
Comparative Studies of Various NNRTIs in the Active Site of Different HIV-1RT Receptors †

Department of Pharmaceutical Sciences and Technology, Birla Institute of Technology, Mesra, Ranchi 835215, India
*
Author to whom correspondence should be addressed.
†
Presented at the 24th International Electronic Conference on Synthetic Organic Chemistry, 15 November–15 December 2020; Available online: https://ecsoc-24.sciforum.net/.
Chem. Proc. 2021, 3(1), 33; https://doi.org/10.3390/ecsoc-24-08313
Published: 14 November 2020
(This article belongs to the Proceedings of The 24th International Electronic Conference on Synthetic Organic Chemistry)
Abstract
:HIV is one of the most deadly viruses known to humans and causes a disease, known as Acquired Immunodeficiency Syndrome (or, AIDS). There are only a handful of drugs which are totally effective against the virus. This is due to the enzyme reverse transcriptase present within the virus. Due to various mutations in the enzyme, the virus becomes unresponsive towards the drugs. In the present study, docking studies of the standard non-nucleoside reverse transcriptase inhibitors were performed in the non-nucleoside inhibitory binding pocket of reverse transcriptase enzymes of wild type and the resistant strains of HIV-1RT virus with PDB IDs 1RT2, 1KLM, 3BGR, and 1JLB, respectively, by using Autodock version 4.5.6. A comparison of different compounds docked into the active site of various HIV-1RT strains was carried out. The obtained results indicate that most of the compounds docked into the active site of the different receptors, such as 1RT2, 1KLM, 3BGR, and 1JLB, with good docking scores, are comparable to that of the internal standard (TNK 651) of the wild type strain of HIV-1 virus. A comparison was made based on the binding modes of the compounds in the active site of all the four receptors.
1. Introduction
Human immunodeficiency virus contains a single strand of RNA belonging to the retroviridae family. It can cause the deadliest disease of the century called AIDS [1]. The enzyme reverse transcriptase helps in reverse transcription of cDNA (formation of double-stranded DNA from single-stranded RNA) and thus plays a crucial role in the life cycle of the virus. HIV can be categorized into two subtypes: HIV-1 (causes infections worldwide) and HIV-2 (confined to the western part of Africa) [2]. The infections caused by HIV can be blocked by targeting various steps of the life cycle of the virus, like attachment of the virus to the human cell, entry of the virus, and uncoating of the virus. Various enzymes like reverse transcriptase, protease, and integrase play a vital role in different processes of the viral life cycle, and various classes of drugs help inhibiting these enzymes such as non-nucleoside reverse transcriptase inhibitors (NNRTIs), nucleoside reverse transcriptase inhibitors (NRTIs), protease inhibitors, and nucleotide reverse transcriptase inhibitors (NtRTIs) [3,4].
The mode of action of NNRTIs is different from NRTIs because they directly bind with the active site of the reverse transcriptase enzyme and inhibit the movable proteins responsible for DNA synthesis and thus block the activity of the enzyme. This ultimately leads to the termination of the chain elongation process [5,6,7].
Although NNRTIs are chemically diversified in nature, their binding site is the same in the enzyme. Their binding site is an allosteric site containing a hydrophobic pocket, which is situated 10 Å (approximately) from the catalytic site of reverse transcriptase [8].
The butterfly-like form is important for the binding of NNRTIs. Chemically diversified drugs of this class assume a butterfly-like form which is found to be similar in all cases. Two hydrophobic aromatic rings are found in NNRTIs. They mimic a butterfly’s wings, whereas the body of the butterfly structure is represented by a hydrophilic moiety [9]. One of the wings contain a heteroaromatic ring that can act as hydrogen bond acceptor and/or donator to the main chain of amino acids, whereas the other wing is capable of forming pi–pi interactions with a hydrophobic moiety.
The keystone of highly active antiretroviral therapy (HAART) are three drugs of the class NNRTIs that have been approved by the FDA for HIV infection treatment: nevirapine, delavirdine, and efavirenz. These drugs were approved in the years 1996, 1997, and 1998, respectively. Often, they are used along with protease inhibitors and NRTIs for an effective treatment of HIV infection [10].
Docking Strategies: In case of structure-based drug designing of drugs in pharmaceutical industries, molecular docking is a very commonly used method because it can predict the binding of various conformations of ligands to the target binding site. Binding activity characterization plays an important role in rational drug designing and in the elucidation of essential biochemical processes [11,12].
The non-competitive binding of NNRTIs causes a change in conformation of the 3D structure of reverse transcriptase and leads to the formation of a non-nucleoside inhibitory binding pocket (NNIBP). It is elastic in nature and various characteristics of NNRTIs such as size, chemical composition, and binding mode affect its conformation. Various amino acid residues are present in NNIBP for interactions with NNRTIs [13].
2. Materials and Methods
2.1. Software
Autodock v 4.5.6 was used for carrying out the computational studies [14], installed in a HP Precision workstation (Radeon Graphics) with an Intel Core 3 quad processor and 8 GB of RAM with Windows 10 as operating system.
2.2. Molecular Modelling Studies
2.2.1. Protein Preparation
The X-ray co-crystallized structures of all of the protein molecules (PDB ID: 1RT2, 1JLB, 1KLM, 3BGR) used in the study were retrieved from the Research Collaboratory for Structural Bioinformatics (RCSB) [15]. From every protein molecule, the co-crystallized water molecule was deleted and polar hydrogens were added as well as Gasteiger charges assigned, and it was saved in PDBQT format using the Autodock v 4.5.6 software.
2.2.2. Ligand Preparation
All of the ligands were prepared by minimizing their energies using the PRODRG 2 server [16]. The PDBQT format of all of the ligands were saved.
2.2.3. Receptor Grid Generation
Autogrid was used to generate specific grid maps for each and every ligand.
The generation of the grid box was done by taking the dimensions of the three coordinates (X, Y, and Z) at 24 × 24 × 24, with grid spacing of 0.100 Å. The values of X, Y, and Z centers were taken according to the crystallographic positions of the native ligand of each and every receptor.
2.2.4. Docking Protocol Validation
For computational studies, Autodock v 4.5.6 was used. This software was used to predict the different binding modes of co-crystallized ligands as well as test molecules with all of the receptors taken to carry out the study.
To carry out the docking procedure, the method was validated to check the robustness of the software. The extracted ligand (previously mentioned) was corrected and then it was redocked using the same protein. Other test molecules were docked using the same procedure. Next, their conformations were compared with the co-crystallized one. The generated docking scores of the co-crystallized ligand were compared with the docking scores of other test molecules to choose the best molecule.
3. Results and Discussions
There Are Many HIV-1 Protein Crystal Structures Available in the Literature
In this work we have considered four crystal structures (PDB ID 1KLM, 3BGR, 1JLB, and 1RT2) co-crystallized with the ligands BHAP U-90152, rilpivirine, nevirapine, and TNK 651, respectively. Docking studies were performed using Autodock Tools (v-4.5.6) on four high resolution crystal structures of the HIV protein. The NNRTIs were studied in the non-nucleoside inhibitory binding pocket of the four receptors. The docking scores and the binding poses of the different NNRTIs were studied, and the results are given in Table 1.
The software used for docking purpose was validated at first to check its reliability for further docking procedures. The internal ligands were removed from the receptors and were redocked into the active site of the protein. Root mean square deviation (RMSD) values were obtained for the internal ligands—BHAP U-90152, rilpivirine, nevirapine, and TNK 651 for the HIV-1 proteins with PDB ID 1KLM, 3BGR, 1JLB, and 1RT2, respectively. As the RMSD values were within the standard limits (i.e., 0.2 Å), the software was used for further docking procedures.
In the receptor (PDB ID 1KLM), the docking score of the internal ligand was found to be −8.3. In the same active site, among the NNRTIs, doravirine had a comparable score with that of the internal ligand, (i.e., −8.6), whereas nevirapine, delaviridine, rilpivirine, and dapivirine had docking scores higher than that of the internal ligand, (i.e., −10.2, −9.5, −9.9, and −10.7, respectively). From the binding mode analysis, it was found that among them the best docking pose was obtained for dapivirine, in the NNIBP, 2-π(pi) bond interactions were obtained between the pyrimidine ring of the compound and the indole ring of the amino acid, with a bond length of 2.239 Å, i.e., dapivirine pyrimidine ring indole ring TRP229 = 2.239 Å (Figure 1).
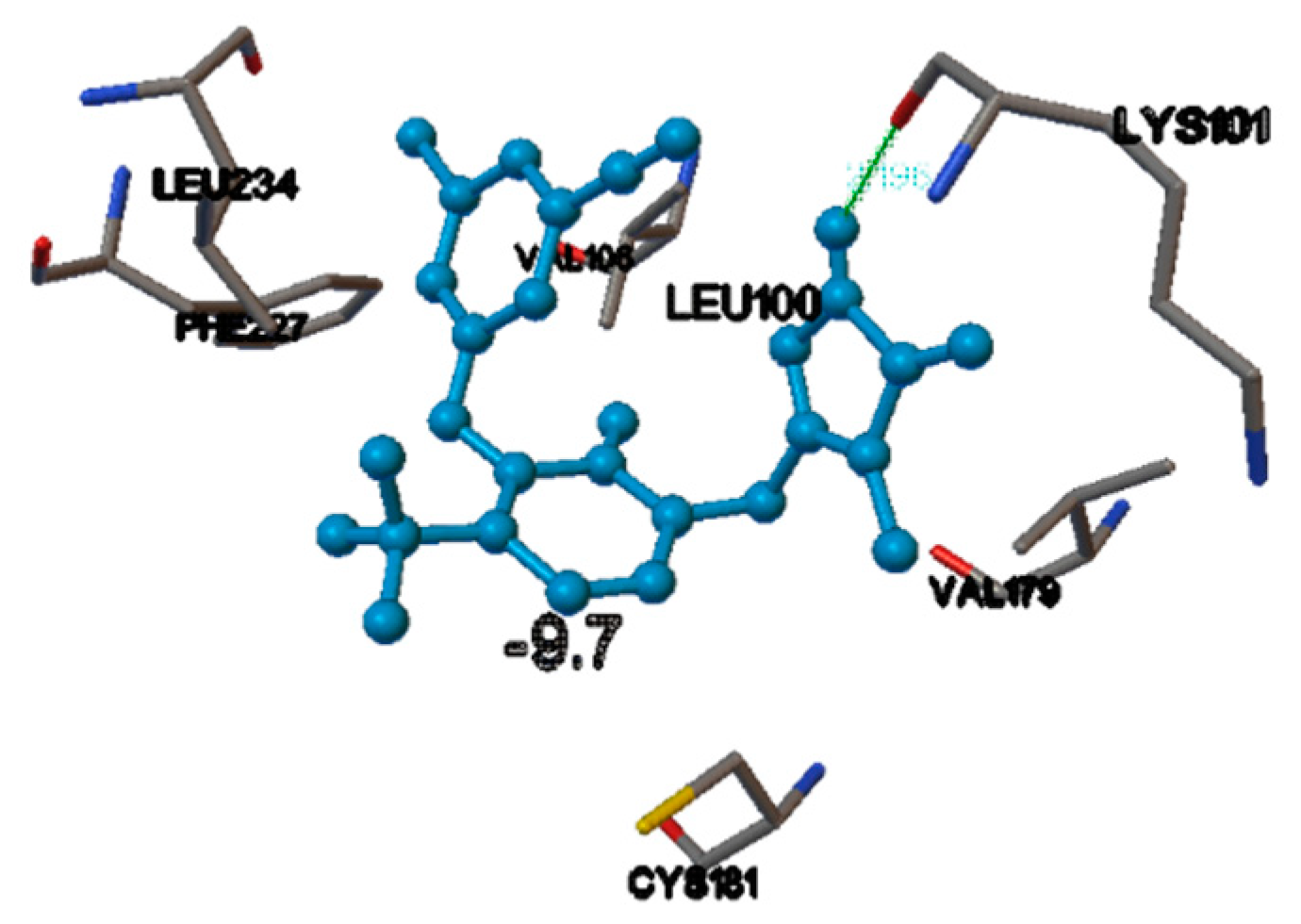
In the receptor (PDB ID 1JLB), the docking score of the internal ligand was found to be −9.7. In the same active site, among the NNRTIs, doravirine and efavirenz had a comparable score with that of the internal ligand, i.e., −9.7 and −9.4, respectively. From the binding mode analysis, it was found that among them, the best docking pose was obtained for doravirine, in the NNIBP, 1-Hydrogen bond interaction was obtained between the Hydrogen atom of the triazole ring of the compound and the oxygen atom of the amino acid, with a bond length of 2.196 Å, i.e., doravirine NH-C=O LYS101 = 2.196 Å (Figure 2).
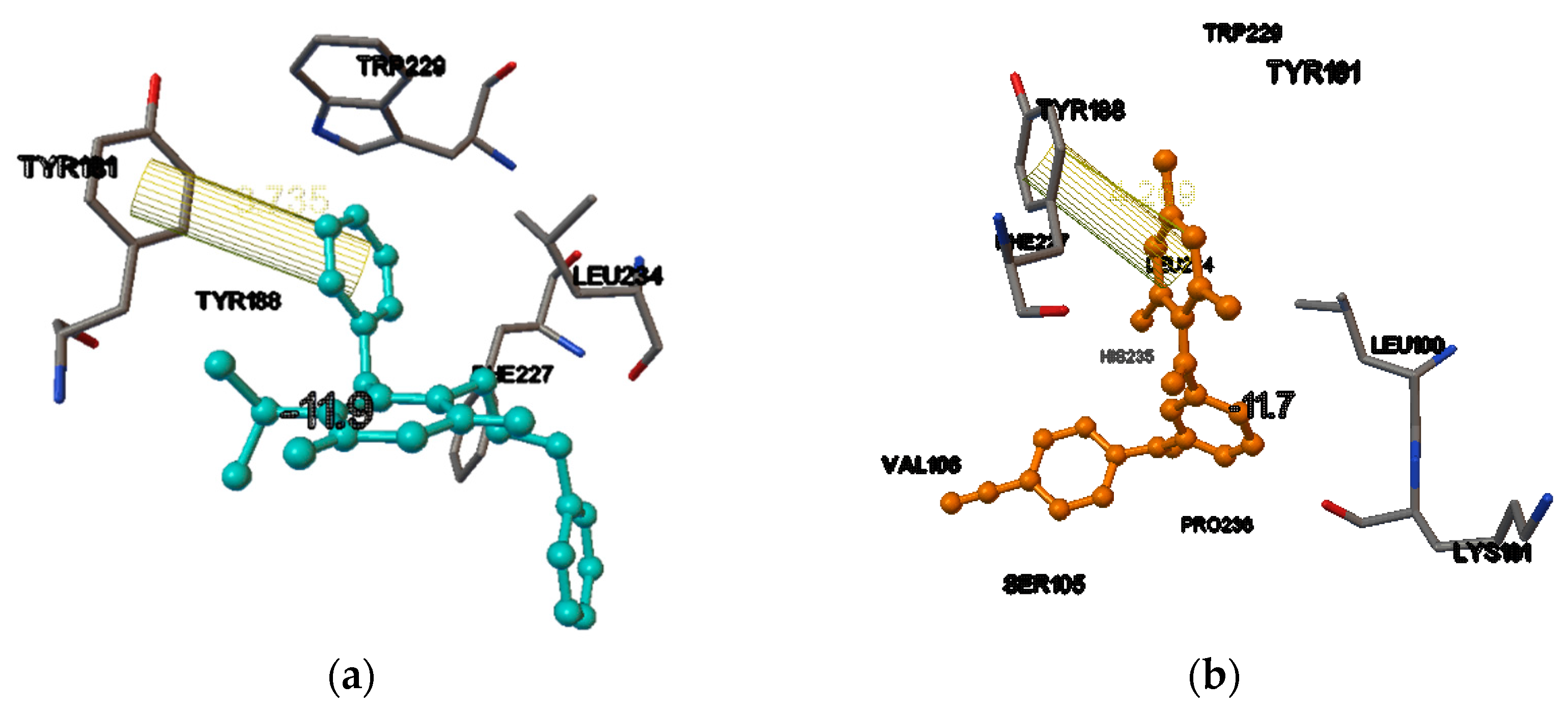
In the receptor (PDB ID 1RT2), the docking score of the internal ligand was found to be −11.9. In the same active site, among the NNRTIs, dapivirine had a comparable score with that of the internal ligand, (i.e., −11.7). From the binding mode analysis, it was found that among them, the best docking pose obtained for dapivirine was in the NNIBP, and the 1-π(pi) bond interaction were obtained between the trimethyl substituted phenyl ring of the compound and the phenyl ring of the amino acid, with a bond length of 4.269 Å, i.e., dapivirine trisubstituted phenyl ring phenyl ring TYR188 = 4.269 Å (Figure 3a,b).
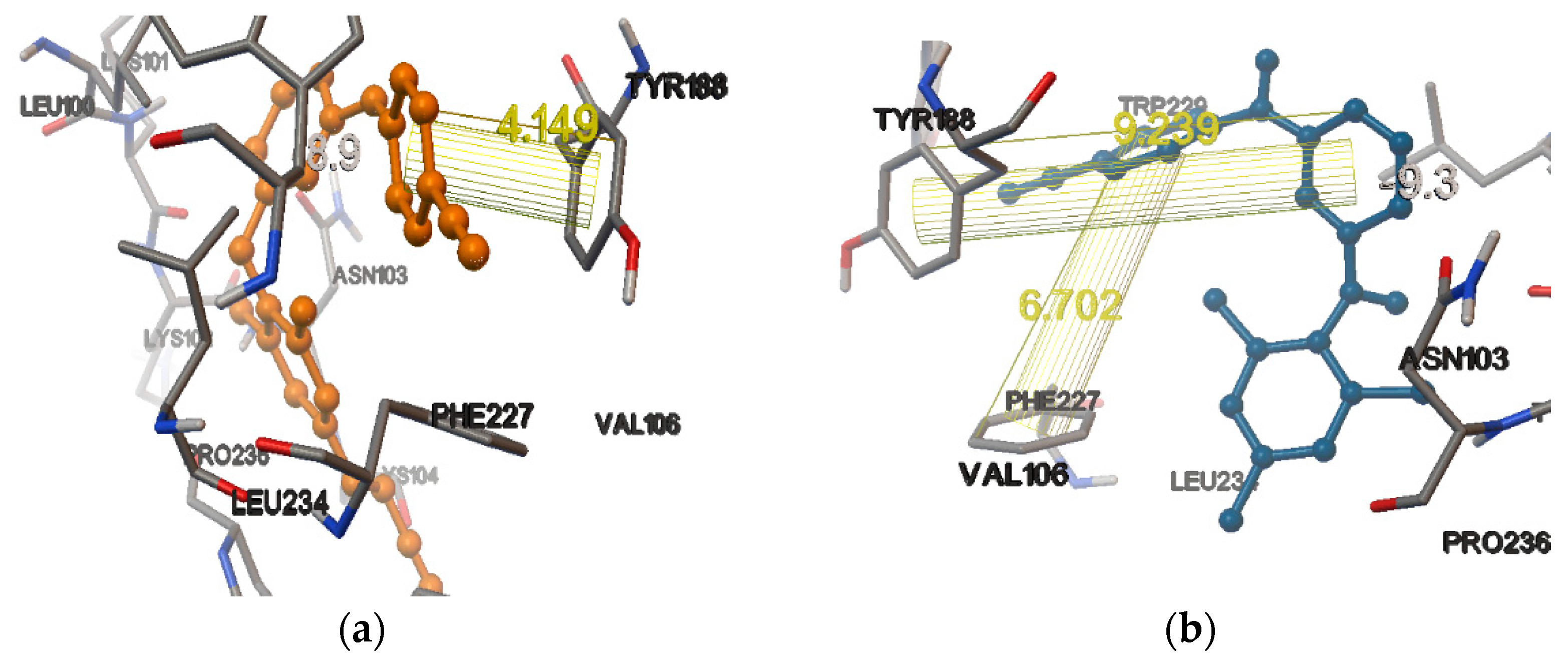
In the receptor (PDB ID 3BGR), the docking score of the internal ligand was found to be −8.9. In the same active site, among the NNRTIs, rilpivirine had a comparable score with that of the internal ligand, (i.e., −8.7), whereas dapivirine, efavirenz, and nevirapine had higher docking scores than that of the internal ligand, i.e., −9.3, −9.3, and −9.5, respectively. From the binding mode analysis, it was found that among them, the best docking pose was obtained for dapivirine. In the NNIBP, 2-π(pi) bond interactions were obtained between the pyrimidine ring of the compound and the phenyl ring of the amino acid TYR188, with a bond length of 9.239 Å, and the other one was obtained for the benzonitrile ring and the phenyl ring of the amino acid PHE227, with a bond length of 6.702 Å, i.e., dapivirine pyrimidine ring phenyl ring TYR188 = 9.239 Å and dapivirine benzonitrile ring phenyl ring PHE227 = 6.702 Å (Figure 4a,b).
4. Conclusions
The NNRTIs were docked into the active site of four different HIV-1RT receptors. Among them, dapivirine, doravirine and efavirenz were found to have good docking scores and the binding mode analysis of these compounds revealed that they have similar interactions (pi-bond interactions) with the amino acid residues TYR188, TRP229, and LYS101 of the NNIBP. Therefore, it can be predicted that these have the best activity and are the most effective among other NNRTIs.
Author Contributions
All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to acknowledge the Department of Pharmaceutical Sciences and Technology, Birla Institute of Technology, Mesra, Ranchi.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Turner, B.G.; Summers, M.F. Structural biology of HIV 1 1Edited by P. E. Wright. J. Mol. Biol. 1999, 285, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Gupta, S.; Singh, J.; Arsi, T. Azoles as non-nucleoside reverse transcriptase inhibitors (NNRTIs): Mini review. Int. J. Pharm. Sci. Res. 2017, 8, 29. [Google Scholar]
- De Clercq, E. New approaches toward anti-HIV chemotherapy. J. Med. Chem. 2005, 48, 1297–1313. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E. The design of drugs for HIV and HCV. Nat. Rev. Drug Discov. 2007, 6, 1001–1018. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, D.; Park, C.; So, W.; Jo, M.; Ok, T.; Kwon, J.; Kong, S.; Jo, S.; Kim, Y.; et al. Discovery of Phenylaminopyridine Derivatives as Novel HIV-1 Non-nucleoside Reverse Transcriptase Inhibitors. ACS Med. Chem. Lett. 2012, 3, 678–682. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Milton, J.; Weaver, K.L.; Short, S.A.; Stuart, D.I.; Stammers, D.K. Structural Basis for the Resilience of Efavirenz (DMP-266) to Drug Resistance Mutations in HIV-1 Reverse Transcriptase. Structure 2000, 8, 1089–1094. [Google Scholar] [CrossRef]
- Marongiu, M.E.; Pani, A.; Artico, M.; Massa, S.; Mai, A.; La Colla, P. Selective inhibition of HIV-1 replication by a novel series of 2-substituted 6-benzyl-pyrimidines. In Proceedings of the VIII International Conference on AIDS, Amsterdam, The Netherlands, 19–24 July 1992; pp. 19–24. [Google Scholar]
- Marongiu, M.E.; Pani, A.; Musiu, C.; LA COLLA, P.; Mai, A.; Sbardella, G.; Artico, M. 3,4-Dihydro-2-Alkoxy-6-Benzyl-Oxopyrimidines [DABOs]: Development of a Potent Class of Non-Nucleoside Reverse Transcriptase Inhibitors. J. Med. Chem. 2001, 65–92. [Google Scholar] [CrossRef]
- Pedersen, O.S.; Pedersen, E.B. Non-Nucleoside Reverse Transcriptase Inhibitors: The NNRTI Boom. Antivir. Chem. Chemother. 1999, 10, 285–314. [Google Scholar] [CrossRef] [PubMed]
- Artico, M. Selected non-nucleoside reverse transcriptase inhibitors (NNRTIs): The DABOs family. Drugs Future 2002, 27, 159. [Google Scholar] [CrossRef]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Mostashari-Rad, T.; Arian, R.; Sadri, H.; Mehridehnavi, A.; Mokhtari, M.; Ghasemi, F.; Fassihi, A. Study of CXCR4 chemokine receptor inhibitors using QSPR and molecular docking methodologies. J. Comput. Chem. 2019, 18, 1950018. [Google Scholar] [CrossRef]
- Das, K.; Lewi, P.J.; Hughes, S.H.; Arnold, E. Crystallography and the design of anti-AIDS drugs: Conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors. Prog. Biophys. Mol. Biol. 2005, 88, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Lindstrom, W.; Morris, G.M.; Weber, C.; Huey, R. AutoDock 4; The Scripps Research Institute, Molecular Graphics Laboratory: La Jolla, CA, USA, 2008. [Google Scholar]
- The Research Collaboratory for Structural Bioinformatics (RCSB). Protein Data Bank. Available online: http://www.rcsb.org/pdb (accessed on 1 May 2012).
- Prodrg 2 Server. Available online: http://prodrg1.dyndns.org/ (accessed on 15 August 2020).
Figure 1.
The binding pocket of HIV-1 reverse transcriptase (1KLM), showing the docking of dapivirine. The blue ball and stick model is the ligand (dapivirine) and the conventionally colored model are the amino acid residues interacting with the ligands. The lines are the π-(pi) bond interactions with amino acid residue of HIV-1 reverse transcriptase.
Figure 1.
The binding pocket of HIV-1 reverse transcriptase (1KLM), showing the docking of dapivirine. The blue ball and stick model is the ligand (dapivirine) and the conventionally colored model are the amino acid residues interacting with the ligands. The lines are the π-(pi) bond interactions with amino acid residue of HIV-1 reverse transcriptase.

Figure 2.
The binding pocket of HIV-1 reverse transcriptase (1JLB), showing the docking of doravirine. The blue ball and stick model is the ligand (doravirine) and the conventionally colored model are the amino acid residues interacting with the ligands. The green line is the hydrogen-bond interaction with amino acid residue of HIV-1 reverse transcriptase.
Figure 2.
The binding pocket of HIV-1 reverse transcriptase (1JLB), showing the docking of doravirine. The blue ball and stick model is the ligand (doravirine) and the conventionally colored model are the amino acid residues interacting with the ligands. The green line is the hydrogen-bond interaction with amino acid residue of HIV-1 reverse transcriptase.

Figure 3.
(a) The binding pocket of HIV-1 reverse transcriptase (1RT2), showing the redocking of co-crystallized ligand TNK651. The blue ball and stick model is the ligand and the conventionally colored model is the amino acid residues interacting with the ligands. The lines are the π-(pi) bond interactions with amino acid residue of HIV-1 reverse transcriptase. (b) The binding pocket of HIV-1 reverse transcriptase (1RT2), showing the docking of dapivirine. The orange ball and stick model is the ligand (dapivirine) and the conventionally colored model is the amino acid residues interacting with the ligands. The lines are the π-(pi) bond interactions with amino acid residue of HIV-1 reverse transcriptase.
Figure 3.
(a) The binding pocket of HIV-1 reverse transcriptase (1RT2), showing the redocking of co-crystallized ligand TNK651. The blue ball and stick model is the ligand and the conventionally colored model is the amino acid residues interacting with the ligands. The lines are the π-(pi) bond interactions with amino acid residue of HIV-1 reverse transcriptase. (b) The binding pocket of HIV-1 reverse transcriptase (1RT2), showing the docking of dapivirine. The orange ball and stick model is the ligand (dapivirine) and the conventionally colored model is the amino acid residues interacting with the ligands. The lines are the π-(pi) bond interactions with amino acid residue of HIV-1 reverse transcriptase.

Figure 4.
(a) The binding pocket of HIV-1 reverse transcriptase (3BGR), showing the redocking of co-crystallized ligand T27. The orange colored ball and stick model is the ligand (T27) and the conventionally colored model is the amino acid residues interacting with the ligands. The lines are the π-(pi) bond interactions with amino acid residue of HIV-1 reverse transcriptase. (b) The binding pocket of HIV-1 reverse transcriptase (3BGR), showing the docking of dapivirine. The orange ball and stick model is the ligand (dapivirine) and the conventionally colored model is the amino acid residues interacting with the ligands. The lines are the π-(pi) bond interactions with amino acid residue of HIV-1 reverse transcriptase.
Figure 4.
(a) The binding pocket of HIV-1 reverse transcriptase (3BGR), showing the redocking of co-crystallized ligand T27. The orange colored ball and stick model is the ligand (T27) and the conventionally colored model is the amino acid residues interacting with the ligands. The lines are the π-(pi) bond interactions with amino acid residue of HIV-1 reverse transcriptase. (b) The binding pocket of HIV-1 reverse transcriptase (3BGR), showing the docking of dapivirine. The orange ball and stick model is the ligand (dapivirine) and the conventionally colored model is the amino acid residues interacting with the ligands. The lines are the π-(pi) bond interactions with amino acid residue of HIV-1 reverse transcriptase.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Docking scores of the non-nucleoside reverse transcriptase inhibitors (NNRTIs) in various HIV-1 RT receptors.
Table 1.
Docking scores of the non-nucleoside reverse transcriptase inhibitors (NNRTIs) in various HIV-1 RT receptors.
| Compound | Docking Scores On | |||
|---|---|---|---|---|
| 1KLM | 1JLB | 1RT2 | 3BGR | |
| Nevirapine | −10.2 | −9.7 | −9.5 | −9.5 |
| Efavirenz | −7.6 | −9.4 | −9.4 | −9.3 |
| Delaviridine | −9.5 | −6.9 | −8.5 | −6.8 |
| Rilpivirine | −9.9 | −8.4 | −9.2 | −8.7 |
| Doravirine | −8.6 | −9.7 | −11.2 | −7.1 |
| Etravirine | −8.8 | −6.9 | −9.6 | −6.9 |
| Lersivirine | −6.5 | −8.3 | −6.5 | −8.3 |
| Dapivirine | −10.7 | −8.9 | −11.7 | −9.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Chandra, P.; Ganguly, S.; Karmakar, S. Comparative Studies of Various NNRTIs in the Active Site of Different HIV-1RT Receptors. Chem. Proc. 2021, 3, 33. https://doi.org/10.3390/ecsoc-24-08313
AMA Style
Chandra P, Ganguly S, Karmakar S. Comparative Studies of Various NNRTIs in the Active Site of Different HIV-1RT Receptors. Chemistry Proceedings. 2021; 3(1):33. https://doi.org/10.3390/ecsoc-24-08313
Chicago/Turabian StyleChandra, Priyanka, Swastika Ganguly, and Soikata Karmakar. 2021. "Comparative Studies of Various NNRTIs in the Active Site of Different HIV-1RT Receptors" Chemistry Proceedings 3, no. 1: 33. https://doi.org/10.3390/ecsoc-24-08313