
Intertwining Neuropathogenic Impacts of Aberrant Circadian Rhythm and Impaired Neuroregenerative Plasticity in Huntington’s Disease: Neurotherapeutic Significance of Chemogenetics

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
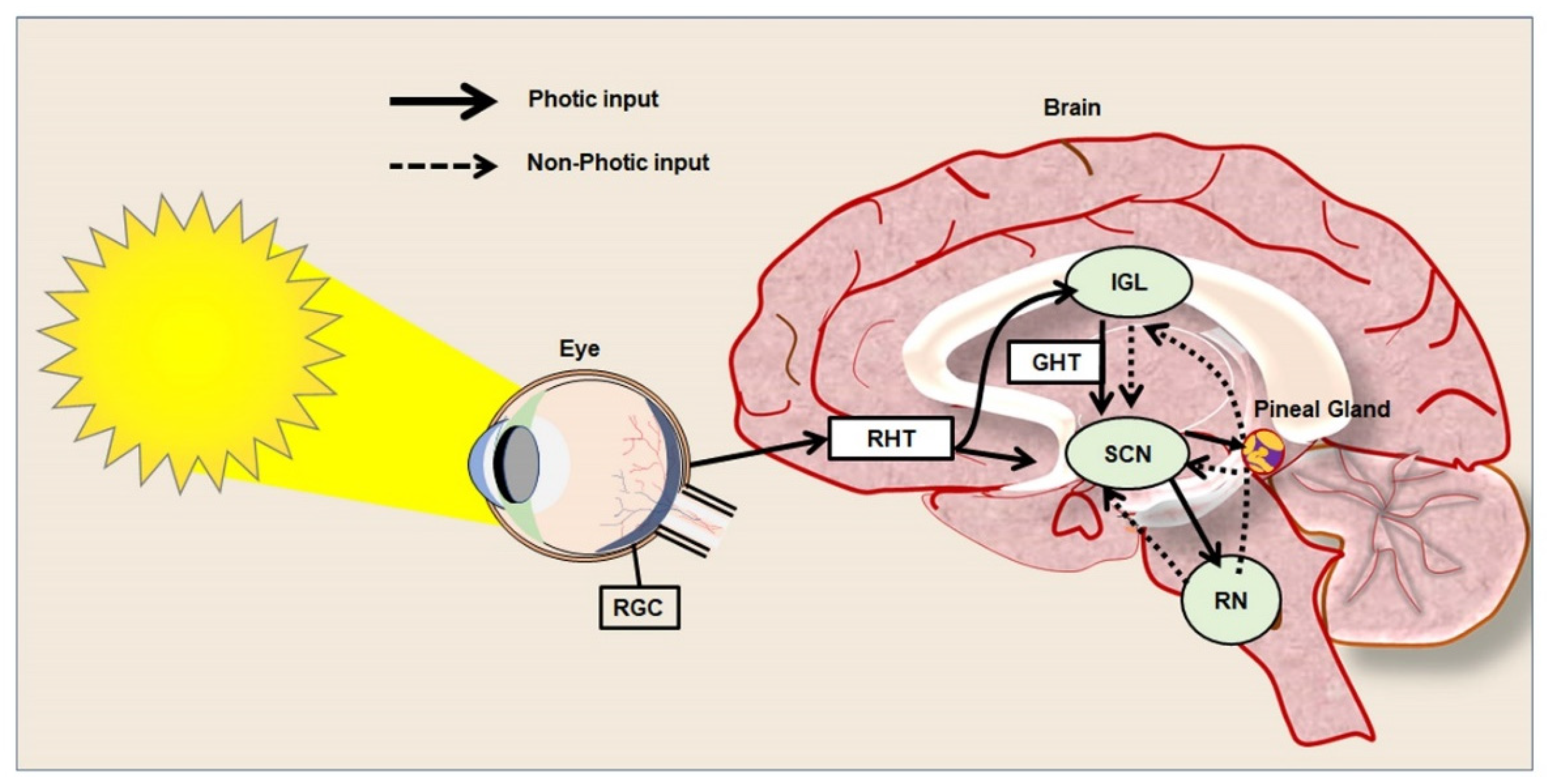
2. Regulation of Circadian Rhythm in Physiological State
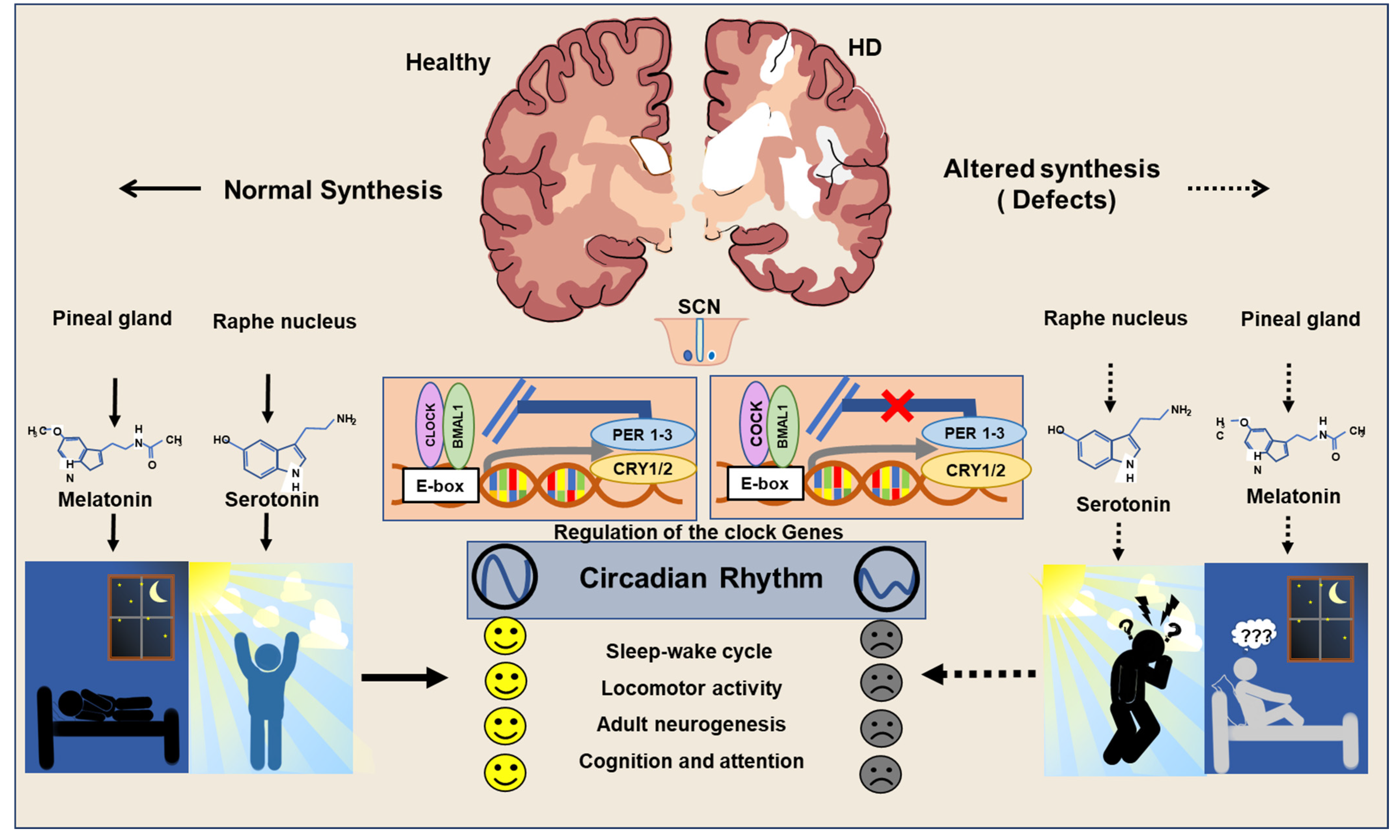
3. Neuropathogenic Input of Abnormal Regulation of Clock Genes in HD
4. Potential Overlap between Altered Clock Gene Pathway and Dysregulation of Neuroregenerative Plasticity in HD
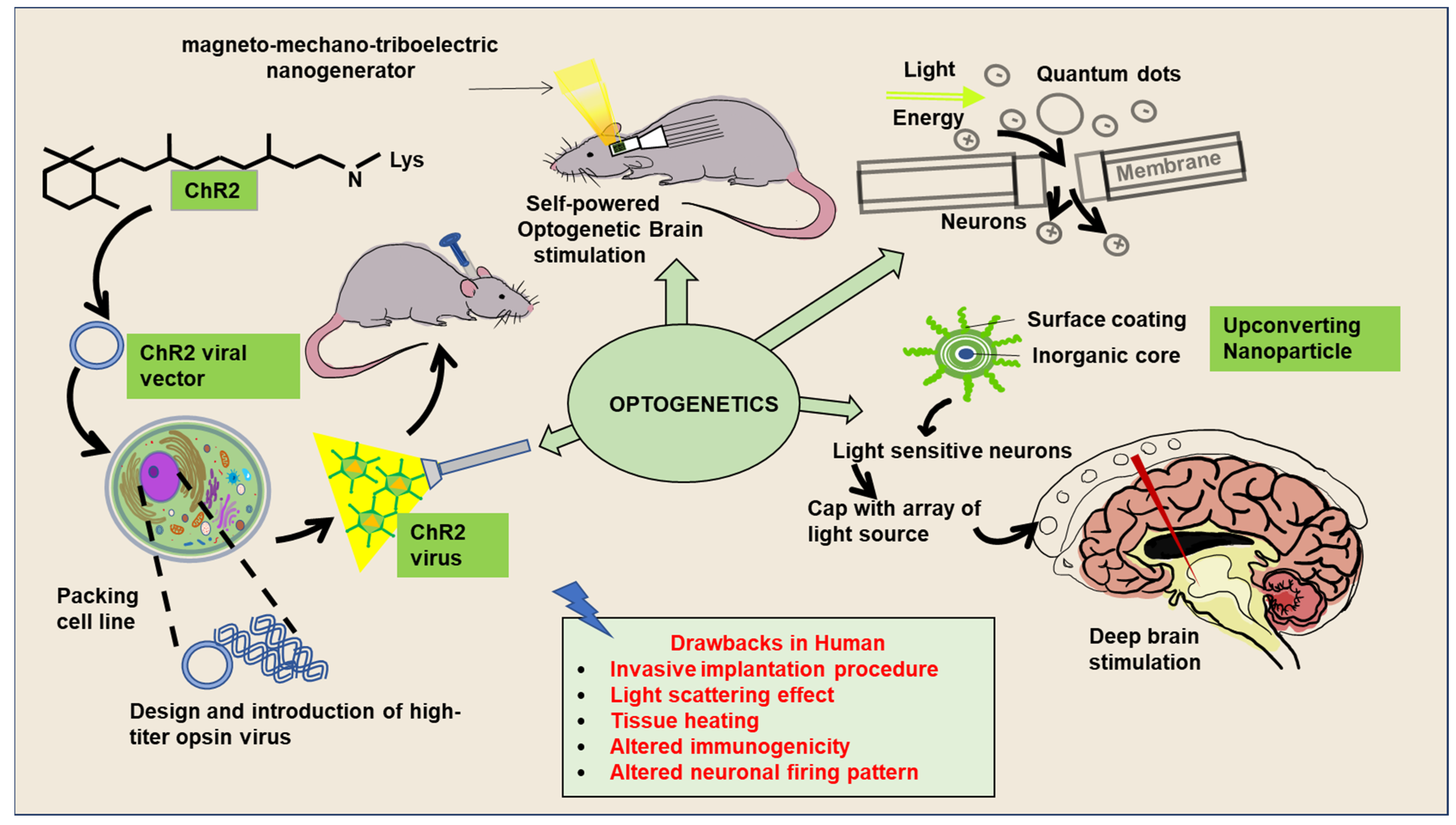
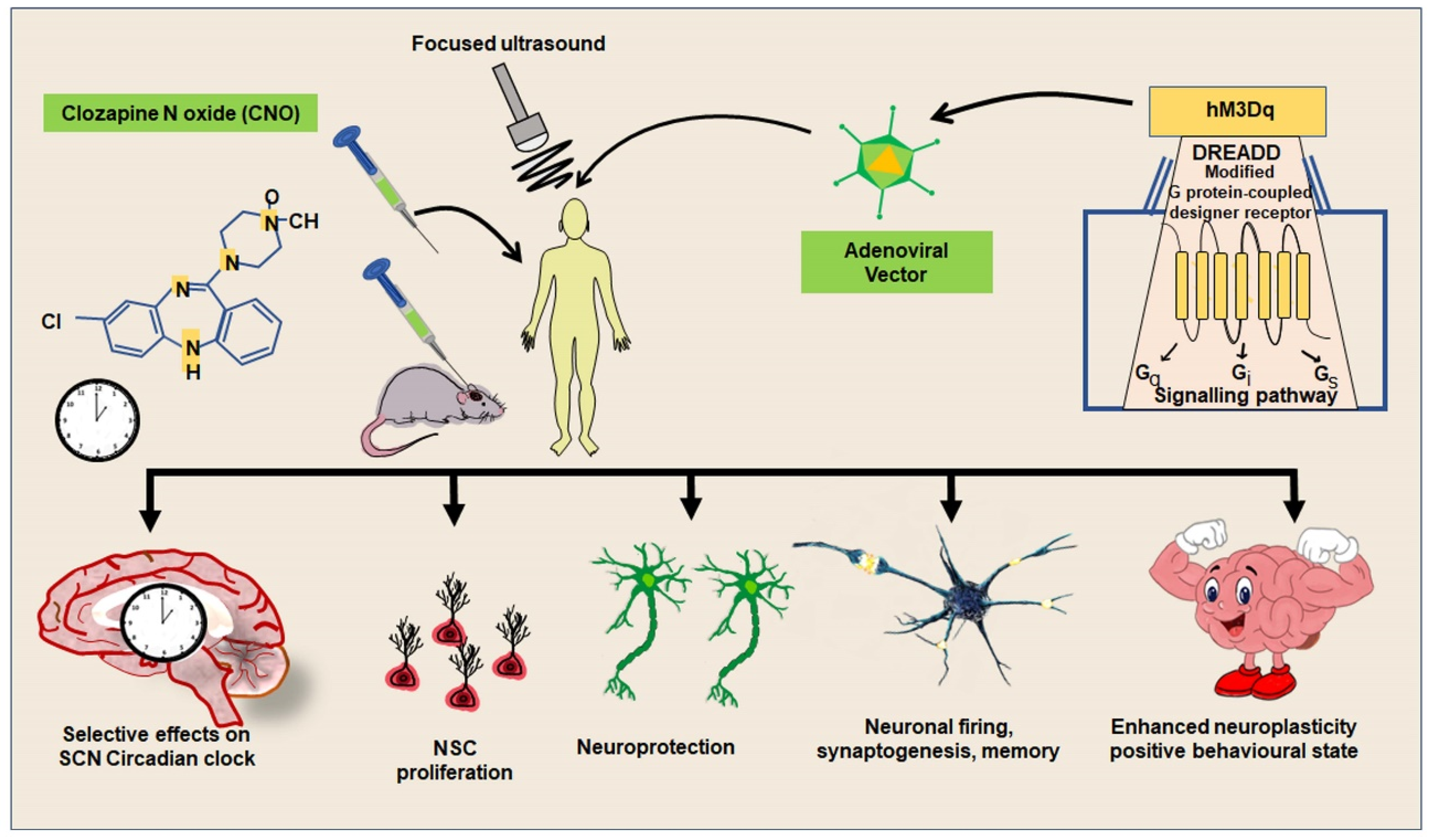
5. An Overview and Significance of Optogenetics and Chemogenetics-Based Experimental Interventions for Neuronal Activities
6. Discussion
7. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gusella, J.F.; Wexler, N.S.; Conneally, P.M.; Naylor, S.L.; Anderson, M.A.; Tanzi, R.E.; Watkins, P.C.; Ottina, K.; Wallace, M.R.; Sakaguchi, A.Y.; et al. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature 1983, 306, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Moily, N.S.; Kota, L.N.; Anjanappa, R.M.; Venugopal, S.; Vaidyanathan, R.; Pal, P.; Purushottam, M.; Jain, S.; Kandasamy, M. Trinucleotide repeats and haplotypes at the huntingtin locus in an Indian sample overlaps with European haplogroup A. PLoS Curr. 2014, 6. [Google Scholar] [CrossRef]
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington’s Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harb. Perspect. Med. 2017, 7, a024240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harper, P.S. The epidemiology of Huntington’s disease. Hum. Genet. 1992, 89, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.O. Huntington’s disease. Lancet 2007, 369, 218–228. [Google Scholar] [CrossRef] [PubMed]
- Herzog–Krzywoszanska, R.; Krzywoszanski, L. Sleep Disorders in Huntington’s Disease. Front. Psychiatry 2019, 10, 221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Voysey, Z.; Fazal, S.V.; Lazar, A.S.; Barker, R.A. The sleep and circadian problems of Huntington’s disease: When, why and their importance. J. Neurol. 2021, 268, 2275–2283. [Google Scholar] [CrossRef]
- Banks, S.; Dinges, D.F. Behavioral and Physiological Consequences of Sleep Restriction. J. Clin. Sleep Med. 2007, 3, 519–528. [Google Scholar] [CrossRef] [Green Version]
- Toda, T.; Parylak, S.; Linker, S.B.; Gage, F.H. The role of adult hippocampal neurogenesis in brain health and disease. Mol. Psychiatry 2019, 24, 67–87. [Google Scholar] [CrossRef]
- Kandasamy, M.; Anusuyadevi, M.; Aigner, K.M.; Unger, M.S.; Kniewallner, K.M.; de Sousa, D.M.B.; Altendorfer, B.; Mrowetz, H.; Bogdahn, U.; Aigner, L. TGF-β Signaling: A Therapeutic Target to Reinstate Regenerative Plasticity in Vascular Dementia? Aging Dis. 2020, 11, 828–850. [Google Scholar] [CrossRef]
- Kandasamy, M.; Reilmann, R.; Winkler, J.; Bogdahn, U.; Aigner, L. Transforming Growth Factor-Beta Signaling in the Neural Stem Cell Niche: A Therapeutic Target for Huntington’s Disease. Neurol. Res. Int. 2011, 2011, 124256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandasamy, M.; Couillard-Despres, S.; Raber, K.A.; Stephan, M.; Lehner, B.; Winner, B.; Kohl, Z.; Rivera, F.J.; Nguyen, H.P.; Riess, O.; et al. Stem cell quiescence in the hippocampal neurogenic niche is associated with elevated transforming growth factor-beta signaling in an animal model of Huntington disease. J. Neuropathol. Exp. Neurol. 2010, 69, 717–728. [Google Scholar] [CrossRef] [Green Version]
- Kohl, Z.; Kandasamy, M.; Winner, B.; Aigner, R.; Gross, C.; Couillard-Despres, S.; Bogdahn, U.; Aigner, L.; Winkler, J. Physical activity fails to rescue hippocampal neurogenesis deficits in the R6/2 mouse model of Huntington’s disease. Brain Res. 2007, 1155, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Kandasamy, M.; Aigner, L. Reactive Neuroblastosis in Huntington’s Disease: A Putative Therapeutic Target for Striatal Regeneration in the Adult Brain. Front. Cell. Neurosci. 2018, 12, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandasamy, M.; Rosskopf, M.; Wagner, K.; Klein, B.; Couillard-Despres, S.; Reitsamer, H.A.; Stephan, M.; Nguyen, H.P.; Riess, O.; Bogdahn, U.; et al. Reduction in subventricular zone-derived olfactory bulb neurogenesis in a rat model of Huntington’s disease is accompanied by striatal invasion of neuroblasts. PLoS ONE 2015, 10, e0116069. [Google Scholar] [CrossRef] [PubMed]
- Lazic, S.E.; Grote, H.; Armstrong, R.J.E.; Blakemore, C.; Hannan, A.J.; van Dellen, A.; Barker, R.A. Decreased hippocampal cell proliferation in R6/1 Huntington’s mice. Neuroreport 2004, 15, 811–813. [Google Scholar] [CrossRef]
- Curtis, M.A.; Penney, E.B.; Pearson, A.G.; van Roon-Mom, W.M.C.; Butterworth, N.J.; Dragunow, M.; Connor, B.; Faull, R.L.M. Increased cell proliferation and neurogenesis in the adult human Huntington’s disease brain. Proc. Natl. Acad. Sci. USA 2003, 100, 9023–9027. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.A.H.; von Gall, C. Adult Neurogenesis under Control of the Circadian System. Cells 2022, 11, 764. [Google Scholar] [CrossRef]
- Malik, A.; Kondratov, R.V.; Jamasbi, R.J.; Geusz, M.E. Circadian Clock Genes Are Essential for Normal Adult Neurogenesis, Differentiation, and Fate Determination. PLoS ONE 2015, 10, e0139655. [Google Scholar] [CrossRef]
- Faragó, A.; Zsindely, N.; Bodai, L. Mutant huntingtin disturbs circadian clock gene expression and sleep patterns in Drosophila. Sci. Rep. 2019, 9, 7174. [Google Scholar] [CrossRef]
- Loh, D.H.; Kudo, T.; Truong, D.; Wu, Y.; Colwell, C.S. The Q175 Mouse Model of Huntington’s Disease Shows Gene Dosage- and Age-Related Decline in Circadian Rhythms of Activity and Sleep. PLoS ONE 2013, 8, e69993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, J.; Zeng, H.; Olson, D.P.; Huber, K.M.; Gibson, J.R.; Takahashi, J.S. Vasoactive Intestinal Polypeptide (VIP)-Expressing Neurons in the Suprachiasmatic Nucleus Provide Sparse GABAergic Outputs to Local Neurons with Circadian Regulation Occurring Distal to the Opening of Postsynaptic GABAA Ionotropic Receptors. J. Neurosci. 2015, 35, 1905–1920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, D.; Honma, K.; Honma, S. Roles of Neuropeptides, VIP and AVP, in the Mammalian Central Circadian Clock. Front. Neurosci. 2021, 15, 650154. Available online: https://www.frontiersin.org/articles/10.3389/fnins.2021.650154 (accessed on 26 September 2022). [CrossRef] [PubMed]
- Ono, D.; Honma, K.-I.; Yanagawa, Y.; Yamanaka, A.; Honma, S. Role of GABA in the regulation of the central circadian clock of the suprachiasmatic nucleus. J. Physiol. Sci. 2018, 68, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Groen, M.R.; Paulsen, O.; Pérez-Garci, E.; Nevian, T.; Wortel, J.; Dekker, M.P.; Mansvelder, H.D.; van Ooyen, A.; Meredith, R.M. Development of dendritic tonic GABAergic inhibition regulates excitability and plasticity in CA1 pyramidal neurons. J. Neurophysiol. 2014, 112, 287–299. [Google Scholar] [CrossRef] [Green Version]
- Turi, G.F.; Li, W.-K.; Chavlis, S.; Pandi, I.; O’Hare, J.; Priestley, J.B.; Grosmark, A.D.; Liao, Z.; Ladow, M.; Zhang, J.F.; et al. Vasoactive Intestinal Polypeptide-Expressing Interneurons in the Hippocampus Support Goal-Oriented Spatial Learning. Neuron 2019, 101, 1150–1165. [Google Scholar] [CrossRef] [Green Version]
- Campbell, E.J.; Marchant, N.J. The use of chemogenetics in behavioural neuroscience: Receptor variants, targeting approaches and caveats. Br. J. Pharmacol. 2018, 175, 994–1003. [Google Scholar] [CrossRef] [Green Version]
- Magnus, C.J.; Lee, P.H.; Bonaventura, J.; Zemla, R.; Gomez, J.L.; Ramirez, M.H.; Hu, X.; Galvan, A.; Basu, J.; Michaelides, M.; et al. Ultrapotent chemogenetics for research and potential clinical applications. Science 2019, 364, eaav5282. [Google Scholar] [CrossRef]
- Rodriguez, G.A.; Barrett, G.M.; Duff, K.E.; Hussaini, S.A. Chemogenetic attenuation of neuronal activity in the entorhinal cortex reduces Aβ and tau pathology in the hippocampus. PLoS Biol. 2020, 18, e3000851. [Google Scholar] [CrossRef]
- Reddy, S.; Reddy, V.; Sharma, S. Physiology, Circadian Rhythm. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2022. Available online: http://www.ncbi.nlm.nih.gov/books/NBK519507/ (accessed on 26 September 2022).
- Czeisler, C.A.; Gooley, J.J. Sleep and circadian rhythms in humans. Cold Spring Harb. Symp. Quant. Biol. 2007, 72, 579–597. [Google Scholar] [CrossRef]
- Ayyar, V.S.; Sukumaran, S. Circadian rhythms: Influence on physiology, pharmacology, and therapeutic interventions. J. Pharmacokinet. Pharmacodyn. 2021, 48, 321–338. [Google Scholar] [CrossRef] [PubMed]
- Challet, E.; Pévet, P. Interactions between photic and nonphotic stimuli to synchronize the master circadian clock in mammals. Front. Biosci. 2003, 8, s246–s257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Morgia, C.; Carelli, V.; Carbonelli, M. Melanopsin Retinal Ganglion Cells and Pupil: Clinical Implications for Neuro-Ophthalmology. Front. Neurol. 2018, 9, 1047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, M.T.H.; Yau, K.-W. Intrinsically Photosensitive Retinal Ganglion Cells. Physiol. Rev. 2010, 90, 1547–1581. [Google Scholar] [CrossRef]
- Reghunandanan, V.; Reghunandanan, R. Neurotransmitters of the suprachiasmatic nuclei. J. Circadian Rhythms 2006, 4, 2. [Google Scholar] [CrossRef] [Green Version]
- Patton, A.P.; Hastings, M.H. The suprachiasmatic nucleus. Curr. Biol. 2018, 28, R816–R822. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, M.; Akiba, Y.; Kaunitz, J.D. Recent advances in vasoactive intestinal peptide physiology and pathophysiology: Focus on the gastrointestinal system. F1000Research 2019, 8, 1629. [Google Scholar] [CrossRef] [Green Version]
- Gozes, I.; Brenneman, D.E. VIP: Molecular biology and neurobiological function. Mol. Neurobiol. 1989, 3, 201–236. [Google Scholar] [CrossRef]
- Cunha-Reis, D.; Caulino-Rocha, A. VIP Modulation of Hippocampal Synaptic Plasticity: A Role for VIP Receptors as Therapeutic Targets in Cognitive Decline and Mesial Temporal Lobe Epilepsy. Front. Cell. Neurosci. 2020, 14, 153. Available online: https://www.frontiersin.org/articles/10.3389/fncel.2020.00153 (accessed on 26 September 2022). [CrossRef]
- Welsh, D.K.; Takahashi, J.S.; Kay, S.A. Suprachiasmatic Nucleus: Cell Autonomy and Network Properties. Annu. Rev. Physiol. 2010, 72, 551–577. [Google Scholar] [CrossRef]
- Jones, J.R.; Simon, T.; Lones, L.; Herzog, E.D. SCN VIP Neurons Are Essential for Normal Light-Mediated Resetting of the Circadian System. J. Neurosci. 2018, 38, 7986–7995. [Google Scholar] [CrossRef] [PubMed]
- Herzog, E.D.; Hermanstyne, T.; Smyllie, N.J.; Hastings, M.H. Regulating the Suprachiasmatic Nucleus (SCN) Circadian Clockwork: Interplay between Cell-Autonomous and Circuit-Level Mechanisms. Cold Spring Harb. Perspect. Biol. 2017, 9, a027706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aulinas, A. Physiology of the Pineal Gland and Melatonin. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dungan, K., Grossman, A., Hershman, J.M., Hofland, H.J., Kaltsas, G., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. Available online: http://www.ncbi.nlm.nih.gov/books/NBK550972/ (accessed on 8 November 2020).
- Piano, C.; Losurdo, A.; Della Marca, G.; Solito, M.; Calandra-Buonaura, G.; Provini, F.; Bentivoglio, A.R.; Cortelli, P. Polysomnographic Findings and Clinical Correlates in Huntington Disease: A Cross-Sectional Cohort Study. Sleep 2015, 38, 1489–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, N.A.; Pijl, H.; Frölich, M.; Schröder-van der Elst, J.P.; van der Bent, C.; Roelfsema, F.; Roos, R.A.C. Delayed onset of the diurnal melatonin rise in patients with Huntington’s disease. J. Neurol. 2009, 256, 1961–1965. [Google Scholar] [CrossRef] [Green Version]
- Gonzales, E.; Yin, J. Drosophila Models of Huntington’s Disease Exhibit Sleep Abnormalities. PLoS Curr. 2010, 2, RRN1185. [Google Scholar] [CrossRef]
- Kuljis, D.; Schroeder, A.M.; Kudo, T.; Loh, D.H.; Willison, D.L.; Colwell, C.S. Sleep and circadian dysfunction in neurodegenerative disorders: Insights from a mouse model of Huntington’s disease. Minerva Pneumol. 2012, 51, 93–106. [Google Scholar]
- Kuljis, D.A.; Gad, L.; Loh, D.H.; MacDowell Kaswan, Z.; Hitchcock, O.N.; Ghiani, C.A.; Colwell, C.S. Sex Differences in Circadian Dysfunction in the BACHD Mouse Model of Huntington’s Disease. PLoS ONE 2016, 11, e0147583. [Google Scholar] [CrossRef] [Green Version]
- Maywood, E.S.; Fraenkel, E.; McAllister, C.J.; Wood, N.; Reddy, A.B.; Hastings, M.H.; Morton, A.J. Disruption of Peripheral Circadian Timekeeping in a Mouse Model of Huntington’s Disease and Its Restoration by Temporally Scheduled Feeding. J. Neurosci. 2010, 30, 10199–10204. [Google Scholar] [CrossRef] [Green Version]
- Aziz, N.A.; Anguelova, G.V.; Marinus, J.; Lammers, G.J.; Roos, R.A.C. Sleep and circadian rhythm alterations correlate with depression and cognitive impairment in Huntington’s disease. Parkinsonism Relat. Disord. 2010, 16, 345–350. [Google Scholar] [CrossRef]
- Andreani, T.S.; Itoh, T.Q.; Yildirim, E.; Hwangbo, D.-S.; Allada, R. Genetics of Circadian Rhythms. Sleep Med. Clin. 2015, 10, 413–421. [Google Scholar] [CrossRef] [Green Version]
- Charrier, A.; Olliac, B.; Roubertoux, P.; Tordjman, S. Clock Genes and Altered Sleep–Wake Rhythms: Their Role in the Development of Psychiatric Disorders. Int. J. Mol. Sci. 2017, 18, 938. [Google Scholar] [CrossRef] [PubMed]
- Mazzoccoli, G.; Pazienza, V.; Vinciguerra, M. Clock genes and clock-controlled genes in the regulation of metabolic rhythms. Chronobiol. Int. 2012, 29, 227–251. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhao, X.; Zhang, Y.; Tan, H.; Qiu, B.; Ma, T.; Zeng, J.; Tao, D.; Liu, Y.; Lu, Y.; et al. RAE1 promotes BMAL1 shuttling and regulates degradation and activity of CLOCK: BMAL1 heterodimer. Cell Death Dis. 2019, 10, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, R.; Selby, C.P.; Chiou, Y.-Y.; Ozkan-Dagliyan, I.; Gaddameedhi, S.; Sancar, A. Dual modes of CLOCK:BMAL1 inhibition mediated by Cryptochrome and Period proteins in the mammalian circadian clock. Genes Dev. 2014, 28, 1989–1998. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ikeda, R.; Tsuchiya, Y.; Koike, N.; Umemura, Y.; Inokawa, H.; Ono, R.; Inoue, M.; Sasawaki, Y.; Grieten, T.; Okubo, N.; et al. REV-ERBα and REV-ERBβ function as key factors regulating Mammalian Circadian Output. Sci. Rep. 2019, 9, 10171. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2017, 18, 164–179. [Google Scholar] [CrossRef] [Green Version]
- Cox, K.H.; Takahashi, J.S. Circadian clock genes and the transcriptional architecture of the clock mechanism. J. Mol. Endocrinol. 2019, 63, R93–R102. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, N.; Rakshit, K.; Chow, E.S.; Wentzell, J.S.; Kretzschmar, D.; Giebultowicz, J.M. Loss of circadian clock accelerates aging in neurodegeneration-prone mutants. Neurobiol. Dis. 2012, 45, 1129–1135. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.C. Tumor suppression by the mammalian Period genes. Cancer Causes Control 2006, 17, 525–530. [Google Scholar] [CrossRef]
- Snider, K.H.; Dziema, H.; Aten, S.; Loeser, J.; Norona, F.E.; Hoyt, K.; Obrietan, K. Modulation of learning and memory by the targeted deletion of the circadian clock gene Bmal1 in forebrain circuits. Behav. Brain Res. 2016, 308, 222–235. [Google Scholar] [CrossRef] [Green Version]
- Rijo-Ferreira, F.; Takahashi, J.S. Genomics of circadian rhythms in health and disease. Genome Med. 2019, 11, 82. [Google Scholar] [CrossRef] [PubMed]
- Leng, Y.; Musiek, E.S.; Hu, K.; Cappuccio, F.P.; Yaffe, K. Association between circadian rhythms and neurodegenerative diseases. Lancet Neurol. 2019, 18, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Videnovic, A.; Zee, P.C. Consequences of Circadian Disruption on Neurologic Health. Sleep Med. Clin. 2015, 10, 469–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moumné, L.; Betuing, S.; Caboche, J. Multiple Aspects of Gene Dysregulation in Huntington’s Disease. Front. Neurol. 2013, 4, 127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, K.E.; van Duijn, E.; Craufurd, D.; Drazinic, C.; Edmondson, M.; Goodman, N.; van Kammen, D.; Loy, C.; Priller, J.; Goodman, L.V. Clinical Management of Neuropsychiatric Symptoms of Huntington Disease: Expert-Based Consensus Guidelines on Agitation, Anxiety, Apathy, Psychosis and Sleep Disorders. J. Huntingt. Dis. 2018, 7, 355–366. [Google Scholar] [CrossRef] [Green Version]
- Van Duijn, E.; Craufurd, D.; Hubers, A.A.M.; Giltay, E.J.; Bonelli, R.; Rickards, H.; Anderson, K.E.; van Walsem, M.R.; van der Mast, R.C.; Orth, M.; et al. Neuropsychiatric symptoms in a European Huntington’s disease cohort (REGISTRY). J. Neurol. Neurosurg. Psychiatry 2014, 85, 1411–1418. [Google Scholar] [CrossRef] [Green Version]
- Fahrenkrug, J.; Popovic, N.; Georg, B.; Brundin, P.; Hannibal, J. Decreased VIP and VPAC2 receptor expression in the biological clock of the R6/2 Huntington’s disease mouse. J. Mol. Neurosci. 2007, 31, 139–148. [Google Scholar] [CrossRef]
- Musiek, E.S. Circadian clock disruption in neurodegenerative diseases: Cause and effect? Front. Pharmacol. 2015, 6, 29. [Google Scholar] [CrossRef] [Green Version]
- Musiek, E.S.; Lim, M.M.; Yang, G.; Bauer, A.Q.; Qi, L.; Lee, Y.; Roh, J.H.; Ortiz-Gonzalez, X.; Dearborn, J.T.; Culver, J.P.; et al. Circadian clock proteins regulate neuronal redox homeostasis and neurodegeneration. J. Clin. Investig. 2013, 123, 5389–5400. [Google Scholar] [CrossRef] [Green Version]
- Pallier, P.N.; Maywood, E.S.; Zheng, Z.; Chesham, J.E.; Inyushkin, A.N.; Dyball, R.; Hastings, M.H.; Morton, A.J. Pharmacological Imposition of Sleep Slows Cognitive Decline and Reverses Dysregulation of Circadian Gene Expression in a Transgenic Mouse Model of Huntington’s Disease. J. Neurosci. 2007, 27, 7869–7878. [Google Scholar] [CrossRef] [Green Version]
- Pallier, P.N.; Morton, A.J. Management of sleep/wake cycles improves cognitive function in a transgenic mouse model of Huntington’s disease. Brain Res. 2009, 1279, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Rocha, N.B.F.; Rocha, S.; Herrera-Solís, A.; Salas-Pacheco, J.; García-García, F.; Murillo-Rodríguez, E.; Yuan, T.-F.; Machado, S.; Arias-Carrión, O. Detrimental role of prolonged sleep deprivation on adult neurogenesis. Front. Cell. Neurosci. 2015, 9, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tamai, S.; Sanada, K.; Fukada, Y. Time-of-Day-Dependent Enhancement of Adult Neurogenesis in the Hippocampus. PLoS ONE 2008, 3, e3835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandasamy, M.; Aigner, L. Neuroplasticity, limbic neuroblastosis and neuro-regenerative disorders. Neural Regen. Res. 2018, 13, 1322–1326. [Google Scholar] [CrossRef]
- Feliciano, D.M.; Bordey, A.; Bonfanti, L. Noncanonical Sites of Adult Neurogenesis in the Mammalian Brain. Cold Spring Harb. Perspect. Biol. 2015, 7, a018846. [Google Scholar] [CrossRef] [Green Version]
- Ming, G.; Song, H. Adult Neurogenesis in the Mammalian Brain: Significant Answers and Significant Questions. Neuron 2011, 70, 687–702. [Google Scholar] [CrossRef] [Green Version]
- Yoo, S.; Blackshaw, S. Regulation and function of neurogenesis in the adult mammalian hypothalamus. Prog. Neurobiol. 2018, 170, 53–66. [Google Scholar] [CrossRef]
- Gabery, S.; Halliday, G.; Kirik, D.; Englund, E.; Petersén, Å. Selective loss of oxytocin and vasopressin in the hypothalamus in early Huntington disease: A case study. Neuropathol. Appl. Neurobiol. 2015, 41, 843–848. [Google Scholar] [CrossRef]
- Hult Lundh, S.; Nilsson, N.; Soylu, R.; Kirik, D.; Petersén, Å. Hypothalamic expression of mutant huntingtin contributes to the development of depressive-like behavior in the BAC transgenic mouse model of Huntington’s disease. Hum. Mol. Genet. 2013, 22, 3485–3497. [Google Scholar] [CrossRef] [Green Version]
- Pla, P.; Orvoen, S.; Saudou, F.; David, D.J.; Humbert, S. Mood disorders in Huntington’s disease: From behavior to cellular and molecular mechanisms. Front. Behav. Neurosci. 2014, 8, 135. [Google Scholar] [CrossRef] [Green Version]
- Ali, A.A.H.; Schwarz-Herzke, B.; Mir, S.; Sahlender, B.; Victor, M.; Görg, B.; Schmuck, M.; Dach, K.; Fritsche, E.; Kremer, A.; et al. Deficiency of the clock gene Bmal1 affects neural progenitor cell migration. Brain Struct. Funct. 2019, 224, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Bouchard-Cannon, P.; Mendoza-Viveros, L.; Yuen, A.; Kærn, M.; Cheng, H.-Y.M. The Circadian Molecular Clock Regulates Adult Hippocampal Neurogenesis by Controlling the Timing of Cell-Cycle Entry and Exit. Cell Rep. 2013, 5, 961–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlasov, K.; van Dort, C.J.; Solt, K. Optogenetics and Chemogenetics. Methods Enzymol. 2018, 603, 181–196. [Google Scholar] [CrossRef] [PubMed]
- Boesmans, W.; Hao, M.M.; Vanden Berghe, P. Optogenetic and chemogenetic techniques for neurogastroenterology. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.P.; Fong, M.; Millard, D.C.; Whitmire, C.J.; Stanley, G.B.; Potter, S.M. Optogenetic feedback control of neural activity. eLife 2015, 4, e07192. [Google Scholar] [CrossRef] [Green Version]
- Klapper, S.D.; Swiersy, A.; Bamberg, E.; Busskamp, V. Biophysical Properties of Optogenetic Tools and Their Application for Vision Restoration Approaches. Front. Syst. Neurosci. 2016, 10, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holley, S.M.; Galvan, L.; Kamdjou, T.; Dong, A.; Levine, M.S.; Cepeda, C. Major Contribution of Somatostatin-Expressing Interneurons and Cannabinoid Receptors to Increased GABA Synaptic Activity in the Striatum of Huntington’s Disease Mice. Front. Synaptic Neurosci. 2019, 11, 14. [Google Scholar] [CrossRef] [Green Version]
- Cepeda, C.; Galvan, L.; Holley, S.M.; Rao, S.P.; André, V.M.; Botelho, E.P.; Chen, J.Y.; Watson, J.B.; Deisseroth, K.; Levine, M.S. Multiple Sources of Striatal Inhibition Are Differentially Affected in Huntington’s Disease Mouse Models. J. Neurosci. 2013, 33, 7393–7406. [Google Scholar] [CrossRef] [Green Version]
- Hristova, K.; Martinez-Gonzalez, C.; Watson, T.C.; Codadu, N.K.; Hashemi, K.; Kind, P.C.; Nolan, M.F.; Gonzalez-Sulser, A. Medial septal GABAergic neurons reduce seizure duration upon optogenetic closed-loop stimulation. Brain 2021, 144, 1576–1589. [Google Scholar] [CrossRef]
- Owen, S.F.; Liu, M.H.; Kreitzer, A.C. Thermal constraints on in vivo optogenetic manipulations. Nat. Neurosci. 2019, 22, 1061–1065. [Google Scholar] [CrossRef]
- Malyshev, A.; Goz, R.; LoTurco, J.J.; Volgushev, M. Advantages and Limitations of the Use of Optogenetic Approach in Studying Fast-Scale Spike Encoding. PLoS ONE 2015, 10, e0122286. [Google Scholar] [CrossRef]
- Berthoud, M.C.; Reilly, C.S. Adverse effects of general anaesthetics. Drug Saf. 1992, 7, 434–459. [Google Scholar] [CrossRef]
- Armbruster, B.N.; Li, X.; Pausch, M.H.; Herlitze, S.; Roth, B.L. Evolving the lock to fit the key to create a family of G protein-coupled receptors potently activated by an inert ligand. Proc. Natl. Acad. Sci. USA 2007, 104, 5163–5168. [Google Scholar] [CrossRef] [Green Version]
- Keifer, O.; Kambara, K.; Lau, A.; Makinson, S.; Bertrand, D. Chemogenetics a robust approach to pharmacology and gene therapy. Biochem. Pharmacol. 2020, 175, 113889. [Google Scholar] [CrossRef]
- Conklin, B.R.; Hsiao, E.C.; Claeysen, S.; Dumuis, A.; Srinivasan, S.; Forsayeth, J.R.; Guettier, J.-M.; Chang, W.C.; Pei, Y.; McCarthy, K.D.; et al. Engineering GPCR signaling pathways with RASSLs. Nat. Methods 2008, 5, 673–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, B.L. DREADDs for Neuroscientists. Neuron 2016, 89, 683–694. [Google Scholar] [CrossRef] [Green Version]
- Sternson, S.M.; Roth, B.L. Chemogenetic tools to interrogate brain functions. Annu. Rev. Neurosci. 2014, 37, 387–407. [Google Scholar] [CrossRef] [PubMed]
- Whissell, P.D.; Tohyama, S.; Martin, L.J. The Use of DREADDs to Deconstruct Behavior. Front. Genet. 2016, 7, 70. [Google Scholar] [CrossRef] [Green Version]
- Assaf, F.; Schiller, Y. A chemogenetic approach for treating experimental Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2019, 34, 469–479. [Google Scholar] [CrossRef]
- Yuan, P.; Grutzendler, J. Attenuation of β-Amyloid Deposition and Neurotoxicity by Chemogenetic Modulation of Neural Activity. J. Neurosci. 2016, 36, 632–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Milosavljevic, N.; Allen, A.E.; Cehajic-Kapetanovic, J.; Lucas, R.J. Chemogenetic Activation of ipRGCs Drives Changes in Dark-Adapted (Scotopic) Electroretinogram. Investig. Ophthalmol. Vis. Sci. 2016, 57, 6305–6312. [Google Scholar] [CrossRef] [PubMed]
- Gomez, J.L.; Bonaventura, J.; Lesniak, W.; Mathews, W.B.; Sysa-Shah, P.; Rodriguez, L.A.; Ellis, R.J.; Richie, C.T.; Harvey, B.K.; Dannals, R.F.; et al. Chemogenetics revealed: DREADD occupancy and activation via converted clozapine. Science 2017, 357, 503–507. [Google Scholar] [CrossRef] [Green Version]
- Baseri, B.; Choi, J.J.; Tung, Y.-S.; Konofagou, E.E. Multi-Modality Safety Assessment of Blood-Brain Barrier Opening Using Focused Ultrasound and Definity Microbubbles: A Short-Term Study. Ultrasound Med. Biol. 2010, 36, 1445–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaspar, B.K.; Erickson, D.; Schaffer, D.; Hinh, L.; Gage, F.H.; Peterson, D.A. Targeted retrograde gene delivery for neuronal protection. Mol. Ther. 2002, 5, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Ono, D.; Honma, K.-I.; Yanagawa, Y.; Yamanaka, A.; Honma, S. GABA in the suprachiasmatic nucleus refines circadian output rhythms in mice. Commun. Biol. 2019, 2, 232. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.A.; Morrison, E.H. Neuroanatomy, Nucleus Suprachiasmatic. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. Available online: http://www.ncbi.nlm.nih.gov/books/NBK546664/ (accessed on 8 November 2020).
- Van Wamelen, D.J.; Aziz, N.A.; Anink, J.J.; van Steenhoven, R.; Angeloni, D.; Fraschini, F.; Jockers, R.; Roos, R.A.C.; Swaab, D.F. Suprachiasmatic Nucleus Neuropeptide Expression in Patients with Huntington’s Disease. Sleep 2013, 36, 117–125. [Google Scholar] [CrossRef] [Green Version]
- Delgado, M.; Ganea, D. Neuroprotective effect of vasoactive intestinal peptide (VIP) in a mouse model of Parkinson’s disease by blocking microglial activation. FASEB J. 2003, 17, 944–946. [Google Scholar] [CrossRef] [Green Version]
- Deng, G.; Jin, L. The effects of vasoactive intestinal peptide in neurodegenerative disorders. Neurol. Res. 2017, 39, 65–72. [Google Scholar] [CrossRef]
- Eva, C.; Meek, J.L.; Costa, E. Vasoactive intestinal peptide which coexists with acetylcholine decreases acetylcholine turnover in mouse salivary glands. J. Pharmacol. Exp. Ther. 1985, 232, 670–674. [Google Scholar]
- Liu, C.; Gillette, M.U. Cholinergic regulation of the suprachiasmatic nucleus circadian rhythm via a muscarinic mechanism at night. J. Neurosci. 1996, 16, 744–751. [Google Scholar] [CrossRef]
- Alcacer, C.; Andreoli, L.; Sebastianutto, I.; Jakobsson, J.; Fieblinger, T.; Cenci, M.A. Chemogenetic stimulation of striatal projection neurons modulates responses to Parkinson’s disease therapy. J. Clin. Investig. 2017, 127, 720–734. [Google Scholar] [CrossRef] [PubMed]
- Kätzel, D.; Nicholson, E.; Schorge, S.; Walker, M.C.; Kullmann, D.M. Chemical-genetic attenuation of focal neocortical seizures. Nat. Commun. 2014, 5, 3847. [Google Scholar] [CrossRef] [PubMed]
- Lieb, A.; Weston, M.; Kullmann, D.M. Designer receptor technology for the treatment of epilepsy. eBioMedicine 2019, 43, 641–649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daya, S.; Berns, K.I. Gene Therapy Using Adeno-Associated Virus Vectors. Clin. Microbiol. Rev. 2008, 21, 583–593. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ravichandran, S.; Suhasini, R.; Madheswaran Deepa, S.; Selvaraj, D.B.; Vergil Andrews, J.F.; Thiagarajan, V.; Kandasamy, M. Intertwining Neuropathogenic Impacts of Aberrant Circadian Rhythm and Impaired Neuroregenerative Plasticity in Huntington’s Disease: Neurotherapeutic Significance of Chemogenetics. J. Mol. Pathol. 2022, 3, 355-371. https://doi.org/10.3390/jmp3040030
Ravichandran S, Suhasini R, Madheswaran Deepa S, Selvaraj DB, Vergil Andrews JF, Thiagarajan V, Kandasamy M. Intertwining Neuropathogenic Impacts of Aberrant Circadian Rhythm and Impaired Neuroregenerative Plasticity in Huntington’s Disease: Neurotherapeutic Significance of Chemogenetics. Journal of Molecular Pathology. 2022; 3(4):355-371. https://doi.org/10.3390/jmp3040030
Chicago/Turabian StyleRavichandran, Sowbarnika, Ramalingam Suhasini, Sudhiksha Madheswaran Deepa, Divya Bharathi Selvaraj, Jemi Feiona Vergil Andrews, Viruthachalam Thiagarajan, and Mahesh Kandasamy. 2022. "Intertwining Neuropathogenic Impacts of Aberrant Circadian Rhythm and Impaired Neuroregenerative Plasticity in Huntington’s Disease: Neurotherapeutic Significance of Chemogenetics" Journal of Molecular Pathology 3, no. 4: 355-371. https://doi.org/10.3390/jmp3040030