
Theoretical Study of the Aza˗Wittig Reaction, Me3P=NR (R = Methyl or Phenyl) with Aldehyde Using the DFT and DFT-D Methods (Dispersion Correction) †


Abstract
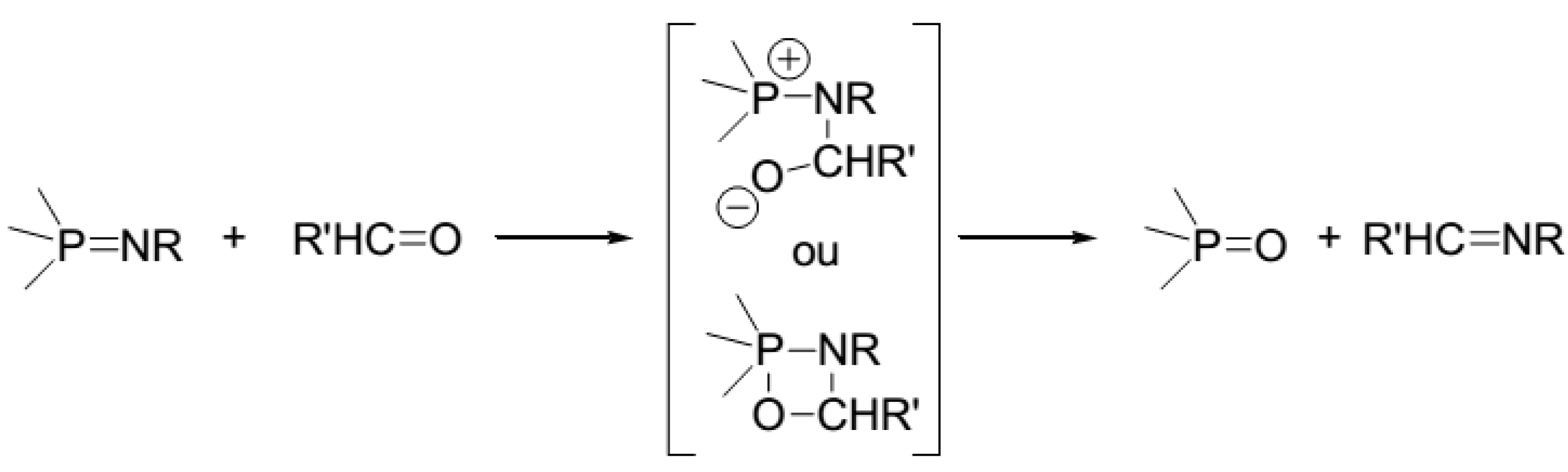
:1. Introduction
2. Computational Methods
3. Results and Discussion
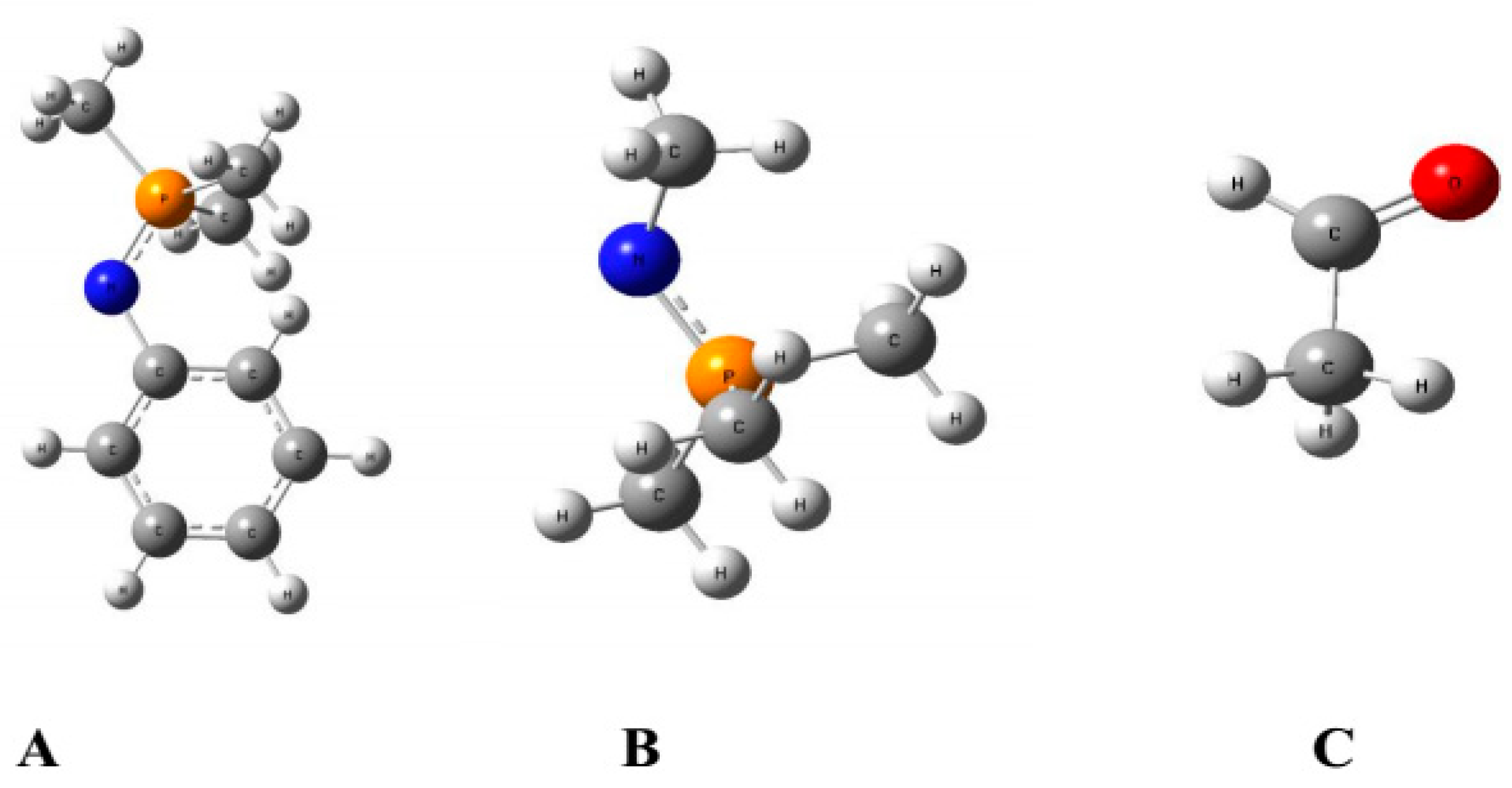
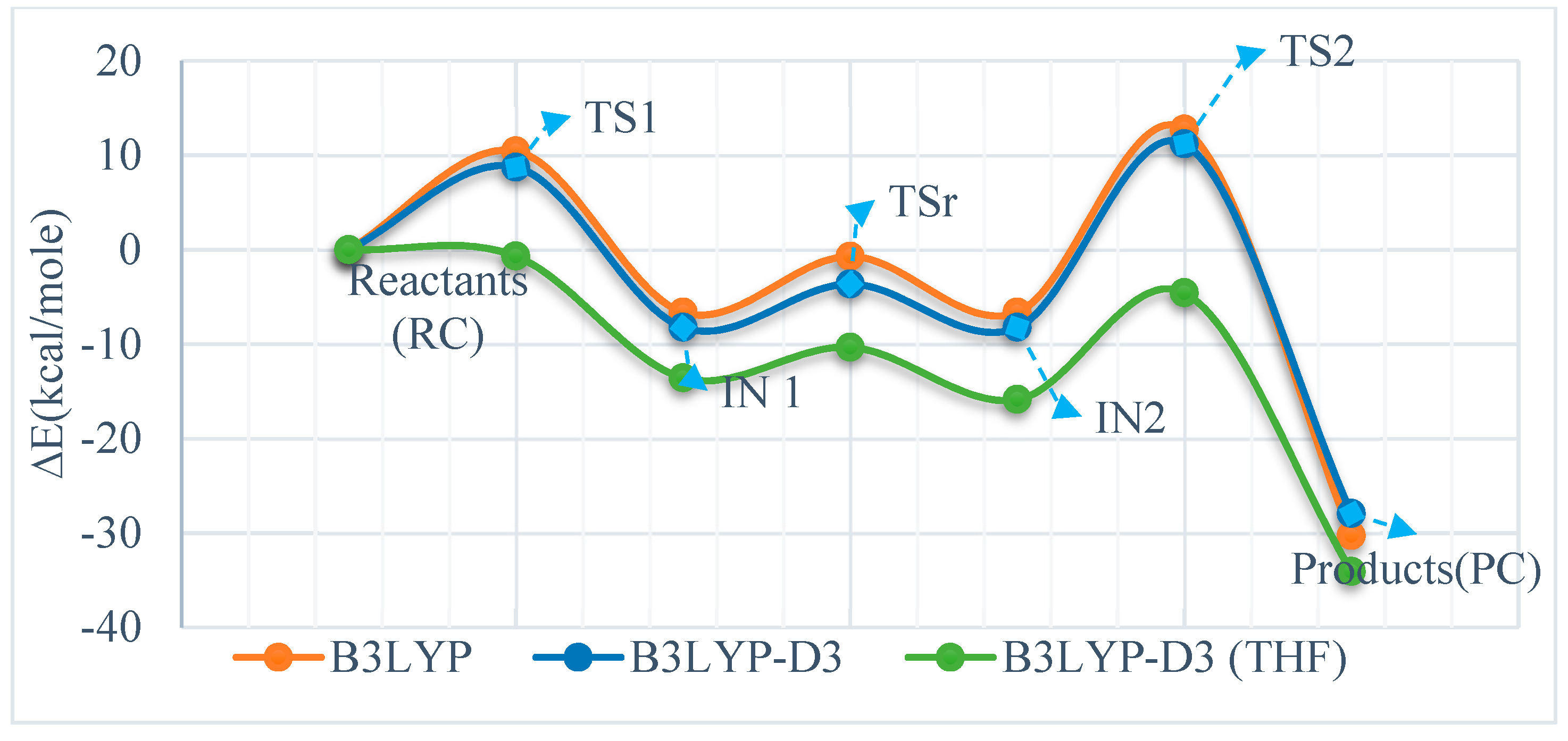
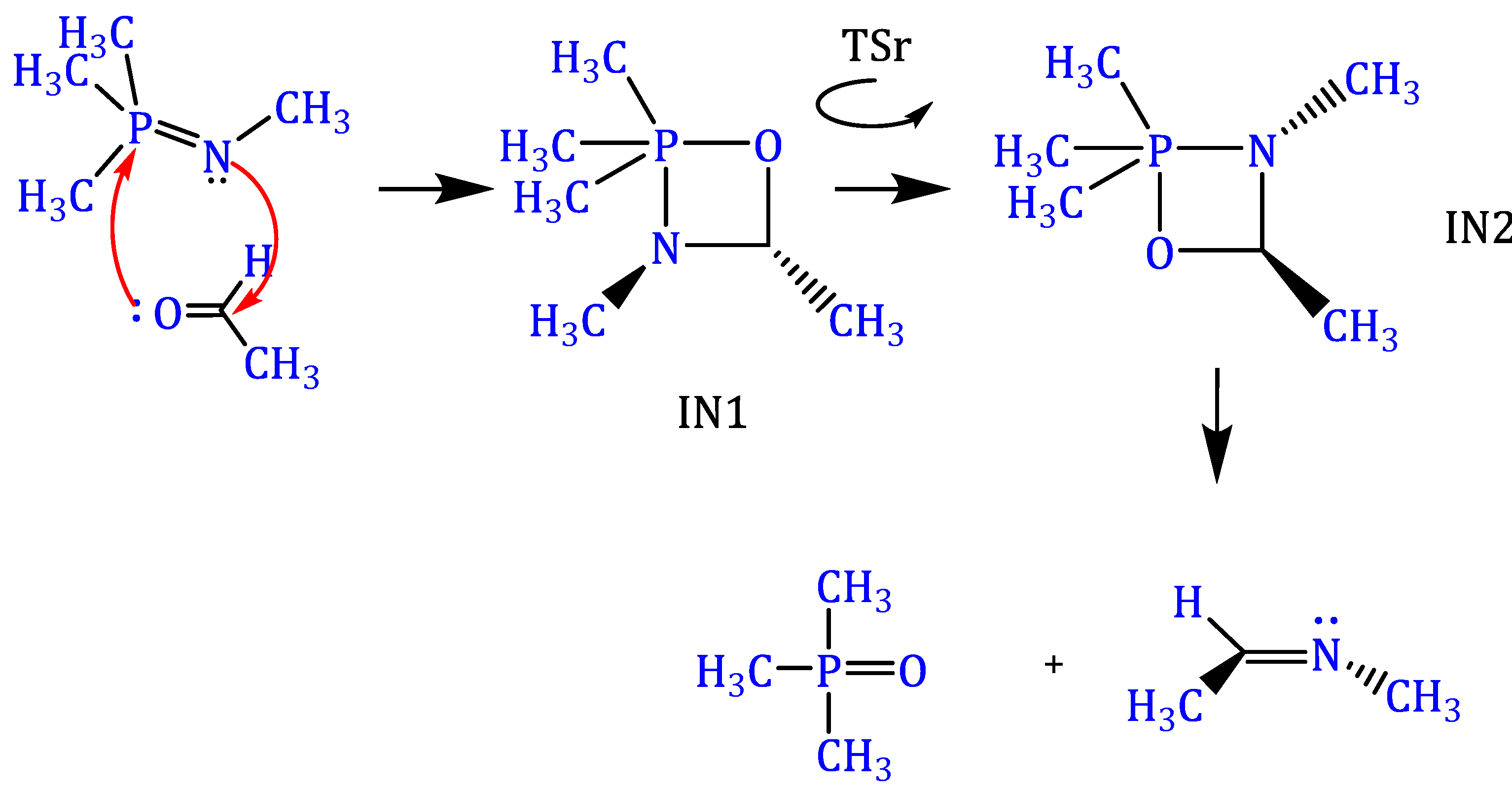
3.1. Reaction of Methylimino(trimethyl)phosphorane with Acetaldehyde
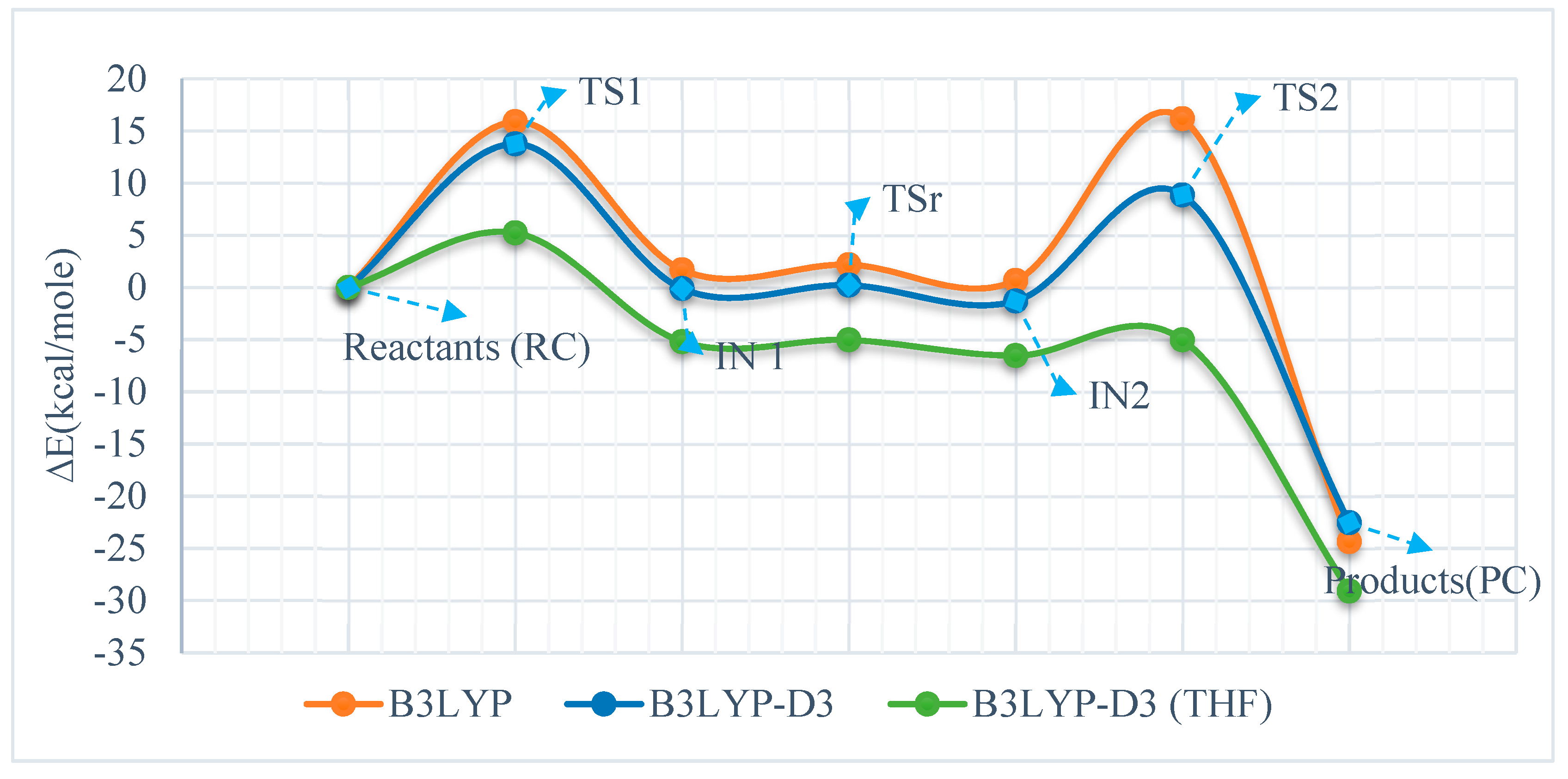
3.2. Reaction of Phenylimino(trimethyl)phosphorane with Acetaldehyde
4. Conclusions
Author Contributions
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
References
- Staudinger, H. New organic compounds of phosphorus. III. Phosphine-methylene derivatives and phosphinimines. Helv. Chim. Acta 1919, 2, 635–646. [Google Scholar] [CrossRef]
- Naito, T.; Nagase, S.; Yamataka, H. Theoretical study of the structure and reactivity of ylides of N, P, As, Sb, and Bi. J. Am. Chem. Soc. 1994, 116, 10080–10088. [Google Scholar] [CrossRef]
- Volatron, F.; Eisenstein, O. Wittig versus Corey-Chaykovsky Reaction. Theoretical study of the reactivity of phosphonium methylide and sulfonium methylide with formaldehyde. J. Am. Chem. Soc. 1987, 109, 1–14. [Google Scholar] [CrossRef]
- Streitwieser Jr, A.; Rajca, A.; McDowell, R.S.; Glaser, R. Semipolar phosphorus-oxygen and phosphorus-carbon bonds. A theoretical study of hypophosphite and related methylenephosphoranes. J. Am. Chem. Soc. 1987, 109, 4184–4188. [Google Scholar] [CrossRef]
- Dixon, D.A.; Smart, B.E. The structures and energetics of fluorine-substituted phosphonium ylides. J. Am. Chem. Soc. 1986, 108, 7172–7177. [Google Scholar] [CrossRef]
- Eades, R.A.; Gassman, P.G.; Dixon, D.A. The conformations and energetics of simple ylides. J. Am. Chem. Soc. 1981, 103, 1066–1068. [Google Scholar] [CrossRef]
- Vincent, M.A.; Schaefer III, H.F.; Schier, A.; Schmidbaur, H. Molecular and electronic structure of phosphonium cyclopropylide: A theoretical study. J. Am. Chem. Soc. 1983, 105, 3806–3811. [Google Scholar] [CrossRef]
- Hoeller, R.; Lischka, H. A theoretical investigation on the model Wittig reaction PH3CH2+ CH2O. fwdarw. PH3O+ C2H4. J. Am. Chem. Soc. 1980, 102, 4632–4635. [Google Scholar] [CrossRef]
- Maryanoff, B.E.; Reitz, A.B. The Wittig olefination reaction and modifications involving phosphoryl-stabilized carbanions. Stereochemistry, mechanism, and selected synthetic aspects. Chem. Rev. 1989, 89, 863–927. [Google Scholar] [CrossRef]
- Lischka, H. Electronic structure and proton affinity of methylenephosphorane by ab initio methods including electron correlation. J. Am. Chem. Soc. 1977, 99, 353–360. [Google Scholar] [CrossRef]
- Nguyen, M.T.; Hegarty, A.F. An Ab initio study of the diadic prototropic tautomerism H3PX⇄ H2PXH (X= O, NH, CH2). J. Chem. Soc. Perkin Trans. 1987, 2, 47–54. [Google Scholar] [CrossRef]
- Bachrach, S.M. Molecular structure of phosphonium ylides. J. Org. Chem. 1992, 57, 4367–4373. [Google Scholar] [CrossRef]
- Sasaki, T.; Eguchi, S.; Okano, T. Novel synthesis and reactivity of 4-azahomoadamant-3-ene and 4-aza-4-homobrend-3-enes via intramolecular aza-Wittig reactions. J. Am. Chem. Soc. 1983, 105, 5912–5913. [Google Scholar] [CrossRef]
- Sheldrick, W.S.; Schomburg, D.; Schmidpeter, A.; von Criegern, T. Four-And Five-Membered Phosphorus Heterocycles. 39. Structural Changes in The (2+ 2) Cycloaddition of Ketones to Azaphospholes. Chem. Inf. 1980, 11, 304. [Google Scholar] [CrossRef]
- Molina, P.; Alajarin, M.; Lopez Leonardo, C.; Claramunt, R.M.; Foces-Foces, M.D.L.C.; Hernandez Cano, F.; Catalan, J.; De Paz, J.L.G.; Elguero, J. Experimental and theoretical study of the R3P+-X-bond. Case of betaines derived from N-iminophosphoranes and alkyl isocyanates. J. Am. Chem. Soc. 1989, 111, 355–363. [Google Scholar] [CrossRef]
- Lu, W.C.; Liu, C.B.; Sun, C.C. Theoretical study of the H3PNH+ H2CO reaction mechanism via five reaction channels. J. Phys. Chem. A 1999, 103, 1078–1083. [Google Scholar] [CrossRef]
- Lu, W.C.; Sun, C.C.; Zang, Q.J.; Liu, C.B. Theoretical study of the aza-Wittig reaction X3P = NH + O = CHCOOH → X3P = O+ HN= CHCOOH for X = Cl, H and CH3. Chem. Phys. Lett. 1999, 311, 491–498. [Google Scholar] [CrossRef]
- Koketsu, J.; Ninomiya, Y.; Suzuki, Y.; Koga, N. Theoretical study on the structures of iminopnictoranes and their reactions with formaldehyde. Inorganic Chemistry 1997, 36, 694–702. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef]
- Hariharan, P.C.; Pople, J.A. The influence of polarization functions on molecular orbital hydrogenation energies. Theor. Chim. Acta 1973, 28, 213–222. [Google Scholar] [CrossRef]
- Marí, F.; Lahti, P.M.; McEwen, W.E. Molecular modeling of the Wittig olefination reaction: Part 2: A molecular orbital approach at the MNDO-PM3 level. Heteroat. Chem. 1991, 2, 265–276. [Google Scholar] [CrossRef]
- Mari, F.; Lahti, P.M.; McEwen, W.E. Molecular modeling of the Wittig reaction. 3. A theoretical study of the Wittig olefination reaction: MNDO-PM3 treatment of the Wittig half-reaction of unstabilized ylides with aldehydes. J. Am. Chem. Soc. 1992, 114, 813–821. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structures | (B3LYP) | (B3LYP-D3) | ||||
|---|---|---|---|---|---|---|
| ∆E (kcal/mol) | ∆G (kcal/mol) | ∆H (kcal/mol) | ∆E (kcal/mol) | ∆G (kcal/mol) | ∆H (kcal/mol) | |
| Reactants (RC) | 0.00 | 000 | 0.00 | 0.00 | 0.00 | 0.00 |
| TS1 | 10.44 | 14.77 | 10.28 | 8.73 | 12.17 | 8.52 |
| IN 1 | −6.63 | −0.05 | −5.01 | −8.23 | −2.46 | −6.62 |
| TSr | −0.71 | 6.44 | −0.98 | −3.63 | 2.75 | −2.85 |
| IN2 | −6.63 | −0.05 | −5.01 | −8.23 | −2.46 | −6.62 |
| TS2 | 12.75 | 17.97 | 12.80 | 11.20 | 16.06 | 11.32 |
| Products (PC) | −30.27 | −29.31 | −29.53 | −27.94 | −26.90 | −27.15 |
| B3LYP/6-31G** B3LYP-D3/6-31G** | dP–N Ångström | dC–O Ångström | dP–O Ångström | dN–C Ångström | ΦPNCO |
|---|---|---|---|---|---|
| Reactants (RC) | 1.58 1.58 | 1.22 1.22 | 3.63 3.45 | 3.07 2.84 | −2.42 −0.50 |
| TS1 | 1.64 1.63 | 1.28 1.28 | 2.69 2.70 | 1.76 1.76 | −7.30 −6.75 |
| IN1 | 1.72 1.71 | 1.40 1.40 | 1.84 1.84 | 1.47 1.46 | −6.73 −6.95 |
| TSr | 1.81 1.81 | 1.42 1.43 | 1.73 1.73 | 1.45 1.45 | −11.76 −11.66 |
| IN2 | 1.72 1.71 | 1.40 1.40 | 1.84 1.84 | 1.47 1.46 | −6.75 −6.95 |
| TS2 | 2.42 2.41 | 1.69 1.69 | 1.59 1.59 | 1.36 1.36 | 8.58 10.20 |
| Products (PC) | 4.13 3.89 | 3.76 3.53 | 1.51 1.51 | 1.27 1.27 | −2.11 −2.01 |
 |  |
| Reactants (RC) | TS1 |
 |  |
| IN1 | TSr |
 |  |
| IN2 | TS2 |
 | |
| Products (PC) | |
| RC → TS1 | 14.77 | 12.17 |
| IN2 → TS2 | 18.01 | 18.52 |
| TS1 |  |
| IN1 |  |
| B3LYP/6-31G** B3LYP-D3/6-31G** | dP–N Ångström | dC–O Ångström | dP–O Ångström | dN–C Ångström | ΦPNCO |
|---|---|---|---|---|---|
| Reactants (RC) | 1.59 1.59 | 1.22 1.22 | 3.60 3.46 | 3.12 2.89 | −3.38 0.72 |
| TS1 | 1.65 1.64 | 1.29 1.29 | 2.55 2.57 | 1.74 1.73 | −7.29 −8.33 |
| IN 1 | 1.73 1.72 | 1.40 1.40 | 1.84 1.84 | 1.47 1.47 | −5.42 −5.35 |
| TSr | 1.93 1.91 | 1.45 1.45 | 1.68 1.68 | 1.44 1.43 | −4.88 −5.37 |
| IN2 | 1.92 1.90 | 1.44 1.44 | 1.68 1.69 | 1.44 1.44 | −3.55 −3.71 |
| TS2 | 2.60 2.50 | 1.62 1.58 | 1.59 1.60 | 1.38 1.39 | 1.77 8.90 |
| Products (PC) | 4.05 3.75 | 3.21 2.97 | 1.51 1.51 | 1.28 1.28 | 9.05 11.62 |
 |  |
| Reactants (RC) | TS1 |
 |  |
| IN1 | TSr |
 |  |
| IN2 | TS2 |
 | |
| Products (PC) | |
| Structures | (B3LYP) | (B3LYP-D3) | ||||
|---|---|---|---|---|---|---|
| ∆E (kcal/mol) | ∆G (kcal/mol) | ∆H (kcal/mol) | ∆E (kcal/mol) | ∆G (kcal/mol) | ∆H (kcal/mol) | |
| Reactants (RC) | 0 | 0 | 0 | 0.00 | 0.00 | 0.00 |
| TS1 | 15.88 | 19.05 | 15.59 | 13.76 | 16.30 | 13.40 |
| IN 1 | 1.69 | 7.36 | 3.03 | −0.11 | 3.69 | 1.15 |
| TSr | 2.18 | 9.21 | 3.00 | 0.23 | 6.08 | 0.99 |
| IN2 | 0.66 | 7.51 | 2.15 | −1.30 | 4.37 | 0.14 |
| TS2 | 10.23 | 14.87 | 10.24 | 8.85 | 13.31 | 8.98 |
| Products (PC) | −24.35 | −23.29 | −23.67 | −22.55 | −21.48 | −21.89 |
| RC → TS1 | 19.05 | 16.30 |
| IN2 → TS2 | 7.35 | 8.93 |
| TS1 |  |
| IN1 |  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adda, A.; Sediki, H. Theoretical Study of the Aza˗Wittig Reaction, Me3P=NR (R = Methyl or Phenyl) with Aldehyde Using the DFT and DFT-D Methods (Dispersion Correction). Chem. Proc. 2021, 3, 47. https://doi.org/10.3390/ecsoc-24-08349
Adda A, Sediki H. Theoretical Study of the Aza˗Wittig Reaction, Me3P=NR (R = Methyl or Phenyl) with Aldehyde Using the DFT and DFT-D Methods (Dispersion Correction). Chemistry Proceedings. 2021; 3(1):47. https://doi.org/10.3390/ecsoc-24-08349
Chicago/Turabian StyleAdda, Abdelghani, and Hayat Sediki. 2021. "Theoretical Study of the Aza˗Wittig Reaction, Me3P=NR (R = Methyl or Phenyl) with Aldehyde Using the DFT and DFT-D Methods (Dispersion Correction)" Chemistry Proceedings 3, no. 1: 47. https://doi.org/10.3390/ecsoc-24-08349