Ig) variable heavy (VH) gene repertoire homologous to RF [73,74]. It is believed that the locally produced IgG autoantibodies in the SGs of pSS patients may form immune complexes, which in turn chronically stimulate B-cells expressing RF B-cell receptors (BCRs) (as reviewed by Stergiou et al. [2]). The majority of patients with pSS-associated SG MALT lymphoma express somatically mutated BCRs that are selected for the monoreactive, high-affinity binding of IgG-Fc [75], while SG lymphomas seem to express BCRs with strong RF homology more frequently compared to MALT lymphomas arising in other anatomical sites (i.e., stomach, lung) [76]. The RF-expressing B-cell clones display antigen-dependent affinity maturation leading to lymphomagenesis [77]. Bende et al. demonstrated, though, that the BCR repertoire strongly biased towards stereotypic RFs in SG MALT lymphoma does not correspond to a similar repertoire in the inflamed SGs of pSS patients, concluding that, in the latter, the repertoire is based on a strong selection advantage of incidental stereotypic RF-expressing B-cells [78]. Of note, it has recently been demonstrated that lymphoma driver mutations are present in B-cells producing pathogenetic antibodies with RF activity [79]. The presence of RF-expressing B-cells may reflect a pre-lymphomatous condition in the setting of pSS [49,80,81]. However, given the observation that B-cell clones are found in approximately 50% of sialadenitis lesions without morphological or clinical evidence of lymphoma [73,82], further research is required to determine which of the patients with RF-expressing B-cell presence in their SGs are those at a higher risk for lymphoma development.
Though the presence of germinal center (GC)-like structures reflects high pSS disease activity [83], its prognostic value as a lymphoma predisposing factor remains controversial. A subset of studies supports the notion of increased lymphoma risk in pSS patients with GC structures in their SG biopsies [54,84,85]. Theander et al. reported that for pSS patients developing lymphoma, the median time from pSS to lymphoma diagnosis was 7 years for those presenting GC-like structures in their diagnostic SG biopsy [54]. On the contrary, other study groups demonstrated no association between ectopic GCs and risk for lymphoma development [66,83,86]. This discrepancy could be attributed to the underestimation of small GCs when an extensive immunohistochemical study is not applied in SG biopsies [87]. On the other hand, the overestimation of GC-like structures may result from the reliance on CD21 staining alone for the detection of follicular dendritic cell (FDC) networks [88,89,90,91]. At this point, we should highlight that the histopathological evaluation both of GCs and FS needs to be standardized when assessed in clinical trials in order to have more subjective calculations and to avoid possible discrepancies in the reported results [90,92]. Moreover, we should always consider the different sizes and characteristics of each study population which could confer to discrepancies in the reported results.
Investigating the role of ectopic GCs in class switch recombination, Bombardieri et al. described different activation-induced cytidine deaminase (AID) distribution patterns in SG biopsies of pSS patients with and without lymphoma. In the SG of pSS patients without MALT lymphoma, AID was expressed by follicular dendritic cells within ectopic GCs and large B-cells residing in a T-cell-rich zone outside the ectopic GC. On the other hand, in the SG of pSS patients with MALT lymphoma, AID expression was retained in residual GCs, whereas neoplastic marginal zone-like B-cells were consistently AID-negative [89]. This finding possibly reflects the multistep process toward lymphomagenesis.
The lymphoid organization in the SGs of pSS patients is mediated by lymphoid chemokines. Barone et al. demonstrated in the SGs of pSS patients with MALT lymphoma the lymphoid chemokines C-X-C motif chemokine ligand 13 (CXCL13) and C-C motif ligand 21 (CCL21) are selectively associated with areas of reactive lymphoid proliferation, while C-C motif ligand 21 (CXCL12) is observed predominantly in infiltrated ducts and malignant B-cells. These findings suggest that in SG MALT lymphoma, the lymphoid chemokines CXCL13 and CCL21 participate in the reactive lymphoid tissue organization, whereas CXCL12 is possibly involved in the regulation of malignant B-cell survival [93].
2.6. Disease Activity as a Predisposing Factor
The quantification of pSS disease by the EULAR SS disease activity index (ESSDAI) score incorporates a sum of domains, many of which have been shown to be independent prognostic factors for lymphoma development. A characteristic example is that of cryoglobulinemia, a validated strong predictor of pSS-related lymphoma. Cryoglobulin-positive patients present significantly higher ESSDAI scores compared to those without cryoglobulinemia [62,94]. This prompted researchers to investigate the effect of pSS activity on the risk for lymphomagenesis. The ESSDAI score (after excluding the lymphadenopathy domain) calculated at least 6 months before lymphoma diagnosis has been reported as an independent predictor of lymphoma development [41]. Brito-Zerón et al. demonstrated that the baseline ESSDAI score is associated with a higher risk of MALT lymphoma but not for non-MALT B-NHL [23]. In accordance, Chatzis et al. identified the ESSDAI score (after excluding the lymphadenopathy domain) calculated at pSS diagnosis as an independent predictor for MALT lymphoma development [11]. On the contrary, De Vita et al. observed no significant difference of the baseline ESSDAI score between pSS patients who developed lymphoma and those who did not [40]. The glandular domain of the baseline ESSDAI has been correlated with MALT lymphoma [23], while the glandular domain of the baseline clinical ESSDAI has been associated with any type of lymphoma [13]. In a cohort of pSS patients from Argentina, glandular and cutaneous ESSDAI domains have been associated with increased lymphoma risk [13]. Non-MALT B-NHL development has been shown to correlate with increased activity of the biological ESSDAI domain [10]. Of note, an increase in the median ESSDAI score (after excluding the effect of the domain lymphadenopathy and lymphoma) has been documented from baseline at pSS diagnosis (median 9, range 0–44) to the time of lymphoma diagnosis (median 18, range 2–50), with glandular and biological domains showing a statistically significant increase in their value [11].
2.7. Molecular and Genetic Predisposing Factors
More recent research focuses on the identification of molecular and genetic predisposing factors that could be used to discriminate pSS patients at a higher risk for lymphoma development. These newly proposed lymphoma biomarkers relate in terms of pathophysiology to B-cell differentiation, lymphoid organization, and immune responses.
2.7.1. Genetics
B-cell activating factor (BAFF) has been implicated in the pathogenesis of pSS, mainly due to its essential role in B-cell regulation and proliferation [95]. Certain BAFF gene polymorphisms have been associated with an increased risk for lymphomagenesis in pSS patients [96]. Furthermore, the His159Tyr mutation of the BAFF receptor (BAFF-R) shows increased prevalence in pSS-patients, particularly in those who develop MALT lymphoma [97]. Of note, Papageorgiou et al. showed that this mutation was detected in more than two-thirds of pSS patients who developed lymphoma with pSS diagnosis between the ages of 31 and 40 years [97]. Pathophysiologically, it has been postulated that the BAFF-R His159Tyr mutation confers a high risk for lymphoproliferation through the activation of the Nuclear Factor-κB (NF-κΒ) signaling pathway [98].
Another protein implicated in NF-κB signaling regulation and associated with pSS-related lymphomagenesis is A20, an enzyme with ubiquitination activity encoded by the Tumor Necrosis Factor (TNF) Alpha Induced Protein 3 (TNFAIP3) gene. Somatic and germline mutations of TNFAIP3 that lead to functional A20 protein abnormalities have been detected in as many as 77% of pSS patients who developed MALT lymphoma [99]. Specifically, the rs2230926 exonic variant has been reported to confer increased lymphoma risk [99], while one-fifth of pSS-lymphoma patients with disease onset at an age of ≤40 years were shown to carry this variant [100].
The activation of the G Protein-Coupled Receptor 34 (GPR34) through the phospholipase A1 (PLA1) pathway in lymphoepithelial lesions may promote B-cell survival and proliferation [101,102]. GPR34 translocations and mutations are specifically associated with SG MALT lymphoma [103,104]. It has therefore been proposed that GPR34 activation might constitute a link between LELs and SG MALT lymphomagenesis. The decreased prevalence of the rs11797 A minor allele of Three Prime Repair Exonuclease 1 (TREX1), an exonuclease involved in DNA repair and degradation, possibly resulting in a dampened type I IFN production, has been described in pSS patients who developed non-MALT lymphomas [105]. The functional Leukocyte immunoglobulin-like receptor A3 (LILRA3) gene variant was shown to confer increased susceptibility to pSS-related lymphoma development in patients with a disease onset of <40 years old [106]. Finally, the HLA complex P5 (HCP5) rs3099844 variant has been reported to associate not only with anti-Ro/SSA and anti-La/SSB positivity in the setting of pSS but also with lymphoma development [107].
2.7.2. Epigenetics
MicroRNAs (miRNAs) of the miR200 family targeting the Ro/SSA and La/SSB autoantigens have been implicated in pSS pathogenesis [108,109]. Downregulated miR200b-5p expression in minor SGs has been shown to be a strong predictive biomarker for pSS-related lymphoma, detected long before clinically overt lymphoma [110]. Apart from its predictive value, miR200b-5p expression could be useful for monitoring and/or predicting the therapeutic response to lymphoma treatment, since it seems to remain stable or decrease for refractory to treat lymphomas and also reduce in the relapse setting [110].
Single-nucleotide polymorphisms (SNPs) of the methylene-tetrahydrofolate reductase (MTHFR) gene, an enzyme essential in DNA synthesis and methylation, have been associated with susceptibility to non-MALT NHL development in SS patients [111]. Reduced levels of long interspersed nuclear elements (LINE-1) retroelements’ promoter methylation along with increased DNA methyltransferase (DNMT) 3B, DNMT1, and methyl CpG binding protein 2 (MeCp2), but reduced lymphoid-specific helicase (LSH) levels were detected in pSS-low risk patients compared to pSS-lymphoma patients [112].
2.7.3. Gene Expression
SG epithelial cells from pSS patients exhibit down-regulated TNFAIP3 expression and increased NF-κB activities compared to controls [113]. Moreover, the A20 protein shows weaker staining in minor SGs with pSS-related lymphoma compared to pSS SGs without lymphoma [114]. This observation implicates lymphocytic A20 down-regulation in the pathogenesis of pSS-related lymphoma development.
Interferon (IFN) signaling aberrations have also been investigated as possible predisposing factors for pSS-related lymphomagenesis. A high IFNγ/IFNα mRNA ratio in diagnostic SG biopsies has been proposed as a novel histopathological predictive biomarker of in situ lymphoma development [115]. High expression of IFN-stimulated gene 15 (ISG-15), both in SGs and peripheral blood, has been shown to discriminate pSS patients with lymphoma from those without [116].
The P2X7 receptor (P2X7R), which promotes inflammatory responses via the NLPR3 inflammasome, was found up-regulated in SG biopsies of patients with SS, stimulating IL-18 production. This up-regulation was restricted to epithelial cells and correlated with the presence of GC-like structures and an increased risk of MALT lymphoma development [117]. More recently, Vakrakou et al. demonstrated that SS patients at high risk for lymphoma development and patients with pSS-associated lymphoma displayed a unique NLRP3 inflammasome gene signature in peripheral blood mononuclear cells and increased levels of IL-18 [118]. Chronic inflammation and macrophages in SS minor salivary glands have been previously suggested as significant predictors for lymphoma development among pSS patients [119]. Lipoprotein-associated phospholipase A2 (Lp-PLA2) is a product mainly of tissue macrophages. Serum Lp-PLA2 activity and Lp-PLA2 expression in MSG tissues are increased in pSS-lymphoma patients compared to pSS patients without lymphoma [120].
2.7.4. Proteins
BAFF levels could be used for the identification of prelymphomatous stages in pSS, since increased BAFF levels discriminate clonal B-cell expansion from polyclonal B-cell proliferation, while even higher levels characterize pSS patients with active lymphoma [52,121]. Interestingly, for pSS patients who have developed lymphoma and received treatment, BAFF levels remain high even years after lymphoma remission [55], a finding suggestive of a genetic origin of such a persistent increase.
Fms-like tyrosine kinase 3 (Flt-3) is a membrane-bound tyrosine kinase receptor, that, upon binding to an Flt-3 ligand (Flt-3L), is activated and mediates the survival, proliferation, and differentiation of hematopoietic progenitor cells [122]. In the setting of pSS, elevated serum Flt-3L levels have been associated with disease activity and lymphoma development [123,124]. Of note, Flt-3L levels are elevated long before lymphoma diagnosis (up to 94 months, mean 46 months) [125]. CXCL13, a chemokine expressed by FDC, stromal cells, monocytes, and macrophages, holds a pivotal role in lymphoid development and organization, mainly in secondary lymphoid tissues [126]. Several studies report that serum CXCL13 levels correlate with pSS disease activity and are increased in pSS patients with lymphoma [52,125,127]. One study also demonstrated a trend towards a higher serum CCL11 level for pSS-lymphoma patients [52]. Thymic stromal lymphopoietin (TSLP) is an epithelial cell-derived cytokine implicated in immune response regulation, which also demonstrates B-cell growth factor activity [128]. In pSS, the transition from benign to malignant lymphoproliferation is characterized by a progressive increase in serum TSLP levels [129]. Gandolfo et al. validated in a multicenter study that serum TSLP levels increase in pSS pre-lymphomatous conditions, reaching their peak values in pSS patients with active NHL [130].
A schematic presentation of the newly proposed molecular and genetic predisposing factors for pSS-related lymphoma development and their pathophysiologic role is presented in Figure 1.
2.8. Imaging-Defined Predisposing Factors
The effective detection of typical pSS SG structural abnormalities through SG ultrasonography (SGUS) has recently gained attention [131]. The retrospective studies of Theander et al. and Coiffier et al. reported that SGUS can identify patients at risk of developing lymphoma [132,133]. Lorenzon et al. prospectively assessed the OMERACT score in pSS patients with clinical findings suspicious for lymphoma development, who subsequently underwent US-guided core-needle biopsy. A higher OMERACT score and a more inhomogenous glandular pattern were documented in pSS patients diagnosed with lymphoma after the core-needle biopsy. For focal lesions, the authors identified eight suspicious lymphoma features (OMERACT grade 3, very hypoechoic, homogenous, oval shape, well-defined margins, presence of septa, color-Doppler vascularization, posterior acoustic enhancement), showing that the simultaneous presence of 6/8 and 7/8 features was significantly higher among pSS-lymphoma patients compared to pSS patients without lymphoma [134]. Overall, SGUS is considered to be the imaging modality of choice for the assessment of the SG parenchyma in patients with pSS, as well as for the identification of lesions suspicious for lymphoproliferative disease that need further histological assessment [135].
Magnetic resonance imaging (MRI) findings display good agreement with SGUS for pSS diagnostic work-up [136,137]. It should be noted, though, that the features of benign and malignant SG lesions in pSS seem to overlap in MRI [136,137], rendering this imaging technique ineffective for the identification of lymphoma. A single study reports that parotid gland solid cystic features could discriminate MALT from non-MALT lymphomas [138].
Bădărînză et al. investigated the role of bidimensional shear wave elastography in the identification of pSS parotid NHL, identifying features characteristic of parotid gland MALT lymphoma (hyperechoic bands in more than half of the glandular parenchyma, large hypoechoic area > 20 mm, traced gland area over 5 cm2, parotid US score greater than 13, and high stiffness, all p < 0.001). These features were shown to have high sensitivity (92.3%), specificity (100%), and positive (100%) and negative predictive values (98.3%) for NHL identification [139].
Since lymphadenopathy is a rather common clinical finding in pSS patients, contemporary imaging modalities could be used to distinguish patients with active lymphoma. To this end, Cohen et al. demonstrated that positron emission tomography/computed tomography (PET/CT) could be a useful diagnostic tool in the setting of pSS lymphadenopathy, since lymph node FDG uptake is marginally higher in pSS patients with lymphoma compared to those without lymphoma [maximum standardized uptake value (SUVmax) = 5.4 vs. 3.2, p = 0.05] [140]. In a more recent retrospective study by Keraen et al. a parotid gland SUVmax of ≥4.7 and/or the presence of focal lung lesions were associated with lymphoma diagnosis [141].
A summary of the proposed predisposing factors for B-cell NHL development in the setting of pSS is described in Table 1.
2.9. Predictive Models for Lymphoma Development
To improve the lymphoma risk stratification of pSS patients, models combining independent predisposing factors have been developed. Ioannidis et al. first proposed that the simultaneous presence of palpable purpura and low C4 levels at the initial evaluation could be used to distinguish pSS patients at high risk for lymphoma development from those at low risk [24]. Baimpa et al. constructed a prognostic model for pSS-associated MALT lymphoma based on the presence of five features, clinical, serological, and hematological as follows: cryoglobulinemia, neutropenia, low C4 levels, lymphadenopathy, and splenomegaly. The proportion of pSS patients who were diagnosed with MALT lymphoma by the number of predisposing features was 3.62%, 11.96%, 34.78%, 80%, and 100%, for patients with 0, 1, 2, 3, and 4 features, respectively [9]. Baldini et al. also developed a “bedside” clinical prediction model for the identification of high risk for lymphoma development in pSS patients, based on the presence of SGE, low C4 and/or C3 levels, and disease duration [32]. A predictive risk score for pSS-related NHL was developed by Fragkioudaki et al., including those features shown to be independent predictors for lymphoma development in their study: SGE, lymphadenopathy, Raynaud’s phenomenon, anti-Ro/SSA or/and anti-La/SSB positivity, RF positivity, the presence of monoclonal immunoglobulins, and C4 hypocomplementemia. The probability for NHL development was 3.8% for patients with fewer than two features, and 39.9% for those with three to six features, while the corresponding probability reached 100% in the presence of all seven features [29]. More recently, an attempt to combine classical clinical and serological predisposing factors for pSS-related lymphomagenesis with genetic variants has been undertaken [144].
2.10. “One Size Fits All?”: Distinct Predisposing Factors for Different Patient Subgroups and Lymphoma Subtypes, Predisposing Factors Detected at Different Time-Points before Lymphoma Development
Different predictive factors for lymphoma development have been shown to apply to pSS patients of different ages at disease onset. Regarding classical predisposing factors, cryoglobulinemia, C4 hypocomplementemia, lymphadenopathy, and SGE have been independently related to lymphomagenesis for pSS patients with disease onset at an age ≤ 35 years, while SGE, C4 hypocomplementemia, and the male gender were independently associated with lymphoma development for those with pSS onset at an age ≥ 65 years [31]. As for the more recently proposed lymphoma biomarkers, the TNFAIP3 rs2230926 variant and the functional LILRA3 gene variant could apply for the pSS patient subgroup with disease onset at an age ≤ 40 years [100,106], while BAFF-R His159Tyr mutation for the subgroup with disease onset between 31 and 40 years of age [97]. No significant differences in lymphoma predisposing factors have been so far described between female and male patients, despite the tendency of men to present more often SGE and lymphadenopathy [33,34].
Since the different pSS-related B-cell NHL subtypes are characterized by significantly different disease severity and prognosis, it is important to define subtype-specific predisposing factors, especially given the dismal outcomes of pSS patients with DLBCL. As summarized in Table 2, cryoglobulinemia, neutropenia, low serum C4 levels, low serum C3 levels, lymphadenopathy, splenomegaly, FS at pSS diagnosis, and ESSDAI at pSS diagnosis have been specifically correlated with MALT lymphoma development, while lymphocytopenia, a CD4+/CD8+ T-cell ratio ≤0.8, anemia, and low serum C4 levels have been proposed as predisposing factors for DLBCL development [6,9,10,11,18].
Certain predisposing factors could be detected a long time before lymphoma development (Figure 2). The reported median time from the expression of a feature to lymphoma development could lead to the monitoring of pSS patients. Specifically, the median time to lymphoma diagnosis has been documented to be 108 months for SGE [38], 88 months for CD4+ lymphocytopenia [6], 84 months for lymphadenopathy/splenomegaly [38], 84 months for histologically detected GC-like structures [54], 68 months for an FS ≥ 3 [64], 48 months for an FS ≥ 4 [65], and 8 months for mixed monoclonal cryoglobulinemia [49]. Among the more recently proposed biomarkers, serum Flt-3L levels have been found to increase at a median time of 46 months before lymphoma diagnosis, [124], and low miR200p-5b expression in SGs can be detected at a median time of 36 months before lymphoma [110].
3. Clinical Picture
The different B-cell NHL subtypes arising in patients with underlying pSS display a distinct clinical picture. Though for the majority of the patients, lymphoma diagnosis follows pSS diagnosis [5,11,12,17,145], for a subset of patients, lymphoma diagnosis precedes pSS diagnosis [5,17]. Vasaitis et al. demonstrated in a cohort of 107 patients with pSS-related lymphoma that 17% of them had pre-existing lymphoma before pSS diagnosis and that the features discriminating them from pSS patients with lymphoma following pSS diagnosis were the male gender and lymph node enlargement [146]. In the following sections, we analyze the clinical features of the most frequently reported B-cell NHLs developing in the setting of pSS (summarized in Figure 3).
3.1. MALT Lymphomas
Overall, pSS patients diagnosed with MALT lymphoma are of younger age and have a shorter disease duration compared to pSS patients diagnosed with other lymphoma subtypes [5,11,12,17]. It should be noted, though, that the reported median ages at lymphoma diagnosis vary in different pSS cohorts. Namely, regarding the most recently published studies, in a large single-center Greek cohort of 121 pSS-lymphoma patients, the median age of MALT lymphoma diagnosis was 57 years [11], while in the largest multicenter cohort of 376 pSS-lymphoma patients the median age of MALT lymphoma diagnosis was 49.5 years [5]. In different cohorts, the reported median disease duration from pSS onset to MALT lymphoma diagnosis is approximately 7 years [11,17], and the estimated median disease duration from pSS diagnosis to MALT lymphoma diagnosis ranges from 2 to 5 years [11,12,17]. Immunosuppressive treatment for pSS before lymphoma diagnosis does not seem to affect the risk of MALT lymphoma development [12].
The most common sites of MALT lymphoma development are the minor SGs and the parotid gland [5,11,12,17], leading to the clinical feature of SGE. Other less frequently involved sites are the stomach, the eyelid-ocular adnexa, the lung, the thymus, the soft palate, and the skin [5,11]. For the subset of patients with gastric MALT lymphoma, concurrent H. pylori infection is reported in 42% to 75% of cases [11,15]. The majority (68–77%) of pSS patients diagnosed with MALT lymphomas have stage I disease mainly confined to the SGs [5,11,12,15,17]. Overall, limited stage I and II disease has been reported for 62.8% to 77.2% of MALT lymphoma cases [11,12,15]. In cases with nodal involvement, reported in 20.6% to 30% of pSS-MALT lymphoma patients, it has been demonstrated that mainly cervical lymph nodes are involved [11,15]. Splenomegaly is an infrequent manifestation (5% to 7%), while the reported incidence of bone marrow (BM) involvement shows diverse rates (17% to 29%) in different cohorts [11,12,15]. Advanced stages of disease, namely stage III and IV by Ann-Arbor staging, have been documented in 22.8% to 37.3% of cases of MALT lymphoma patients [11,12]. MALT lymphomas involving more than one extranodal site are also described in about 20% of cases [11,14,15]. The most frequent sites of concomitant extranodal involvement are the minor SG and parotid [11] and the minor SG and stomach [11]. Regarding the laboratory profile, MALT lymphoma cases often present anemia (31–59.7%), lymphopenia (34–41.2%), and monoclonal immunoglobulins (31.4–40.6%) [11,12,15].
3.2. DLBCL
The majority of pSS-related DLBCL cases are of the non-GCB subtype [142,145]. It has been documented that a subset of DLBCLs is the result of the transformation of pre-existing MALT lymphomas [11,16,18]. This prompts us to consider that in the latter case, pSS patients may undergo the necessary work-up for lymphoma diagnosis only when lymphoma symptomatology is rather intense, with cases of asymptomatic lymphomas, mainly of the MALT lymphoma subtype, being underdiagnosed and histologically documented after their transformation to high-grade B-cell NHL. Gorodetskiy et al. addressed the question of the clonal relationship between MALT lymphomas and DLBCL in the setting of pSS, proving that in the majority of cases, the two subtypes are clonally related, demonstrating that DLBCL is the result of the progression/transformation of a pre-existing MALT lymphoma [143]. Comparing the frequency of DLBCLs in a Swedish cohort of pSS patients to the frequency in the general population, Vasaitis et al. report no significant difference [17].
Compared to pSS patients with MALT lymphomas, pSS patients diagnosed with DLBCLs are older (median age 53.3 to 71 years) [5,11,12,15,17], with a longer disease duration from pSS onset to DLBCL diagnosis (median 14 to 20.5 years) [11,17,145] and from pSS diagnosis to DLBCL diagnosis (median 8 to 14 years) [11,12,17,145]. In the case of DLBCL, there is commonly nodal involvement, mainly of the neck and axillae, though more generalized lymphadenopathy involving the mediastinal and abdominal lymph nodes has also been reported [11]. Glandular enlargement is not so common, affecting up to 16% of pSS patients diagnosed with DLBCL [5]. Limited (stage I and II) is reported in 25% to 54.5% of DLBCL cases, while advanced stage disease (stage III and IV) is reported in 45.5% to 75% of the cases [11,12,15]. The concomitant involvement of more than one extranodal site is not so frequently described, ranging from 9% to 16.7% of DLBCL cases [11,12]. pSS patients with DLBCL present more often B symptoms compared to patients with MALT lymphomas [5,11,12]. Compared to other pSS-related lymphoma subtypes, DLBCL patients present more often with anemia [12,127], while cryoglobulinemia has also been reported as a frequent serological feature [145].
DLBCL patients generally present higher IPI scores at lymphoma diagnosis compared to other lymphoma subtypes [11,12,15]. However, in 2015, Papageorgiou et al. documented an intermediate-high and high IPI in 58.3% of pSS-DLBCL patients; more recently, Chatzis et al. reported a lower rate of 36.4% [11,12].
3.3. Nodal Marginal Zone Lymphomas
The third most commonly reported B-cell NHL developing in pSS patients is NMZL, with reported rates ranging from 7% to 15% of all reported lymphoma subtypes [5,11,12,15]. The median age of pSS patients at NMZL diagnosis ranges from 54 to 56 years [5,11,12,15], while the reported median disease duration from pSS to NMZL diagnosis ranges from 6.5 to 12 years [11,12,15]. The main clinical presentation of NMZL is that of peripheral lymphadenopathy [5,11,12,15]. NMZLs are usually of an advanced stage, III 50–75% or IV 25–50% by Ann Arbor staging [11,12,15]. Splenomegaly is rather common, regarding 50–75% of NMZL cases [11,12,15], while BM involvement is not so frequent, with some studies documenting no patients with BM and others documenting a rate of 25% [11,12]. Constitutional symptoms are a common clinical finding in 12.5% [12]. Palpable purpura is reported in up to 50% of patients with NMZL [12,15]. Compared to patients with MALT lymphomas, patients with NMZL present more frequently cryoglobulinemia, anemia, and lymphopenia [15]. Leucopenia and thrombocytopenia are also described in a subset of patients [12,127].
3.4. Other B-Cell NHLs
Less frequently, pSS patients are diagnosed with chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL), FL, and lymphoplasmacytic lymphoma [5,11,12,15]. CLL/SLL diagnosis may be incidental, and the SGs are not so often infiltrated by the neoplastic lymphoid population [5]. Hernandez-Molina et al. reported that CLL and FL diagnosis may precede pSS diagnosis in 9.3% and 7% of patients, respectively [5]. Voulgarelis et al. reported that these lymphoma subtypes are characterized by the relative absence of lymphopenia and the notable absence of cryoglobulinemia and purpura before their diagnosis [15].
4. Outcome
The outcome of pSS patients diagnosed with B-cell NHL varies based on lymphoma subtype and treatment approach. Response rates to treatment also differ among the different B-cell NHL subtypes, while over the years, the addition of rituximab in the applied treatment protocols has conferred better outcomes. Overall, pSS patients with MALT lymphomas demonstrate better OS and event-free survival (EFS) rates compared to patients with DLBCLs.
4.1. Response to Treatment
Data regarding the treatment outcomes of pSS-related lymphomas in the pre-rituximab era are described in the studies of Royer et al. [16] and Voulgarelis et al. [14], with the main treatment modalities being chemotherapy alone or combined with surgical excision and radiotherapy. The latter study provided specific information on the treatment and outcome specifically of MALT lymphomas. pSS-lymphoma patients requiring therapy received chemotherapy alone [Cyclophosphamide, Hydroxydaunorubicin, Oncovin, Prednisone (CHOP) regimen, Cyclophosphamide, Oncovin, Prednisone (COP) regimen, Chlorambucil], chemotherapy in combination with surgical excision or radiotherapy, surgical excision alone, or radiotherapy alone. It has been described that a percentage as high as 80% achieved complete remission (CR), while most of the patients with low-grade lymphoma tended to sustain a long-term remission. The subset of patients that relapsed with low-grade lymphoma achieved a second remission after the second-line treatment. Deaths related to lymphoma were noticed in the high-grade lymphoma group [14].
For pSS patients diagnosed with MALT lymphomas a “watch and wait” approach may be applied [11,15]. Though the majority of these patients eventually require treatment, mainly due to MALT lymphoma progression [11]. Based on the symptomatology and the bulk of the disease, symptomatic patients or patients with advanced-stage disease may receive rituximab monotherapy or a combination of rituximab with chemotherapy [11,12,15]. For local disease radiation or surgical excision, approaches have been applied [11,12,15]. We should note that for the subset of patients presenting with gastric MALT lymphoma and concurrent H. pylori infection, the administration of antibiotic treatment may lead to lymphoma regression in some cases [15,16].
Overall, CR rates after first-line treatment for MALT lymphomas range from 68% to 94% [5,12,15]. Papageorgiou et al. reported that for MALT lymphoma cases, rituximab monotherapy yielded a CR rate of 71.4%, while the combination of rituximab with chemotherapeutic agents (CHOP, COP, Fludarabine, Cyclophosphamide) yielded CR rates ranging from 70% to 100% [12]. It should be noted that first-line treatment with rituximab combined with chemotherapy leads to variable CR rates, depending on the chemotherapeutic agents used (83.3% for R-CHOP, 70% for R-Fludarabine, 100% for R-COP, 50% for R-Cyclophosphamide) [12]. Despite the high CR rates with rituximab monotherapy, a subset of pSS patients with MALT lymphoma may relapse, requiring second-line treatment [11]. In the study of Chatzis et al., of the 27 patients that received only rituximab as the first-line therapy for MALT lymphoma, 8 patients relapsed after achieving CR, 3 patients showed progression requiring treatment after achieving partial remission (PR), and 1 patient showed stable disease (SD) and received second-line treatment [127]. Among the MALT lymphoma patients that required second-line treatment for MALT lymphoma, either due to relapse or disease progression after PR, approximately 84% achieved CR [11].
For pSS patients diagnosed with DLBCL, the usual therapeutic approach is that of chemo-immunotherapy (predominantly R-CHOP) [12,15]. The reported overall CR rates range from 66.7% to 87% [5,12,15]. Relapse after first-line therapy for DLBCL occurs in about one-third of patients [5]. Interestingly, the CR rates reported for pSS patients with NMZL after first-line treatment demonstrate a rather wide range from 33% to 85% [5,12]. Among pSS-associated B-NHL subtypes, NMZLs demonstrate the highest relapse rates [5,15].
Regarding the involved sites, it has recently been reported that patients diagnosed with SG lymphoma demonstrate higher response rates compared to those with extra-SG lymphoma (94% vs. 74%) [5]. This finding could be attributed to the SG involvement in the most frequently observed MALT lymphoma cases, which generally display a more “benign” course compared to other lymphoma subtypes with more frequent extra-SG involvement.
Lymphoma treatment does not only result in lymphoma control, but it also seems to affect the immunological profile of pSS patients. For patients with cryoglobulinemia and the related manifestations of palpable purpura, glomerulonephritis, and peripheral neuropathy at NHL diagnosis, a statistically significant decline in cryoglobulin levels has been reported after NHL treatment, with a concurrent improvement of the related clinical manifestations [15]. It seems, though, that pre- and post-treatment complement levels remain stable in these patients [15].
4.2. Survival
Over the years, survival rates for pSS patients diagnosed with B-NHL lymphomas have improved, probably due to the addition of rituximab in the treatment modalities. In the pre-rituximab era, the reported median survival was as low as 1.83 years for pSS patients with intermediate-high-risk lymphomas and 6.33 years for those with low-risk lymphomas, with a 5-year OS of 48% for the high-risk group and of about 70% for the low-risk group [14]. More recent studies, including patients that have received rituximab-containing regimens, demonstrate a noteworthy improvement in survival rates, with a median OS after lymphoma diagnosis of 6 years for DLBCL and 13 years for MALT lymphoma patients [17].
Papageorgiou et al. demonstrated that the 5-year OS was 90.91% and the EFS was 77.92% [12]. In the largest published cohort of 414 pSS patients who developed hematological neoplasia with a mean follow-up of 8 years, the authors report a death rate of 16% for patients with B-cell NHL and a 5-year OS of 86.5% [5]. Chatzis et al. reported a 5-year OS of 91% for MALT lymphomas, 54.5% for DLBCLs, and 62.5% for NMZLs, while in the same study, the 10-year OS was estimated to be 79% for MALTL, 40,9% for DLBCL, and 46% for NMZL [11]. In the same study, though, the EFS rates documented were rather low, namely 45.5% for MALT lymphomas, 24.2% for DLBCLs, and 31% for NMZLs (with an event being defined as lymphoma progression, lymphoma relapse, treatment failure, histologic transformation, treatment initiation after a “watch and wait” approach, development of a second lymphoma or death by any cause) [11]. Given the above, we should note that, especially for MALT lymphomas, despite the very favorable OS rates, frequent events seem to complicate their course, and this prompts us to consider that these patients should be under close monitoring for further complications. In a cohort of 18 patients with pSS-related DLBCL, Gorodetskiy et al. reported very dismal outcomes, with a median OS of 3 months and a 5-year OS of 37.5% [145]. The main studies reporting the survival rates for pSS patients who developed B-cell NHLs are summarized in Table 3.
Higher ESSDAI at lymphoma diagnosis confers a greater risk for death or an event, while it is also associated with worse 5-year EFS and OS. Patients that do not achieve an ESSDAI improvement post-chemotherapy are more probable to experience an event. Another factor negatively affecting 5-year EFS and OS is IPI, which retains a significant prognostic effect on survival outcomes [12]. An increased risk of death by lymphoma has been reported for pSS patients with low serum C4 levels and purpura [24].
In general, patients diagnosed with SG lymphoma demonstrate a lower mortality rate compared to those with extra-SG lymphoma (3% vs. 18%) [5]. Deaths are mainly attributed to B-cell NHL progression, infections, and chemotherapy-related complications [5,11,12,15]. The transformation of MALT lymphoma to DLBCL is a frequently described cause of death [18].
5. Conclusions
Lymphoma development is recognized as the most serious complication of pSS. Overall, SGE and cryoglobulinemia are the best validated predisposing factors for pSS-associated lymphoma development. The plethora of newly proposed molecular and genetic biomarkers for lymphoma in the setting of pSS definitely needs further validation in future studies. Research should focus on the question of whether different predisposing factors (classical or novel ones) apply to different pSS patient subgroups. Though composite scores for lymphoma prediction have been proposed, their value should be tested, while an effort to create more sophisticated prediction models integrating new molecular biomarkers into classical lymphoma predictors should be undertaken. Moreover, we should also consider the assessment of certain quantifiable disease features (i.e., ESSDAI), not only at pSS diagnosis but at a continuum during patient follow-up since their change over time may indicate an increased risk for lymphomagenesis. We should moreover comment that the proposed predisposing factors for B-cell NHLs associated with pSS probably apply more for the MALT lymphoma subtype since, after all, it is the prevalent one. Given the unfavorable prognosis of high-grade B-cell NHLS, an effort to define distinct biomarkers for distinct pSS-related lymphoma subtypes should be considered, especially in the case of the more aggressive DLBCL subtype. We should note though that this may be practically difficult due to the fewer reported cases compared to MALT lymphoma.
Even though the published literature extensively focuses on predisposing factors for pSS-related lymphoma, few studies describe in detail the clinical picture of the different B-cell NHL subtypes, which irrefutably shows marked differences. A better description of the distinct clinical picture of each B-cell NHL subtyped might lead to increased awareness for clinicians and support more effective lymphoma staging approaches. Furthermore, there is a paucity of data regarding the “standard-of-care” treatment for lymphomas developing in the setting of pSS. Despite the favorable prognosis and satisfactory OS of pSS-lymphoma patients, different therapeutic approaches are currently being applied. This prompts us to consider the need for comparison between therapeutic regimens to conclude on which is the right timing for therapy and the more efficient therapeutic approach in terms both of EFS and OS but also in terms of minimal toxicity and therapy-related complications.
Author Contributions
Conceptualization, A.G.T. and M.V; investigation, I.E.S.; writing—original draft preparation, I.E.S.; writing—review and editing, A.G.T., A.V.G. and M.V.; visualization, I.E.S.; supervision, A.G.T., A.V.G. and M.V. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Goules, A.V.; Tzioufas, A.G. Lymphomagenesis in Sjögren’s syndrome: Predictive biomarkers towards precision medicine. Autoimmun. Rev. 2019, 18, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Stergiou, I.E.; Poulaki, A.; Voulgarelis, M. Pathogenetic Mechanisms Implicated in Sjögren’s Syndrome Lymphomagenesis: A Review of the Literature. J. Clin. Med. 2020, 9, 3794. [Google Scholar] [CrossRef] [PubMed]
- Christodoulou, M.I.; Kapsogeorgou, E.K.; Moutsopoulos, H.M. Characteristics of the minor salivary gland infiltrates in Sjögren’s syndrome. J. Autoimmun. 2010, 34, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Goules, A.V.; Tzioufas, A.G. Primary Sjӧgren’s syndrome: Clinical phenotypes, outcome and the development of biomarkers. Autoimmun. Rev. 2016, 15, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Molina, G.; Kostov, B.; Brito-Zerón, P.; Vissink, A.; Mandl, T.; Hinrichs, A.C.; Quartuccio, L.; Baldini, C.; Seror, R.; Szántó, A.; et al. Characterization and outcomes of 414 patients with primary SS who developed hematological malignancies. Rheumatology 2022. [Google Scholar] [CrossRef]
- Theander, E.; Henriksson, G.; Ljungberg, O.; Mandl, T.; Manthorpe, R.; Jacobsson, L.T. Lymphoma and other malignancies in primary Sjögren’s syndrome: A cohort study on cancer incidence and lymphoma predictors. Ann. Rheum. Dis. 2006, 65, 796–803. [Google Scholar] [CrossRef] [Green Version]
- Stergiou, I.E.; Papageorgiou, A.; Chatzis, L.G.; Tzioufas, A.G.; Voulgarelis, M.; Goules, A. T cell lymphoma in the setting of Sjögren’s syndrome: T cells gone bad? Report of five cases from a single centre cohort. Clin. Exp. Rheumatol. 2020, 38 (Suppl. 126), 125–129. [Google Scholar]
- Swerdlow, S.H.C.E.; Harris, N.L.; Jaffe, E.S.; Pileri, S.A.; Stein, H.; Thiele, J. World Health Organization Classification of Tumours of Haematopoietic and Lymphoid Tissues, 4th ed.; IARC: Lyon, France, 2017. [Google Scholar]
- Baimpa, E.; Dahabreh, I.J.; Voulgarelis, M.; Moutsopoulos, H.M. Hematologic manifestations and predictors of lymphoma development in primary Sjögren syndrome: Clinical and pathophysiologic aspects. Medicine 2009, 88, 284–293. [Google Scholar] [CrossRef]
- Brito-Zerón, P.; Kostov, B.; Fraile, G.; Caravia-Durán, D.; Maure, B.; Rascón, F.-J.; Zamora, M.; Casanovas, A.; Lopez-Dupla, M.; Ripoll, M.; et al. Characterization and risk estimate of cancer in patients with primary Sjögren syndrome. J. Hematol. Oncol. 2017, 10, 90. [Google Scholar] [CrossRef] [Green Version]
- Chatzis, L.G.; Stergiou, I.E.; Goules, A.V.; Pezoulas, V.; Tsourouflis, G.; Fotiadis, D.; Tzioufas, A.G.; Voulgarelis, M. Clinical picture, outcome and predictive factors of lymphoma in primary Sjögren’s syndrome: Results from a harmonized dataset (1981–2021). Rheumatology 2022, 61, 3576–3585. [Google Scholar] [CrossRef]
- Papageorgiou, A.; Ziogas, D.C.; Mavragani, C.P.; Zintzaras, E.; Tzioufas, A.G.; Moutsopoulos, H.M.; Voulgarelis, M. Predicting the outcome of Sjogren’s syndrome-associated non-hodgkin’s lymphoma patients. PLoS ONE 2015, 10, e0116189. [Google Scholar] [CrossRef] [PubMed]
- Schenone, L.N.; Pellet, A.C.; Mamani, M.; Melo, F.; Adrover, M.; Barreira, J.; Dermarchi, J.; Escobar, C.S.; Santiago, L.; Salvatierra, G.; et al. Development of lymphoma in patients with primary Sjögren Syndrome OMICS Publishing Group. Int. J. Clin. Rheumatol. 2019, 14, 69–74. [Google Scholar]
- Voulgarelis, M.; Dafni, U.G.; Isenberg, D.A.; Moutsopoulos, H.M. Malignant lymphoma in primary Sjögren’s syndrome: A multicenter, retrospective, clinical study by the European Concerted Action on Sjögren’s Syndrome. Arthritis Rheum. 1999, 42, 1765–1772. [Google Scholar] [CrossRef]
- Voulgarelis, M.; Ziakas, P.D.; Papageorgiou, A.; Baimpa, E.; Tzioufas, A.G.; Moutsopoulos, H.M. Prognosis and outcome of non-Hodgkin lymphoma in primary Sjögren syndrome. Medicine 2012, 91, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Royer, B.; Cazals-Hatem, D.; Sibilia, J.; Agbalika, F.; Cayuela, J.-M.; Soussi, T.; Maloisel, F.d.r.; Clauvel, J.-P.; Brouet, J.-C.; Mariette, X. Lymphomas in Patients with Sjögren’s Syndrome Are Marginal Zone B-Cell Neoplasms, Arise in Diverse Extranodal and Nodal Sites, and Are Not Associated with Viruses. Blood 1997, 90, 766–775. [Google Scholar] [CrossRef] [Green Version]
- Vasaitis, L.; Nordmark, G.; Theander, E.; Backlin, C.; Smedby, K.E.; Askling, J.; Rönnblom, L.; Sundström, C.; Baecklund, E. Population-based study of patients with primary Sjögren’s syndrome and lymphoma: Lymphoma subtypes, clinical characteristics, and gender differences. Scand. J. Rheumatol. 2020, 49, 225–232. [Google Scholar] [CrossRef]
- Solans-Laqué, R.; López-Hernandez, A.; Bosch-Gil, J.A.; Palacios, A.; Campillo, M.; Vilardell-Tarres, M. Risk, predictors, and clinical characteristics of lymphoma development in primary Sjögren’s syndrome. Semin. Arthritis Rheum. 2011, 41, 415–423. [Google Scholar] [CrossRef]
- Tapinos, N.I.; Polihronis, M.; Moutsopoulos, H.M. Lymphoma development in Sjögren’s syndrome: Novel p53 mutations. Arthritis Rheum. 1999, 42, 1466–1472. [Google Scholar] [CrossRef]
- Pisa, E.K.; Pisa, P.; Kang, H.I.; Fox, R.I. High frequency of t(14;18) translocation in salivary gland lymphomas from Sjögren’s syndrome patients. J. Exp. Med. 1991, 174, 1245–1250. [Google Scholar] [CrossRef] [Green Version]
- Ihrler, S.; Baretton, G.B.; Menauer, F.; Blasenbreu-Vogt, S.; Löhrs, U. Sjögren’s Syndrome and MALT Lymphomas of Salivary Glands: A DNA-Cytometric and Interphase-Cytogenetic Study. Mod. Pathol. 2000, 13, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Alamanos, Y.; Tsifetaki, N.; Voulgari, P.V.; Venetsanopoulou, A.I.; Siozos, C.; Drosos, A.A. Epidemiology of primary Sjögren’s syndrome in north-west Greece, 1982–2003. Rheumatology 2006, 45, 187–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brito-Zerón, P.; Kostov, B.; Solans, R.; Fraile, G.; Suárez-Cuervo, C.; Casanovas, A.; Rascón, F.J.; Qanneta, R.; Pérez-Alvarez, R.; Ripoll, M.; et al. Systemic activity and mortality in primary Sjögren syndrome: Predicting survival using the EULAR-SS Disease Activity Index (ESSDAI) in 1045 patients. Ann. Rheum. Dis. 2016, 75, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, J.P.; Vassiliou, V.A.; Moutsopoulos, H.M. Long-term risk of mortality and lymphoproliferative disease and predictive classification of primary Sjögren’s syndrome. Arthritis Rheum. 2002, 46, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Skopouli, F.N.; Dafni, U.; Ioannidis, J.P.; Moutsopoulos, H.M. Clinical evolution, and morbidity and mortality of primary Sjögren’s syndrome. Semin. Arthritis Rheum. 2000, 29, 296–304. [Google Scholar] [CrossRef]
- Theander, E.; Manthorpe, R.; Jacobsson, L.T. Mortality and causes of death in primary Sjögren’s syndrome: A prospective cohort study. Arthritis Rheum. 2004, 50, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Casals, M.; Cervera, R.; Font, J.; Garcóa-Carrasco, M.; Espinosa, G.; Reino, S.; Pallarés, L.; Ingelmo, M. Young Onset of Primary Sjögren’s syndrome: Clinical and immunological characteristics. Lupus 1998, 7, 202–206. [Google Scholar] [CrossRef]
- Brito-Zerón, P.; Ramos-Casals, M.; Bove, A.; Sentis, J.; Font, J. Predicting adverse outcomes in primary Sjogren’s syndrome: Identification of prognostic factors. Rheumatology 2007, 46, 1359–1362. [Google Scholar] [CrossRef] [Green Version]
- Fragkioudaki, S.; Mavragani, C.P.; Moutsopoulos, H.M. Predicting the risk for lymphoma development in Sjogren syndrome: An easy tool for clinical use. Medicine 2016, 95, e3766. [Google Scholar] [CrossRef]
- Johnsen, S.J.; Brun, J.G.; Gøransson, L.G.; Småstuen, M.C.; Johannesen, T.B.; Haldorsen, K.; Harboe, E.; Jonsson, R.; Meyer, P.A.; Omdal, R. Risk of non-Hodgkin’s lymphoma in primary Sjögren’s syndrome: A population-based study. Arthritis Care Res. 2013, 65, 816–821. [Google Scholar] [CrossRef]
- Goules, A.V.; Argyropoulou, O.D.; Pezoulas, V.C.; Chatzis, L.; Critselis, E.; Gandolfo, S.; Ferro, F.; Binutti, M.; Donati, V.; Zandonella Callegher, S.; et al. Primary Sjögren’s Syndrome of Early and Late Onset: Distinct Clinical Phenotypes and Lymphoma Development. Front. Immunol. 2020, 11, 594096. [Google Scholar] [CrossRef]
- Baldini, C.; Pepe, P.; Luciano, N.; Ferro, F.; Talarico, R.; Grossi, S.; Tavoni, A.; Bombardieri, S. A clinical prediction rule for lymphoma development in primary Sjögren’s syndrome. J. Rheumatol. 2012, 39, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Chatzis, L.; Pezoulas, V.C.; Ferro, F.; Gandolfo, S.; Donati, V.; Binutti, M.; Callegher, S.Z.; Venetsanopoulou, A.; Zampeli, E.; Mavrommati, M.; et al. Sjögren’s Syndrome: The Clinical Spectrum of Male Patients. J. Clin. Med. 2020, 9, 2620. [Google Scholar] [CrossRef] [PubMed]
- Ramírez Sepúlveda, J.I.; Kvarnström, M.; Eriksson, P.; Mandl, T.; Norheim, K.B.; Johnsen, S.J.; Hammenfors, D.; Jonsson, M.V.; Skarstein, K.; Brun, J.G.; et al. Long-term follow-up in primary Sjögren’s syndrome reveals differences in clinical presentation between female and male patients. Biol. Sex Differ. 2017, 8, 25. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.Y.; Huang, Y.T.; Liu, M.F.; Lu, T.H. Incidence of cancer in a nationwide population cohort of 7852 patients with primary Sjogren’s syndrome in Taiwan. Ann. Rheum. Dis. 2012, 71, 524–527. [Google Scholar] [CrossRef] [Green Version]
- Thandra, K.C.; Barsouk, A.; Saginala, K.; Padala, S.A.; Barsouk, A.; Rawla, P. Epidemiology of Non-Hodgkin’s Lymphoma. Med. Sci. 2021, 9, 5. [Google Scholar] [CrossRef] [PubMed]
- Gondran, G.; Fauchais, A.; Lambert, M.; Ly, K.; Launay, D.; Queyrel, V.; Benazahari, H.; Liozon, E.; Loustaud-Ratti, V.; Hachulla, E.; et al. Primary Sjogren’s syndrome in men. Scand. J. Rheumatol. 2008, 37, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Kassan, S.S.; Thomas, T.L.; Moutsopoulos, H.M.; Hoover, R.; Kimberly, R.P.; Budman, D.R.; Costa, J.; Decker, J.L.; Chused, T.M. Increased risk of lymphoma in sicca syndrome. Ann. Intern. Med. 1978, 89, 888–892. [Google Scholar] [CrossRef]
- Abrol, E.; González-Pulido, C.; Praena-Fernández, J.M.; Isenberg, D.A. A retrospective study of long-term outcomes in 152 patients with primary Sjogren’s syndrome: 25-year experience. Clin. Med. 2014, 14, 157–164. [Google Scholar] [CrossRef]
- De Vita, S.; Gandolfo, S.; Zandonella Callegher, S.; Zabotti, A.; Quartuccio, L. The evaluation of disease activity in Sjögren’s syndrome based on the degree of MALT involvement: Glandular swelling and cryoglobulinaemia compared to ESSDAI in a cohort study. Clin. Exp. Rheumatol. 2018, 36 (Suppl. 112), 150–156. [Google Scholar]
- Nocturne, G.; Virone, A.; Ng, W.F.; Le Guern, V.; Hachulla, E.; Cornec, D.; Daien, C.; Vittecoq, O.; Bienvenu, B.; Marcelli, C.; et al. Rheumatoid Factor and Disease Activity Are Independent Predictors of Lymphoma in Primary Sjögren’s Syndrome. Arthritis Rheumatol. 2016, 68, 977–985. [Google Scholar] [CrossRef] [Green Version]
- Quartuccio, L.; Isola, M.; Baldini, C.; Priori, R.; Bartoloni Bocci, E.; Carubbi, F.; Maset, M.; Gregoraci, G.; Della Mea, V.; Salvin, S.; et al. Biomarkers of lymphoma in Sjögren’s syndrome and evaluation of the lymphoma risk in prelymphomatous conditions: Results of a multicenter study. J. Autoimmun. 2014, 51, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Risselada, A.P.; Kruize, A.A.; Bijlsma, J.W. Clinical features distinguishing lymphoma development in primary Sjögren’s Syndrome--a retrospective cohort study. Semin. Arthritis Rheum. 2013, 43, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe, N.; Inanc, M.; Speight, P.; Isenberg, D. Predictors of lymphoma development in primary Sjögren’s syndrome. Semin. Arthritis Rheum. 1998, 28, 80–87. [Google Scholar] [CrossRef]
- Zhang, W.; Feng, S.; Yan, S.; Zhao, Y.; Li, M.; Sun, J.; Zhang, F.C.; Cui, Q.; Dong, Y. Incidence of malignancy in primary Sjogren’s syndrome in a Chinese cohort. Rheumatology 2010, 49, 571–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Vita, S.; Isola, M.; Baldini, C.; Goules, A.V.; Chatzis, L.G.; Quartuccio, L.; Zabotti, A.; Giovannini, I.; Donati, V.; Ferro, F.; et al. Predicting lymphoma in Sjögren’s syndrome and the pathogenetic role of parotid microenvironment through precise parotid swelling recording. Rheumatology 2022. [Google Scholar] [CrossRef]
- Goules, A.; Masouridi, S.; Tzioufas, A.G.; Ioannidis, J.P.; Skopouli, F.N.; Moutsopoulos, H.M. Clinically significant and biopsy-documented renal involvement in primary Sjögren syndrome. Medicine 2000, 79, 241–249. [Google Scholar] [CrossRef]
- Tzioufas, A.G.; Manoussakis, M.N.; Costello, R.; Silis, M.; Papadopoulos, N.M.; Moutsopoulos, H.M. Cryoglobulinemia in autoimmune rheumatic diseases. Evidence of circulating monoclonal cryoglobulins in patients with primary Sjögren’s syndrome. Arthritis Rheum. 1986, 29, 1098–1104. [Google Scholar] [CrossRef]
- Tzioufas, A.G.; Boumba, D.S.; Skopouli, F.N.; Moutsopoulos, H.M. Mixed monoclonal cryoglobulinemia and monoclonal rheumatoid factor cross-reactive idiotypes as predictive factors for the development of lymphoma in primary Sjögren’s syndrome. Arthritis Rheum. 1996, 39, 767–772. [Google Scholar] [CrossRef]
- Baldini, C.; Pepe, P.; Quartuccio, L.; Priori, R.; Bartoloni, E.; Alunno, A.; Gattamelata, A.; Maset, M.; Modesti, M.; Tavoni, A.; et al. Primary Sjogren’s syndrome as a multi-organ disease: Impact of the serological profile on the clinical presentation of the disease in a large cohort of Italian patients. Rheumatology 2014, 53, 839–844. [Google Scholar] [CrossRef] [Green Version]
- Martel, C.; Gondran, G.; Launay, D.; Lalloué, F.; Palat, S.; Lambert, M.; Ly, K.; Loustaud-Ratti, V.; Bezanahary, H.; Hachulla, E.; et al. Active immunological profile is associated with systemic Sjögren’s syndrome. J. Clin. Immunol. 2011, 31, 840–847. [Google Scholar] [CrossRef]
- Nocturne, G.; Seror, R.; Fogel, O.; Belkhir, R.; Boudaoud, S.; Saraux, A.; Larroche, C.; Le Guern, V.; Gottenberg, J.E.; Mariette, X. CXCL13 and CCL11 Serum Levels and Lymphoma and Disease Activity in Primary Sjögren’s Syndrome. Arthritis Rheumatol. 2015, 67, 3226–3233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Argyropoulou, O.D.; Pezoulas, V.; Chatzis, L.; Critselis, E.; Gandolfo, S.; Ferro, F.; Quartuccio, L.; Donati, V.; Treppo, E.; Bassoli, C.R.; et al. Cryoglobulinemic vasculitis in primary Sjögren’s Syndrome: Clinical presentation, association with lymphoma and comparison with Hepatitis C-related disease. Semin. Arthritis Rheum. 2020, 50, 846–853. [Google Scholar] [CrossRef] [PubMed]
- Theander, E.; Vasaitis, L.; Baecklund, E.; Nordmark, G.; Warfvinge, G.; Liedholm, R.; Brokstad, K.; Jonsson, R.; Jonsson, M.V. Lymphoid organisation in labial salivary gland biopsies is a possible predictor for the development of malignant lymphoma in primary Sjögren’s syndrome. Ann. Rheum. Dis. 2011, 70, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Gottenberg, J.E.; Seror, R.; Miceli-Richard, C.; Benessiano, J.; Devauchelle-Pensec, V.; Dieude, P.; Dubost, J.J.; Fauchais, A.L.; Goeb, V.; Hachulla, E.; et al. Serum levels of beta2-microglobulin and free light chains of immunoglobulins are associated with systemic disease activity in primary Sjögren’s syndrome. Data at enrollment in the prospective ASSESS cohort. PLoS ONE 2013, 8, e59868. [Google Scholar] [CrossRef] [Green Version]
- Walters, M.T.; Stevenson, F.K.; Herbert, A.; Cawley, M.I.; Smith, J.L. Urinary monoclonal free light chains in primary Sjögren’s syndrome: An aid to the diagnosis of malignant lymphoma. Ann. Rheum. Dis. 1986, 45, 210–219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatzis, L.G.; Pezoulas, V.; Voulgari, P.V.; Baldini, C.; Exarchos, T.P.; Fotiadis, D.I.; Mavragani, C.P.; Skopouli, F.N.; Moutsopoulos, H.M.; Tzioufas, A.G.; et al. Combined seronegativity in Sjögren’s syndrome. Clin. Exp. Rheumatol. 2021, 39 (Suppl. 133), 80–84. [Google Scholar] [CrossRef]
- Quartuccio, L.; Baldini, C.; Bartoloni, E.; Priori, R.; Carubbi, F.; Corazza, L.; Alunno, A.; Colafrancesco, S.; Luciano, N.; Giacomelli, R.; et al. Anti-SSA/SSB-negative Sjögren’s syndrome shows a lower prevalence of lymphoproliferative manifestations, and a lower risk of lymphoma evolution. Autoimmun. Rev. 2015, 14, 1019–1022. [Google Scholar] [CrossRef]
- Pertovaara, M.; Pukkala, E.; Laippala, P.; Miettinen, A.; Pasternack, A. A longitudinal cohort study of Finnish patients with primary Sjögren’s syndrome: Clinical, immunological, and epidemiological aspects. Ann. Rheum. Dis. 2001, 60, 467–472. [Google Scholar] [CrossRef] [Green Version]
- Agmon-Levin, N.; Kivity, S.; Tzioufas, A.G.; López Hoyos, M.; Rozman, B.; Efes, I.; Shapira, Y.; Shamis, A.; Amital, H.; Youinou, P.; et al. Low levels of vitamin-D are associated with neuropathy and lymphoma among patients with Sjögren’s syndrome. J. Autoimmun. 2012, 39, 234–239. [Google Scholar] [CrossRef]
- Gerli, R.; Muscat, C.; Giansanti, M.; Danieli, M.G.; Sciuto, M.; Gabrielli, A.; Fiandra, E.; Vitali, C. Quantitative assessment of salivary gland inflammatory infiltration in primary Sjögren’s syndrome: Its relationship to different demographic, clinical and serological features of the disorder. Br. J. Rheumatol. 1997, 36, 969–975. [Google Scholar] [CrossRef] [Green Version]
- Brito-Zerón, P.; Retamozo, S.; Ramos-Casals, M. Phenotyping Sjögren’s syndrome: Towards a personalised management of the disease. Clin. Exp. Rheumatol. 2018, 36 (Suppl. 112), 198–209. [Google Scholar] [PubMed]
- Carubbi, F.; Alunno, A.; Cipriani, P.; Bartoloni, E.; Baldini, C.; Quartuccio, L.; Priori, R.; Valesini, G.; De Vita, S.; Bombardieri, S.; et al. A retrospective, multicenter study evaluating the prognostic value of minor salivary gland histology in a large cohort of patients with primary Sjögren’s syndrome. Lupus 2015, 24, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Risselada, A.P.; Kruize, A.A.; Goldschmeding, R.; Lafeber, F.P.; Bijlsma, J.W.; van Roon, J.A. The prognostic value of routinely performed minor salivary gland assessments in primary Sjögren’s syndrome. Ann. Rheum. Dis. 2014, 73, 1537–1540. [Google Scholar] [CrossRef]
- Chatzis, L.; Goules, A.V.; Pezoulas, V.; Baldini, C.; Gandolfo, S.; Skopouli, F.N.; Exarchos, T.P.; Kapsogeorgou, E.K.; Donati, V.; Voulgari, P.V.; et al. A biomarker for lymphoma development in Sjogren’s syndrome: Salivary gland focus score. J. Autoimmun. 2021, 121, 102648. [Google Scholar] [CrossRef] [PubMed]
- Haacke, E.A.; van der Vegt, B.; Vissink, A.; Spijkervet, F.K.L.; Bootsma, H.; Kroese, F.G.M. Germinal centres in diagnostic labial gland biopsies of patients with primary Sjögren’s syndrome are not predictive for parotid MALT lymphoma development. Ann. Rheum. Dis. 2017, 76, 1781–1784. [Google Scholar] [CrossRef] [PubMed]
- Mossel, E.; Delli, K.; van Nimwegen, J.F.; Stel, A.J.; Kroese, F.G.M.; Spijkervet, F.K.L.; Vissink, A.; Arends, S.; Bootsma, H. Ultrasonography of major salivary glands compared with parotid and labial gland biopsy and classification criteria in patients with clinically suspected primary Sjögren’s syndrome. Ann. Rheum. Dis. 2017, 76, 1883–1889. [Google Scholar] [CrossRef] [PubMed]
- Pijpe, J.; Kalk, W.W.I.; van der Wal, J.E.; Vissink, A.; Kluin, P.M.; Roodenburg, J.L.N.; Bootsma, H.; Kallenberg, C.G.M.; Spijkervet, F.K.L. Parotid gland biopsy compared with labial biopsy in the diagnosis of patients with primary Sjögren’s syndrome. Rheumatology 2007, 46, 335–341. [Google Scholar] [CrossRef] [Green Version]
- van Nimwegen, J.F.; van Ginkel, M.S.; Arends, S.; Haacke, E.A.; van der Vegt, B.; Sillevis Smitt-Kamminga, N.; Spijkervet, F.K.L.; Kroese, F.G.M.; Stel, A.J.; Brouwer, E.; et al. Validation of the ACR-EULAR criteria for primary Sjögren’s syndrome in a Dutch prospective diagnostic cohort. Rheumatology 2018, 57, 818–825. [Google Scholar] [CrossRef] [Green Version]
- Voulgarelis, M.; Moutsopoulos, H.M. Lymphoproliferation in autoimmunity and Sjögren’s syndrome. Curr. Rheumatol. Rep. 2003, 5, 317–323. [Google Scholar] [CrossRef]
- Stott, D.I.; Hiepe, F.; Hummel, M.; Steinhauser, G.; Berek, C. Antigen-driven clonal proliferation of B cells within the target tissue of an autoimmune disease. The salivary glands of patients with Sjögren’s syndrome. J. Clin. Investig. 1998, 102, 938–946. [Google Scholar] [CrossRef] [Green Version]
- Jordan, R.C.; Masaki, Y.; Takeshita, S.; Speight, P.M.; Sugai, S. High prevalence of B-cell monoclonality in labial gland biopsies of Japanese Sjögren’s syndrome patients. Int. J. Hematol. 1996, 64, 47–52. [Google Scholar] [CrossRef]
- Miklos, J.A.; Swerdlow, S.H.; Bahler, D.W. Salivary gland mucosa-associated lymphoid tissue lymphoma immunoglobulin VH genes show frequent use of V1-69 with distinctive CDR3 features. Blood 2000, 95, 3878–3884. [Google Scholar] [CrossRef] [PubMed]
- Visser, A.; Verstappen, G.M.; van der Vegt, B.; Vissink, A.; Bende, R.J.; Bootsma, H.; Bos, N.A.; Kroese, F.G.M. Repertoire Analysis of B-Cells Located in Striated Ducts of Salivary Glands of Patients with Sjögren’s Syndrome. Front. Immunol. 2020, 11, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Bende, R.J.; Janssen, J.; Beentjes, A.; Wormhoudt, T.A.M.; Wagner, K.; Haacke, E.A.; Kroese, F.G.M.; Guikema, J.E.J.; van Noesel, C.J.M. Salivary Gland Mucosa-Associated Lymphoid Tissue-Type Lymphoma From Sjögren’s Syndrome Patients in the Majority Express Rheumatoid Factors Affinity-Selected for IgG. Arthritis Rheumatol. 2020, 72, 1330–1340. [Google Scholar] [CrossRef]
- Bende, R.J.; Aarts, W.M.; Riedl, R.G.; de Jong, D.; Pals, S.T.; van Noesel, C.J. Among B cell non-Hodgkin’s lymphomas, MALT lymphomas express a unique antibody repertoire with frequent rheumatoid factor reactivity. J. Exp. Med. 2005, 201, 1229–1241. [Google Scholar] [CrossRef]
- Broeren, M.G.A.; Wang, J.J.; Balzaretti, G.; Groenen, P.; van Schaik, B.D.C.; Chataway, T.; Kaffa, C.; Bervoets, S.; Hebeda, K.M.; Bounova, G.; et al. Proteogenomic analysis of the autoreactive B cell repertoire in blood and tissues of patients with Sjögren’s syndrome. Ann. Rheum. Dis. 2022, 81, 644–652. [Google Scholar] [CrossRef]
- Bende, R.J.; Slot, L.M.; Hoogeboom, R.; Wormhoudt, T.A.; Adeoye, A.O.; Guikema, J.E.; van Noesel, C.J. Stereotypic rheumatoid factors that are frequently expressed in mucosa-associated lymphoid tissue-type lymphomas are rare in the labial salivary glands of patients with Sjögren’s syndrome. Arthritis Rheumatol. 2015, 67, 1074–1083. [Google Scholar] [CrossRef]
- Singh, M.; Jackson, K.J.L.; Wang, J.J.; Schofield, P.; Field, M.A.; Koppstein, D.; Peters, T.J.; Burnett, D.L.; Rizzetto, S.; Nevoltris, D.; et al. Lymphoma Driver Mutations in the Pathogenic Evolution of an Iconic Human Autoantibody. Cell 2020, 180, 878–894.e819. [Google Scholar] [CrossRef]
- De Vita, S.; Boiocchi, M.; Sorrentino, D.; Carbone, A.; Avellini, C.; Dolcetti, R.; Marzotto, A.; Gloghini, A.; Bartoli, E.; Beltrami, C.A.; et al. Characterization of prelymphomatous stages of B cell lymphoproliferation in Sjögren’s syndrome. Arthritis Rheum. 1997, 40, 318–331. [Google Scholar] [CrossRef]
- De Re, V.; De Vita, S.; Gasparotto, D.; Marzotto, A.; Carbone, A.; Ferraccioli, G.; Boiocchi, M. Salivary gland B cell lymphoproliferative disorders in Sjögren’s syndrome present a restricted use of antigen receptor gene segments similar to those used by hepatitis C virus-associated non-Hodgkins’s lymphomas. Eur. J. Immunol. 2002, 32, 903–910. [Google Scholar] [CrossRef]
- Carbone, A.; Gloghini, A.; Ferlito, A. Pathological features of lymphoid proliferations of the salivary glands: Lymphoepithelial sialadenitis versus low-grade B-cell lymphoma of the malt type. Ann. Otol. Rhinol. Laryngol. 2000, 109, 1170–1175. [Google Scholar] [CrossRef] [PubMed]
- Haacke, E.A.; van der Vegt, B.; Vissink, A.; Spijkervet, F.K.L.; Bootsma, H.; Kroese, F.G.M. Germinal Centers in Diagnostic Biopsies of Patients with Primary Sjögren’s Syndrome Are Not a Risk Factor for Non-Hodgkin’s Lymphoma but a Reflection of High Disease Activity: Comment on the Article by Sène et al. Arthritis Rheumatol. 2019, 71, 170–171. [Google Scholar] [CrossRef] [PubMed]
- Risselada, A.P.; Looije, M.F.; Kruize, A.A.; Bijlsma, J.W.; van Roon, J.A. The role of ectopic germinal centers in the immunopathology of primary Sjögren’s syndrome: A systematic review. Semin. Arthritis Rheum. 2013, 42, 368–376. [Google Scholar] [CrossRef] [PubMed]
- Sène, D.; Ismael, S.; Forien, M.; Charlotte, F.; Kaci, R.; Cacoub, P.; Diallo, A.; Dieudé, P.; Lioté, F. Ectopic Germinal Center-Like Structures in Minor Salivary Gland Biopsy Tissue Predict Lymphoma Occurrence in Patients with Primary Sjögren’s Syndrome. Arthritis Rheumatol. 2018, 70, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, S.J.; Berget, E.; Jonsson, M.V.; Helgeland, L.; Omdal, R.; Jonsson, R. Evaluation of germinal center-like structures and B cell clonality in patients with primary Sjögren syndrome with and without lymphoma. J. Rheumatol. 2014, 41, 2214–2222. [Google Scholar] [CrossRef]
- Nakshbandi, U.; Haacke, E.A.; Bootsma, H.; Vissink, A.; Spijkervet, F.K.L.; van der Vegt, B.; Kroese, F.G.M. Bcl6 for identification of germinal centres in salivary gland biopsies in primary Sjögren’s syndrome. Oral Dis. 2020, 26, 707–710. [Google Scholar] [CrossRef]
- Barone, F.; Bombardieri, M.; Manzo, A.; Blades, M.C.; Morgan, P.R.; Challacombe, S.J.; Valesini, G.; Pitzalis, C. Association of CXCL13 and CCL21 expression with the progressive organization of lymphoid-like structures in Sjögren’s syndrome. Arthritis Rheum. 2005, 52, 1773–1784. [Google Scholar] [CrossRef]
- Bombardieri, M.; Barone, F.; Humby, F.; Kelly, S.; McGurk, M.; Morgan, P.; Challacombe, S.; De Vita, S.; Valesini, G.; Spencer, J.; et al. Activation-induced cytidine deaminase expression in follicular dendritic cell networks and interfollicular large B cells supports functionality of ectopic lymphoid neogenesis in autoimmune sialoadenitis and MALT lymphoma in Sjögren’s syndrome. J. Immunol. 2007, 179, 4929–4938. [Google Scholar] [CrossRef] [Green Version]
- Delli, K.; Haacke, E.A.; Ihrler, S.; van der Vegt, B.; Vissink, A.; Bootsma, H.; Spijkervet, F.K.; Kroese, F.G. Need for consensus guidelines to standardise the assessment of germinal centres and other histopathological parameters in salivary gland tissue of patients with primary Sjögren’s syndrome. Ann. Rheum. Dis. 2016, 75, e32. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, M.V.; Skarstein, K. Follicular dendritic cells confirm lymphoid organization in the minor salivary glands of primary Sjögren’s syndrome. J. Oral Pathol. Med. 2008, 37, 515–521. [Google Scholar] [CrossRef]
- Fisher, B.A.; Jonsson, R.; Daniels, T.; Bombardieri, M.; Brown, R.M.; Morgan, P.; Bombardieri, S.; Ng, W.-F.; Tzioufas, A.G.; Vitali, C.; et al. Standardisation of labial salivary gland histopathology in clinical trials in primary Sjögren’s syndrome. Ann. Rheum. Dis. 2017, 76, 1161–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barone, F.; Bombardieri, M.; Rosado, M.M.; Morgan, P.R.; Challacombe, S.J.; De Vita, S.; Carsetti, R.; Spencer, J.; Valesini, G.; Pitzalis, C. CXCL13, CCL21, and CXCL12 expression in salivary glands of patients with Sjogren’s syndrome and MALT lymphoma: Association with reactive and malignant areas of lymphoid organization. J. Immunol. 2008, 180, 5130–5140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quartuccio, L.; Baldini, C.; Bartoloni, E.; Priori, R.; Carubbi, F.; Alunno, A.; Gandolfo, S.; Gattamelata, A.; Giacomelli, R.; Gerli, R.; et al. Correlation between ESSDAI and ClinESSDAI in a real-life cohort of patients with Sjögren’s syndrome. Clin. Exp. Rheumatol. 2017, 35, 546–547. [Google Scholar] [CrossRef] [PubMed]
- Mackay, F.; Groom, J.R.; Tangye, S.G. An important role for B-cell activation factor and B cells in the pathogenesis of Sjögren’s syndrome. Curr. Opin. Rheumatol. 2007, 19, 406–413. [Google Scholar] [CrossRef]
- Nezos, A.; Papageorgiou, A.; Fragoulis, G.; Ioakeimidis, D.; Koutsilieris, M.; Tzioufas, A.G.; Moutsopoulos, H.M.; Voulgarelis, M.; Mavragani, C.P. B-cell activating factor genetic variants in lymphomagenesis associated with primary Sjogren’s syndrome. J. Autoimmun. 2014, 51, 89–98. [Google Scholar] [CrossRef]
- Papageorgiou, A.; Mavragani, C.P.; Nezos, A.; Zintzaras, E.; Quartuccio, L.; De Vita, S.; Koutsilieris, M.; Tzioufas, A.G.; Moutsopoulos, H.M.; Voulgarelis, M. A BAFF receptor His159Tyr mutation in Sjögren’s syndrome-related lymphoproliferation. Arthritis Rheumatol. 2015, 67, 2732–2741. [Google Scholar] [CrossRef]
- Hildebrand, J.M.; Luo, Z.; Manske, M.K.; Price-Troska, T.; Ziesmer, S.C.; Lin, W.; Hostager, B.S.; Slager, S.L.; Witzig, T.E.; Ansell, S.M.; et al. A BAFF-R mutation associated with non-Hodgkin lymphoma alters TRAF recruitment and reveals new insights into BAFF-R signaling. J. Exp. Med. 2010, 207, 2569–2579. [Google Scholar] [CrossRef]
- Nocturne, G.; Boudaoud, S.; Miceli-Richard, C.; Viengchareun, S.; Lazure, T.; Nititham, J.; Taylor, K.E.; Ma, A.; Busato, F.; Melki, J.; et al. Germline and somatic genetic variations of TNFAIP3 in lymphoma complicating primary Sjogren’s syndrome. Blood 2013, 122, 4068–4076. [Google Scholar] [CrossRef]
- Nezos, A.; Gkioka, E.; Koutsilieris, M.; Voulgarelis, M.; Tzioufas, A.G.; Mavragani, C.P. TNFAIP3 F127C Coding Variation in Greek Primary Sjogren’s Syndrome Patients. J. Immunol. Res. 2018, 2018, 6923213. [Google Scholar] [CrossRef] [Green Version]
- Korona, B.; Korona, D.; Zhao, W.; Wotherspoon, A.C.; Du, M.Q. GPR34 activation potentially bridges lymphoepithelial lesions to genesis of salivary gland MALT lymphoma. Blood 2022, 139, 2186–2197. [Google Scholar] [CrossRef]
- Quintanilla-Martinez, L.; Fend, F. Turning up the heat on salivary gland MALT lymphoma. Blood 2022, 139, 2094–2096. [Google Scholar] [CrossRef] [PubMed]
- Mathijs, B.; Julio Finalet, F.; Thomas, T.; Helena, U.; Lucienne, M.; de Laurence, L.; Daan, D.; Pascal, W.; Xavier, S.; Peter, V.; et al. t(X;14)(p11.4;q32.33) is recurrent in marginal zone lymphoma and up-regulates GPR34. Haematologica 2012, 97, 184–188. [Google Scholar] [CrossRef] [Green Version]
- Moody, S.; Thompson, J.S.; Chuang, S.S.; Liu, H.; Raderer, M.; Vassiliou, G.; Wlodarska, I.; Wu, F.; Cogliatti, S.; Robson, A.; et al. Novel GPR34 and CCR6 mutation and distinct genetic profiles in MALT lymphomas of different sites. Haematologica 2018, 103, 1329–1336. [Google Scholar] [CrossRef] [PubMed]
- Nezos, A.; Makri, P.; Gandolfo, S.; De Vita, S.; Voulgarelis, M.; Crow, M.K.; Mavragani, C.P. TREX1 variants in Sjogren’s syndrome related lymphomagenesis. Cytokine 2020, 132, 154781. [Google Scholar] [CrossRef] [PubMed]
- Argyriou, E.; Nezos, A.; Roussos, P.; Venetsanopoulou, A.; Voulgarelis, M.; Boki, K.; Tzioufas, A.G.; Moutsopoulos, H.M.; Mavragani, C.P. Leukocyte Immunoglobulin-Like Receptor A3 (LILRA3): A Novel Marker for Lymphoma Development among Patients with Young Onset Sjogren’s Syndrome. J. Clin. Med. 2021, 10, 644. [Google Scholar] [CrossRef]
- Colafrancesco, S.; Ciccacci, C.; Priori, R.; Latini, A.; Picarelli, G.; Arienzo, F.; Novelli, G.; Valesini, G.; Perricone, C.; Borgiani, P. STAT4, TRAF3IP2, IL10, and HCP5 Polymorphisms in Sjögren’s Syndrome: Association with Disease Susceptibility and Clinical Aspects. J. Immunol. Res. 2019, 2019, 7682827. [Google Scholar] [CrossRef] [Green Version]
- Gourzi, V.C.; Kapsogeorgou, E.K.; Kyriakidis, N.C.; Tzioufas, A.G. Study of microRNAs (miRNAs) that are predicted to target the autoantigens Ro/SSA and La/SSB in primary Sjögren’s Syndrome. Clin. Exp. Immunol. 2015, 182, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Kapsogeorgou, E.K.; Gourzi, V.C.; Manoussakis, M.N.; Moutsopoulos, H.M.; Tzioufas, A.G. Cellular microRNAs (miRNAs) and Sjögren’s syndrome: Candidate regulators of autoimmune response and autoantigen expression. J. Autoimmun. 2011, 37, 129–135. [Google Scholar] [CrossRef]
- Kapsogeorgou, E.K.; Papageorgiou, A.; Protogerou, A.D.; Voulgarelis, M.; Tzioufas, A.G. Low miR200b-5p levels in minor salivary glands: A novel molecular marker predicting lymphoma development in patients with Sjögren’s syndrome. Ann. Rheum. Dis. 2018, 77, 1200–1207. [Google Scholar] [CrossRef]
- Fragkioudaki, S.; Nezos, A.; Souliotis, V.L.; Chatziandreou, I.; Saetta, A.A.; Drakoulis, N.; Tzioufas, A.G.; Voulgarelis, M.; Sfikakis, P.P.; Koutsilieris, M.; et al. MTHFR gene variants and non-MALT lymphoma development in primary Sjogren’s syndrome. Sci. Rep. 2017, 7, 7354. [Google Scholar] [CrossRef] [Green Version]
- Mavragani, C.P.; Nezos, A.; Sagalovskiy, I.; Seshan, S.; Kirou, K.A.; Crow, M.K. Defective regulation of L1 endogenous retroelements in primary Sjogren’s syndrome and systemic lupus erythematosus: Role of methylating enzymes. J. Autoimmun. 2018, 88, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Sisto, M.; Lisi, S.; Lofrumento, D.D.; Ingravallo, G.; Maiorano, E.; D’Amore, M. A failure of TNFAIP3 negative regulation maintains sustained NF-κB activation in Sjögren’s syndrome. Histochem. Cell Biol. 2011, 135, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Johnsen, S.J.; Gudlaugsson, E.; Skaland, I.; Janssen, E.A.; Jonsson, M.V.; Helgeland, L.; Berget, E.; Jonsson, R.; Omdal, R. Low Protein A20 in Minor Salivary Glands is Associated with Lymphoma in Primary Sjögren’s Syndrome. Scand. J. Immunol. 2016, 83, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Nezos, A.; Gravani, F.; Tassidou, A.; Kapsogeorgou, E.K.; Voulgarelis, M.; Koutsilieris, M.; Crow, M.K.; Mavragani, C.P. Type I and II interferon signatures in Sjogren’s syndrome pathogenesis: Contributions in distinct clinical phenotypes and Sjogren’s related lymphomagenesis. J. Autoimmun. 2015, 63, 47–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinoku, I.I.; Verrou, K.M.; Piperi, E.; Voulgarelis, M.; Moutsopoulos, H.M.; Mavragani, C.P. Interferon (IFN)-stimulated gene 15: A novel biomarker for lymphoma development in Sjögren’s syndrome. J. Autoimmun. 2021, 123, 102704. [Google Scholar] [CrossRef]
- Baldini, C.; Santini, E.; Rossi, C.; Donati, V.; Solini, A. The P2X7 receptor-NLRP3 inflammasome complex predicts the development of non-Hodgkin’s lymphoma in Sjogren’s syndrome: A prospective, observational, single-centre study. J. Intern. Med. 2017, 282, 175–186. [Google Scholar] [CrossRef]
- Vakrakou, A.G.; Boiu, S.; Ziakas, P.D.; Xingi, E.; Boleti, H.; Manoussakis, M.N. Systemic activation of NLRP3 inflammasome in patients with severe primary Sjögren’s syndrome fueled by inflammagenic DNA accumulations. J. Autoimmun. 2018, 91, 23–33. [Google Scholar] [CrossRef]
- Manoussakis, M.N.; Boiu, S.; Korkolopoulou, P.; Kapsogeorgou, E.K.; Kavantzas, N.; Ziakas, P.; Patsouris, E.; Moutsopoulos, H.M. Rates of infiltration by macrophages and dendritic cells and expression of interleukin-18 and interleukin-12 in the chronic inflammatory lesions of Sjögren’s syndrome: Correlation with certain features of immune hyperactivity and factors associated with high risk of lymphoma development. Arthritis Rheum. 2007, 56, 3977–3988. [Google Scholar] [CrossRef]
- Nezos, A.; Skarlis, C.; Psarrou, A.; Markakis, K.; Garantziotis, P.; Papanikolaou, A.; Gravani, F.; Voulgarelis, M.; Tzioufas, A.G.; Koutsilieris, M.; et al. Lipoprotein-Associated Phospholipase A2: A Novel Contributor in Sjögren’s Syndrome-Related Lymphoma? Front. Immunol. 2021, 12, 683623. [Google Scholar] [CrossRef]
- Quartuccio, L.; Salvin, S.; Fabris, M.; Maset, M.; Pontarini, E.; Isola, M.; De Vita, S. BLyS upregulation in Sjogren’s syndrome associated with lymphoproliferative disorders, higher ESSDAI score and B-cell clonal expansion in the salivary glands. Rheumatology 2013, 52, 276–281. [Google Scholar] [CrossRef] [Green Version]
- Antonysamy, M.A.; Thomson, A.W. Flt3 ligand (FL) and its influence on immune reactivity. Cytokine 2000, 12, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Tobón, G.J.; Renaudineau, Y.; Hillion, S.; Cornec, D.; Devauchelle-Pensec, V.; Youinou, P.; Pers, J.O. The Fms-like tyrosine kinase 3 ligand, a mediator of B cell survival, is also a marker of lymphoma in primary Sjögren’s syndrome. Arthritis Rheum. 2010, 62, 3447–3456. [Google Scholar] [CrossRef] [PubMed]
- Tobón, G.J.; Saraux, A.; Gottenberg, J.E.; Quartuccio, L.; Fabris, M.; Seror, R.; Devauchelle-Pensec, V.; Morel, J.; Rist, S.; Mariette, X.; et al. Role of Fms-like tyrosine kinase 3 ligand as a potential biologic marker of lymphoma in primary Sjögren’s syndrome. Arthritis Rheum. 2013, 65, 3218–3227. [Google Scholar] [CrossRef] [PubMed]
- Traianos, E.Y.; Locke, J.; Lendrem, D.; Bowman, S.; Hargreaves, B.; Macrae, V.; Tarn, J.R.; Ng, W.F. Serum CXCL13 levels are associated with lymphoma risk and lymphoma occurrence in primary Sjögren’s syndrome. Rheumatol. Int. 2020, 40, 541–548. [Google Scholar] [CrossRef] [Green Version]
- Allen, C.D.; Ansel, K.M.; Low, C.; Lesley, R.; Tamamura, H.; Fujii, N.; Cyster, J.G. Germinal center dark and light zone organization is mediated by CXCR4 and CXCR5. Nat. Immunol. 2004, 5, 943–952. [Google Scholar] [CrossRef]
- Chatzis, L.; Goules, A.V.; Stergiou, I.E.; Voulgarelis, M.; Tzioufas, A.G.; Kapsogeorgou, E.K. Serum, but Not Saliva, CXCL13 Levels Associate with Infiltrating CXCL13+ Cells in the Minor Salivary Gland Lesions and Other Histologic Parameters in Patients with Sjögren’s Syndrome. Front. Immunol. 2021, 12, 705079. [Google Scholar] [CrossRef]
- Ziegler, S.F.; Artis, D. Sensing the outside world: TSLP regulates barrier immunity. Nat. Immunol. 2010, 11, 289–293. [Google Scholar] [CrossRef]
- Gandolfo, S.; Bulfoni, M.; Fabro, C.; Russi, S.; Sansonno, D.; Di Loreto, C.; Cesselli, D.; De Vita, S. Thymic stromal lymphopoietin expression from benign lymphoproliferation to malignant B-cell lymphoma in primary Sjögren’s syndrome. Clin. Exp. Rheumatol. 2019, 37 (Suppl. 118), 55–64. [Google Scholar]
- Gandolfo, S.; Fabro, C.; Kapsogeorgou, E.; Colafrancesco, S.; Ferro, F.; Bartoloni, E.; Quartuccio, L.; Goules, A.; Priori, R.; Alunno, A.; et al. Validation of thymic stromal lymphopoietin as a biomarker of primary Sjögren’s syndrome and related lymphoproliferation: Results in independent cohorts. Clin. Exp. Rheumatol. 2020, 38 (Suppl. 126), 189–194. [Google Scholar]
- Devauchelle-Pensec, V.; Zabotti, A.; Carvajal-Alegria, G.; Filipovic, N.; Jousse-Joulin, S.; De Vita, S. Salivary gland ultrasonography in primary Sjögren’s syndrome: Opportunities and challenges. Rheumatology 2019. [Google Scholar] [CrossRef]
- Coiffier, G.; Martel, A.; Albert, J.D.; Lescoat, A.; Bleuzen, A.; Perdriger, A.; De Bandt, M.; Maillot, F. Ultrasonographic damages of major salivary glands are associated with cryoglobulinemic vasculitis and lymphoma in primary Sjogren’s syndrome: Are the ultrasonographic features of the salivary glands new prognostic markers in Sjogren’s syndrome? Ann. Rheum. Dis. 2021, 80, e111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theander, E.; Mandl, T. Primary Sjögren’s syndrome: Diagnostic and prognostic value of salivary gland ultrasonography using a simplified scoring system. Arthritis Care Res. 2014, 66, 1102–1107. [Google Scholar] [CrossRef] [PubMed]
- Lorenzon, M.; Tulipano Di Franco, F.; Zabotti, A.; Pegolo, E.; Giovannini, I.; Manfrè, V.; Mansutti, E.; De Vita, S.; Zuiani, C.; Girometti, R. Sonographic features of lymphoma of the major salivary glands diagnosed with ultrasound-guided core needle biopsy in Sjögren’s syndrome. Clin. Exp. Rheumatol. 2021, 39 (Suppl. 133), 175–183. [Google Scholar] [CrossRef] [PubMed]
- Lorenzon, M.; Spina, E.; Tulipano Di Franco, F.; Giovannini, I.; De Vita, S.; Zabotti, A. Salivary Gland Ultrasound in Primary Sjögren’s Syndrome: Current and Future Perspectives. Open Access Rheumatol. 2022, 14, 147–160. [Google Scholar] [CrossRef] [PubMed]
- Baldini, C.; Zabotti, A.; Filipovic, N.; Vukicevic, A.; Luciano, N.; Ferro, F.; Lorenzon, M.; De Vita, S. Imaging in primary Sjögren’s syndrome: The ‘obsolete and the new’. Clin. Exp. Rheumatol. 2018, 36 (Suppl. 112), 215–221. [Google Scholar] [PubMed]
- Grevers, G.; Ihrler, S.; Vogl, T.J.; Weiss, M. A comparison of clinical, pathological and radiological findings with magnetic resonance imaging studies of lymphomas in patients with Sjögren’s syndrome. Eur. Arch. Otorhinolaryngol. 1994, 251, 214–217. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, P.; Yang, J.; Yu, Q. Non-Hodgkin lymphoma involving the parotid gland: CT and MR imaging findings. Dentomaxillofac. Radiol. 2013, 42, 20130046. [Google Scholar] [CrossRef] [Green Version]
- Bădărînză, M.; Serban, O.; Maghear, L.; Bocsa, C.; Micu, M.; Damian, L.; Felea, I.; Fodor, D. Shear wave elastography as a new method to identify parotid lymphoma in primary Sjögren Syndrome patients: An observational study. Rheumatol. Int. 2020, 40, 1275–1281. [Google Scholar] [CrossRef]
- Cohen, C.; Mekinian, A.; Uzunhan, Y.; Fauchais, A.L.; Dhote, R.; Pop, G.; Eder, V.; Nunes, H.; Brillet, P.Y.; Valeyre, D.; et al. 18F-fluorodeoxyglucose positron emission tomography/computer tomography as an objective tool for assessing disease activity in Sjögren’s syndrome. Autoimmun. Rev. 2013, 12, 1109–1114. [Google Scholar] [CrossRef]
- Keraen, J.; Blanc, E.; Besson, F.L.; Leguern, V.; Meyer, C.; Henry, J.; Belkhir, R.; Nocturne, G.; Mariette, X.; Seror, R. Usefulness of (18) F-Labeled Fluorodeoxyglucose-Positron Emission Tomography for the Diagnosis of Lymphoma in Primary Sjögren’s Syndrome. Arthritis Rheumatol. 2019, 71, 1147–1157. [Google Scholar] [CrossRef]
- Gorodetskiy, V.; Probatova, N.; Obukhova, T.; Vasilyev, V. Analysis of prognostic factors in diffuse large B-cell lymphoma associated with rheumatic diseases. Lupus Sci. Med. 2021, 8, e000561. [Google Scholar] [CrossRef] [PubMed]
- Gorodetskiy, V.R.; Probatova, N.A.; Radenska-Lopovok, S.G.; Ryzhikova, N.V.; Sidorova, Y.V.; Sudarikov, A.B. Clonal relationship of marginal zone lymphoma and diffuse large B-cell lymphoma in Sjogren’s syndrome patients: Case series study and review of the literature. Rheumatol. Int. 2020, 40, 499–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kourou, K.D.; Pezoulas, V.C.; Georga, E.I.; Exarchos, T.; Papaloukas, C.; Voulgarelis, M.; Goules, A.; Nezos, A.; Tzioufas, A.G.; Moutsopoulos, E.M.; et al. Predicting Lymphoma Development by Exploiting Genetic Variants and Clinical Findings in a Machine Learning-Based Methodology with Ensemble Classifiers in a Cohort of Sjögren’s Syndrome Patients. IEEE Open J. Eng. Med. Biol. 2020, 1, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Gorodetskiy, V.R.; Probatova, N.A.; Vasilyev, V.I. Characteristics of diffuse large B-cell lymphoma in patients with primary Sjögren’s syndrome. Int. J. Rheum. Dis. 2020, 23, 540–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasaitis, L.; Nordmark, G.; Theander, E.; Backlin, C.; Smedby, K.E.; Askling, J.; Rönnblom, L.; Sundström, C.; Baecklund, E. Comparison of patients with and without pre-existing lymphoma at diagnosis of primary Sjögren’s syndrome. Scand. J. Rheumatol. 2019, 48, 207–212. [Google Scholar] [CrossRef]
Figure 1.
Schematic presentation of newly proposed pSS-related lymphoma biomarkers and their pathophysiologic role. BAFF, B-cell Activating Factor; BAFF-R, BAFF Receptor; CXCL13, C-X-C motif chemokine ligand 13; IFN, Interferon; FLT-3L, Fms-like tyrosine kinase 3 Ligand; GC, Germinal Center; GPR34, G Protein-Coupled Receptor 34; HCP5, HLA complex P5; HSC, Hematopoietic Stem Cell; HPC, Hematopoietic Progenitor Cell; ISG-15, IFN-stimulated gene 15; miR, micro-RNA; MZ, Marginal Zone; NF-Κb, Nuclear Factor κΒ’PLA1, Phospholipase A1; P2X7R, P2X7 receptor; SG, Salivary Gland; TNFAIP3, Tumor Necrosis Factor Alpha Induced Protein 3; TREX, Three Prime Repair Exonuclease 1; TSLP, Thymic Stromal Lymphopoietin. (Created with BioRender.com).
Figure 1.
Schematic presentation of newly proposed pSS-related lymphoma biomarkers and their pathophysiologic role. BAFF, B-cell Activating Factor; BAFF-R, BAFF Receptor; CXCL13, C-X-C motif chemokine ligand 13; IFN, Interferon; FLT-3L, Fms-like tyrosine kinase 3 Ligand; GC, Germinal Center; GPR34, G Protein-Coupled Receptor 34; HCP5, HLA complex P5; HSC, Hematopoietic Stem Cell; HPC, Hematopoietic Progenitor Cell; ISG-15, IFN-stimulated gene 15; miR, micro-RNA; MZ, Marginal Zone; NF-Κb, Nuclear Factor κΒ’PLA1, Phospholipase A1; P2X7R, P2X7 receptor; SG, Salivary Gland; TNFAIP3, Tumor Necrosis Factor Alpha Induced Protein 3; TREX, Three Prime Repair Exonuclease 1; TSLP, Thymic Stromal Lymphopoietin. (Created with BioRender.com).

Figure 2.
The median time (months) before lymphoma diagnosis that certain pSS-related lymphoma predisposing factors could be detected. FLT-3L, Fms-like tyrosine kinase 3 Ligand; FS, Focus Score; GC, Germinal Center; miR, micro-RNA; SGE, Salivary Gland Enlargement.
Figure 2.
The median time (months) before lymphoma diagnosis that certain pSS-related lymphoma predisposing factors could be detected. FLT-3L, Fms-like tyrosine kinase 3 Ligand; FS, Focus Score; GC, Germinal Center; miR, micro-RNA; SGE, Salivary Gland Enlargement.

Figure 3.
The clinical picture of the most common pSS-associated B-cell NHL subtypes. BM, Bone Marrow; DLBCL, Diffuse Large B-cell Lymphoma; MALT, Mucosa Associated Lymphoid Tissue; NMZL, Nodal Marginal Zone Lymphoma; SG, Salivary Gland; SGE, Salivary Gland Enlargement (Created with BioRender.com).
Figure 3.
The clinical picture of the most common pSS-associated B-cell NHL subtypes. BM, Bone Marrow; DLBCL, Diffuse Large B-cell Lymphoma; MALT, Mucosa Associated Lymphoid Tissue; NMZL, Nodal Marginal Zone Lymphoma; SG, Salivary Gland; SGE, Salivary Gland Enlargement (Created with BioRender.com).


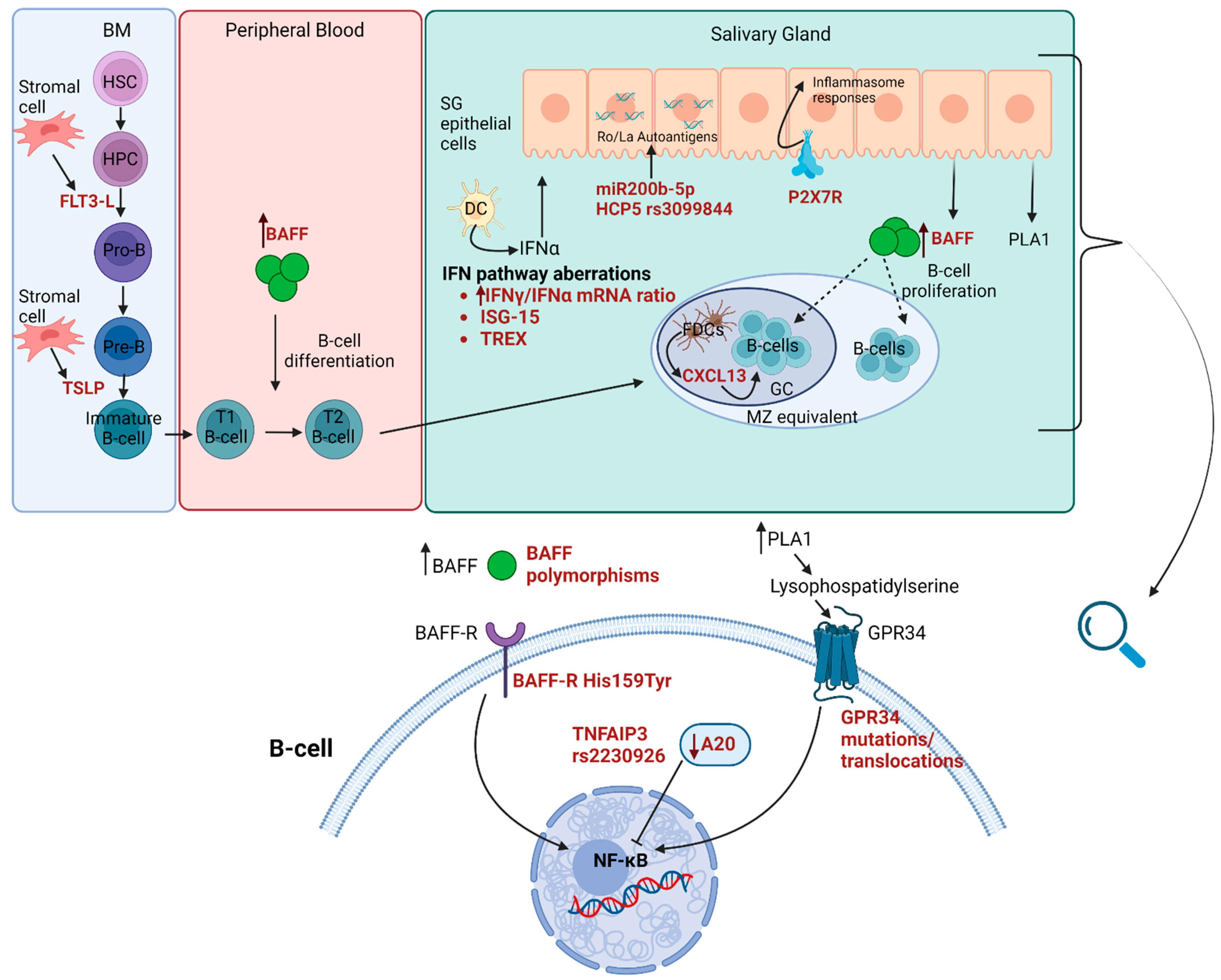{kind=link}
{kind=link}
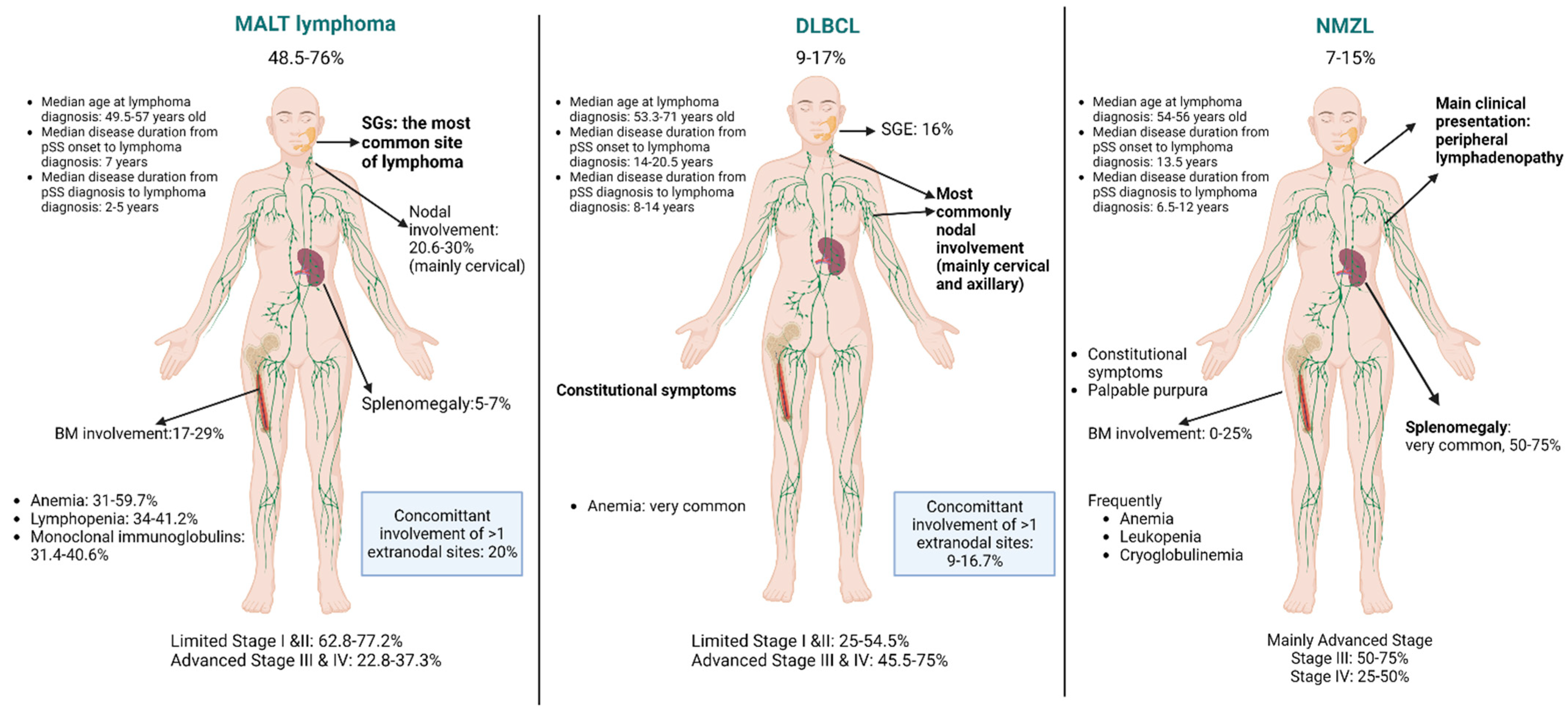{kind=link}
Table 1.
Proposed predisposing factors for B-cell NHL in pSS.
| Classical | Newly Proposed | ||
|---|---|---|---|
| Clinical | SGE [11,18,24,28,29,32,38,39,40,41,42,43,44,45] Skin vasculitis/Palpable purpura [6,14,18,24,25,39,43,44] Lymphadenopathy [9,14,29,38,44,45] Splenomegaly [9,38] Raynaud [29] Peripheral nerve involvement [14,43] Glomerulonephritis [25,43,47] | Disease activity | ESSDAI [11,23,41] |
| Serological | Cryoglobulinemia [9,10,11,25,40,41,42,49,50,51] RF positivity [29,41,49,50] Anti-Ro/SSA [29,142] Anti-La/SSB [29,42,142] Low serum C4 levels [6,9,10,18,24,25,29,32,41,42,43,50] Low serum C3 levels [6,10,18,32,50] Monoclonal Igs [10,29,43,45] Hypergammaglobulinemia [18,45,50] Serum and urine Ig light chains and their kappa/lamda ratio [143] β2 microglobulin [55,59] | Genetics | BAFF polymorphisms [96] BAFF-R His159Tyr mutation [97,98] TNFAIP3 variants [99,100] GPR34 translocations/mutations [103,104] TREX variants [105] LILRA3 variant [106] HCP5 rs3099844 variant [107] |
| Epigenetics | Low miR200b-5p levels in MSGs [110] MTHFR SNPs [111] LINE1-retroelements [112] | ||
| Hematological | Anemia [10,14,18] Leukopenia [18,42] Lymphopenia [9,14,18,41] Neutropenia [9] CD4+ T lymphopenia [6] Low CD4+/CD8+ ratio ≤ 0.8 [6] | Gene expression | Decreased A20 expression in MSGs [113,114] High IFNγ/IFNα mRNA ratio in MSGs [115] High ISG-15 expression [116] P2X7R-inflammasome complex [117] Increased Lp-PLA2 [120] |
| Histological | Ectopic GCs [54,85] MSG FS [11,64,65] AID distribution in ectopic GCs [89] | Proteins | Increased BAFF serum levels [52,55,121] Increased FLT-3L serum levels [123,124] Increased CXCL13 serum levels [52,125] Increased serum TSLP levels [130] |
| Imaging | SGUS: OMERACT score, hypoechoic lesions [132,133,134,135] PET/CT: SUVmax of ≥4.7 and/or focal lung lesions [141] | ||
AID, activation-induced cytidine deaminase; BAFF, B-cell Activating Factor; BAFF-R, BAFF Receptor; CXCL13, C-X-C motif chemokine ligand 13; ESSDAI, EULAR Sjögren Syndrome Disease Activity Index; FLT-3L, Fms-like tyrosine kinase 3 Ligand; FS, Focus Score; GC, Germinal Center; GPR34, G Protein-Coupled Receptor 34; HCP5, HLA complex P5; IFN, interferon; Ig, immunoglobulin; ISG-15, IFN-stimulated gene 15; LINE-1, long interspersed nuclear elements 1; Lp-PLA1, Lipoprotein-associated phospholipase A2; miR, micro-RNA; MSGs, Minor Salivary Glands; MTHFR, Methylene-Tetrahydrofolate reductase; P2X7R, P2X7 receptor; PET-CT, Positron Emission Tomography/computed Tomography; RF, Rheumatoid, Factor; SGE, Salivary Gland Enlargement; SGUS, Salivary Gland Ultrasonography; SUVmax, maximum Standardized Uptake Value; TNFAIP3, Tumor Necrosis Factor Alpha Induced Protein 3; TREX, Three Prime Repair Exonuclease 1; TSLP, Thymic Stromal Lymphopoietin.
Table 2.
Proposed different predisposing factors for pSS-related MALT lymphoma and DLBCL.
| MALT Lymphoma | DLBCL |
|---|---|
| Cryoglobulinemia [9,11] FS [11] ESSDAI at pSS diagnosis [11] Neutropenia [9] Low serum C4 levels [9,18] Low serum C3 levels [10,18] Lymphadenopathy/Splenomegaly [9] | Lymphocytopenia [9,18] CD4+/CD8+ T-cell ratio ≤ 0.8 [6] Anemia [10] Low serum C4 levels [10] |
ESSDAI, EULAR Sjögren Syndrome Disease Activity Index; FS, Focus Score.
Table 3.
Survival rates for pSS-related B-cell NHLs described in different studies over the years.
| Median OS | ||
| Pre-Rituximab era | Low-risk lymphomas: 6.33 years Intermediate/High-risk lymphomas: 1.83 years | [14] |
| Post-Rituximab era | MALT lymphomas: 13 years DLBCLs: 6 years | [17] |
| 5-year OS | ||
| Pre-Rituximab era | Low-risk lymphomas: 70% Intermediate/High-risk lymphomas: 48% | [14] |
| Post-Rituximab era | All lymphoma subtypes: 90.91% | [12] |
| All lymphoma subtypes: 86.5% | [5] | |
| MALT lymphomas: 94.1% DLBCLs: 75% NMZLs: 87.5% | [12] | |
| MALT lymphomas: 91% DLBCL: 54.5% NMZLs: 62.5% | [11] | |
| 5-year EFS | ||
| Post-Rituximab era | MALT lymphomas: 86.2% DLBCLs: 50% NMZL: 62.5% | [12] |
| MALT lymphomas: 63.6% DLBCLs: 45.4% NMZL: 62.5% | [11] | |
| 10-year OS | ||
| Post-Rituximab era | MALT lymphomas: 79% DLBCLs: 40.9% NMZLs: 46% | [11] |
| 10-year EFS | ||
| Post-Rituximab era | MALT lymphomas: 45.5% DLBCLs: 24.2% NMZLs: 31% | [11] |
DLBCL, Diffuse Large B-Cell Lymphoma; EFS, Event-Free Survival; MALT, Mucosa Associated Lymphoid Tissue; NMZL, Nodal Marginal Zone Lymphoma; OS, Overall Survival.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Stergiou, I.E.; Goules, A.V.; Voulgarelis, M.; Tzioufas, A.G. Predisposing Factors, Clinical Picture, and Outcome of B-Cell Non-Hodgkin’s Lymphoma in Sjögren’s Syndrome. Immuno 2022, 2, 584-608. https://doi.org/10.3390/immuno2040037
AMA Style
Stergiou IE, Goules AV, Voulgarelis M, Tzioufas AG. Predisposing Factors, Clinical Picture, and Outcome of B-Cell Non-Hodgkin’s Lymphoma in Sjögren’s Syndrome. Immuno. 2022; 2(4):584-608. https://doi.org/10.3390/immuno2040037
Chicago/Turabian StyleStergiou, Ioanna E., Andreas V. Goules, Michael Voulgarelis, and Athanasios G. Tzioufas. 2022. "Predisposing Factors, Clinical Picture, and Outcome of B-Cell Non-Hodgkin’s Lymphoma in Sjögren’s Syndrome" Immuno 2, no. 4: 584-608. https://doi.org/10.3390/immuno2040037