
A New Fluorogenic Substrate for Granzyme B Based on Fluorescence Resonance Energy Transfer †


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Reagents and Solvents
2.2. Instrumentation
2.3. Methods
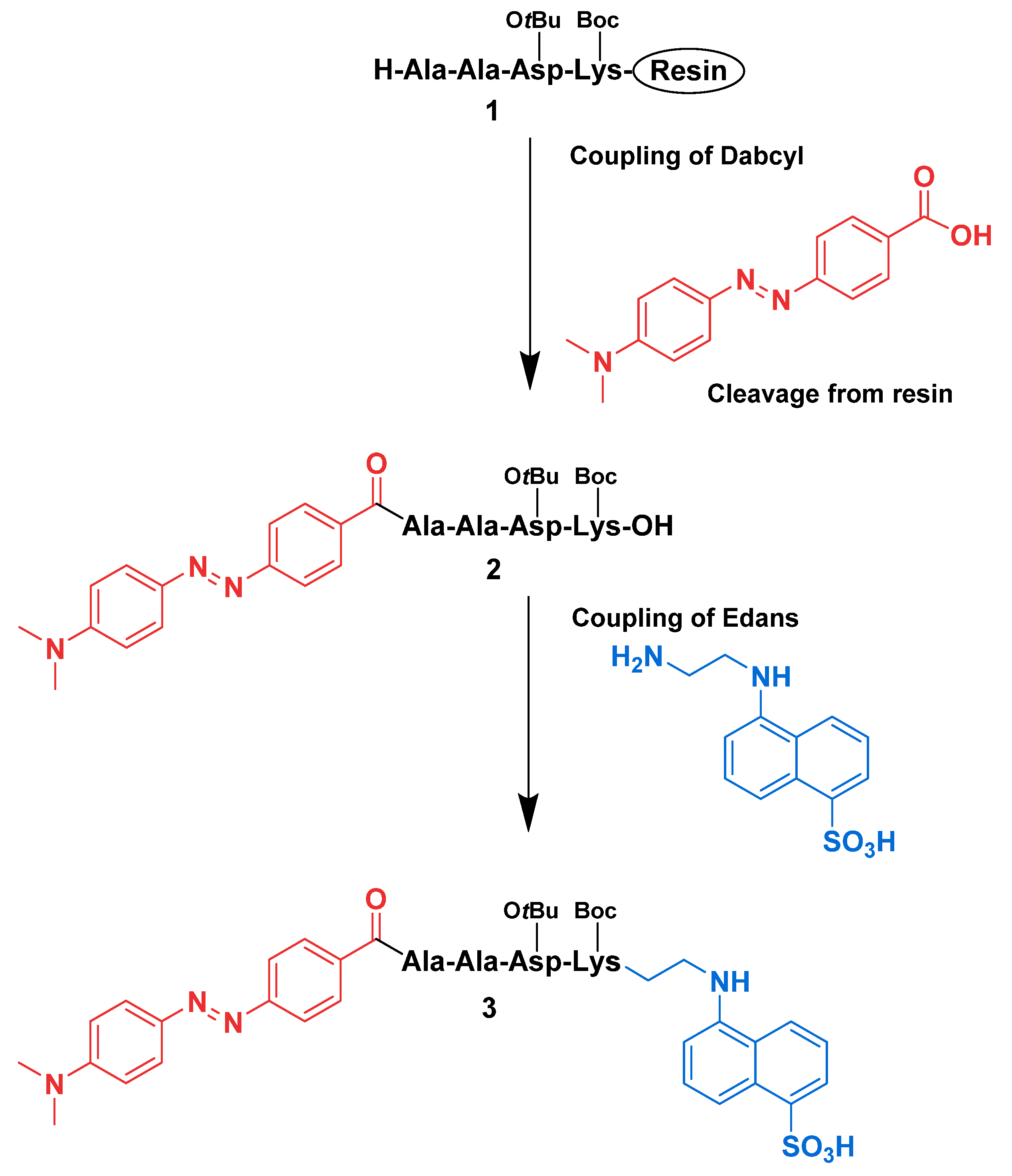
2.3.1. Synthesis of H-Ala-Ala-Asp(OtBu)-Lys(Boc)-OH on Resin (1)
2.3.2. Synthesis of Dabcyl-Ala-Ala-Asp(OtBu)-Lys(Boc)-OH (2)
2.3.3. Synthesis of Dabcyl-Ala-Ala-Asp(OtBu)-Lys(Boc)-Edans (3)
2.3.4. UV/Vis Absorption and Fluorescence Spectroscopy of Peptides 2–3
3. Results and Discussion
3.1. Synthesis of the Fluorogenic Substrate for Granzyme B
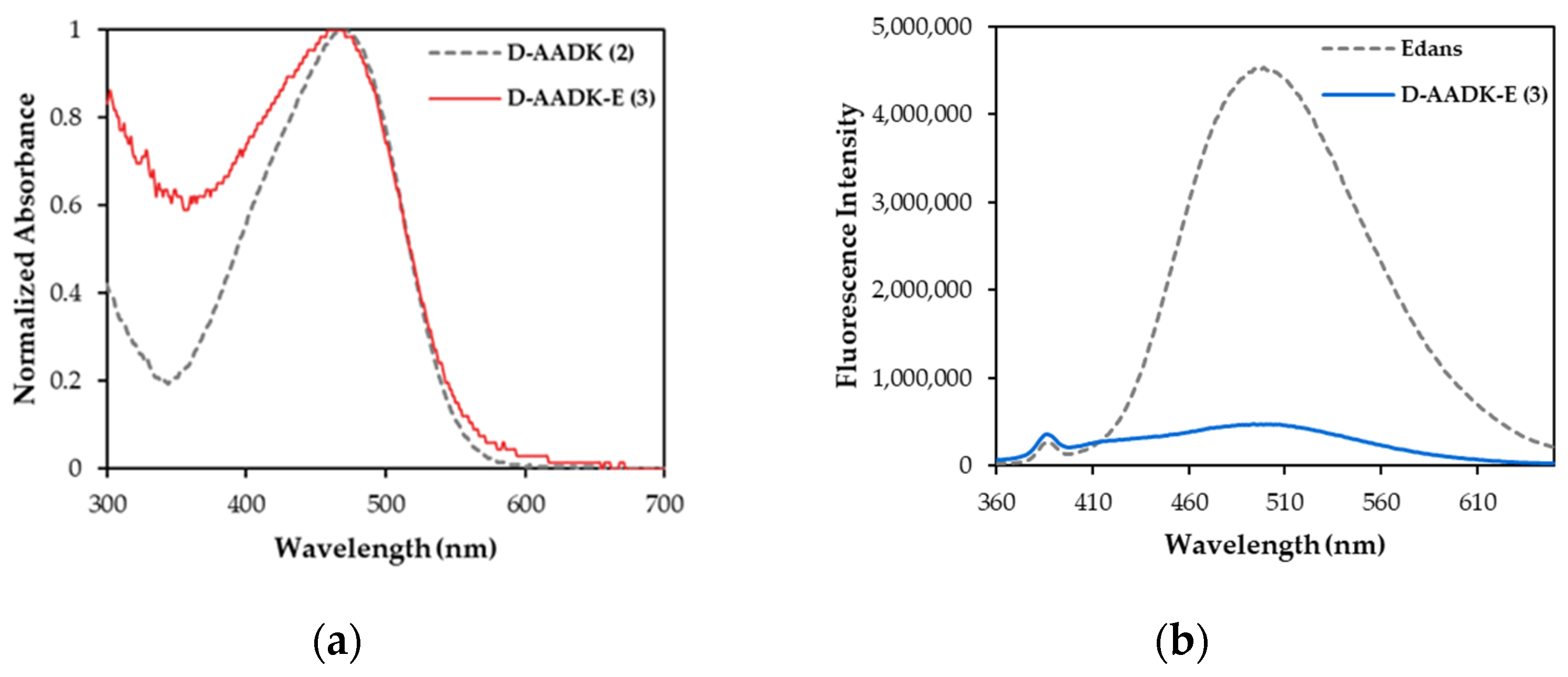
3.2. UV/Vis Absorption and Fluorescence Spectroscopy
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Larimer, B.M.; Wehrenberg-Klee, E.; Dubois, F.; Mehta, A.; Kalomeris, T.; Flaherty, K.; Boland, G.; Mahmood, U. Granzyme B PET Imaging as a Predictive Biomarker of Immunotherapy Response. Cancer Res. 2017, 77, 2318–2327. [Google Scholar] [CrossRef] [PubMed]
- Ida, H.; Utz, P.J.; Anderson, P.; Eguchi, K. Granzyme B and Natural Killer (NK) Cell Death. Mod. Rheumatol. 2005, 15, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Waugh, S.M.; Craik, C.S.; Harris, J.L.; Fletterick, R. The Structure of the Pro-Apoptotic Protease Granzyme B Reveals the Molecular Determinants of Its Specificity. Nat. Struct. Biol. 2000, 7, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Hagn, M.; Sutton, V.R.; Trapani, J.A. A Colorimetric Assay That Specifically Measures Granzyme B Proteolytic Activity: Hydrolysis of Boc-Ala-Ala-Asp-S-Bzl. J. Vis. Exp. 2014, 93, e52419. [Google Scholar] [CrossRef] [PubMed]
- Nie, Z.; Phenix, B.N.; Lum, J.J.; Alam, A.; Lynch, D.H.; Beckett, B.; Krammer, P.H.; Sekaly, R.P.; Badley, A.D. HIV-1 Protease Processes Procaspase 8 to Cause Mitochondrial Release of Cytochrome c, Caspase Cleavage and Nuclear Fragmentation. Cell Death Differ. 2002, 9, 1172–1184. [Google Scholar] [CrossRef] [PubMed]
- Estébanez-Perpiñá, E.; Fuentes-Prior, P.; Belorgey, D.; Braun, M.; Kiefersauer, R.; Maskos, K.; Huber, R.; Rubin, H.; Bode, W. Crystal Structure of the Caspase Activator Human Granzyme B, a Proteinase Highly Specific for an Asp-P1 Residue. Biol. Chem. 2000, 381, 1203–1214. [Google Scholar] [CrossRef] [PubMed]
- Harris, J.L.; Peterson, E.P.; Hudig, D.; Thornberry, N.A.; Craik, C.S. Definition and Redesign of the Extended Substrate Specificity of Granzyme B. J. Biol. Chem. 1998, 273, 27364–27373. [Google Scholar] [CrossRef] [PubMed]
- Drake, C.R.; Miller, D.C.; Jones, E.F. Activatable Optical Probes for the Detection of Enzymes. Curr. Org. Synth. 2011, 8, 498–520. [Google Scholar] [CrossRef] [PubMed]
- Johansson, M.K.; Cook, R.M. Intramolecular Dimers: A New Design Strategy for Fluorescence-Quenched Probes. Chem. Eur. J. 2003, 9, 3466–3471. [Google Scholar] [CrossRef] [PubMed]
- Grahn, S.; Ullmann, D.; Jakubke, H.-D. Design and Synthesis of Fluorogenic Trypsin Peptide Substrates Based on Resonance Energy Transfer. Anal. Biochem. 1998, 265, 225–231. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Chen, L.L.; Luo, H.B.; Sun, T.; Chen, J.; Ye, F.; Cai, J.; Shen, J.; Shen, X.; Jiang, H.L. Enzymatic Activity Characterization of SARS Coronavirus 3C-like Protease by Fluorescence Resonance Energy Transfer Technique. Acta Pharmacol. Sin. 2005, 26, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Windsor, I.W.; Raines, R.T. Fluorogenic Assay for Inhibitors of HIV-1 Protease with Sub-Picomolar Affinity. Sci. Rep. 2015, 5, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, A.V.; Perelshtein, I.; Perkas, N.; Gedanken, A.; Cunha, J.; Cavaco-Paulo, A. Detection of Human Neutrophil Elastase (HNE) on Wound Dressings as Marker of Inflammation. Appl. Microbiol. Biotechnol. 2017, 101, 1443–1454. [Google Scholar] [CrossRef] [PubMed]
- Holskin, B.P.; Bukhtiyarova, M.; Dunn, B.M.; Baur, P.; Dechastonay, J.; Pennington, M.W. A Continuous Fluorescence-Based Assay of Human Cytomegalovirus Protease Using a Peptide Substrate. Anal. Biochem. 1995, 227, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, A.K.; Richardson, P.L.; Kurtz, K.A.; Tripathi, R.; Chen, C.M.; Huang, P.; Randolph, J.; Towne, D.; Donnelly, J.; Warrior, U.; et al. Longer Wavelength Fluorescence Resonance Energy Transfer Depsipeptide Substrates for Hepatitis C Virus NS3 Protease. Anal. Biochem. 2007, 368, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.D.F.; Raposo, M.M.M.; Costa, S.P.G. Intermolecular Quenching of Edans/Dabcyl Donor–Acceptor FRET Pair. Proceedings 2019, 41, 34. [Google Scholar] [CrossRef]
- Hunger, K.; Mischke, P.; Rieper, W.; Raue, R.; Kunde, K.; Engel, A. Azo Dyes. In Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar] [CrossRef]
- Wang, G.T.; Matayoshi, E.; Jan Huffaker, H.; Krafft, G.A. Design and Synthesis of New Fluorogenic HIV Protease Substrates Based on Resonance Energy Transfer. Tetrahedron Lett. 1990, 31, 6493–6496. [Google Scholar] [CrossRef]
- Schechter, I.; Berger, A. On the Size of the Active Site in Proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martins, C.D.F.; Raposo, M.M.M.; Costa, S.P.G. A New Fluorogenic Substrate for Granzyme B Based on Fluorescence Resonance Energy Transfer. Chem. Proc. 2021, 3, 6. https://doi.org/10.3390/ecsoc-24-08311
Martins CDF, Raposo MMM, Costa SPG. A New Fluorogenic Substrate for Granzyme B Based on Fluorescence Resonance Energy Transfer. Chemistry Proceedings. 2021; 3(1):6. https://doi.org/10.3390/ecsoc-24-08311
Chicago/Turabian StyleMartins, Cátia D. F., M. Manuela M. Raposo, and Susana P. G. Costa. 2021. "A New Fluorogenic Substrate for Granzyme B Based on Fluorescence Resonance Energy Transfer" Chemistry Proceedings 3, no. 1: 6. https://doi.org/10.3390/ecsoc-24-08311