
Sustainable Synthesis of Polymeric Materials versus Fine Chemicals via CO2 Addition to Epoxides †

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
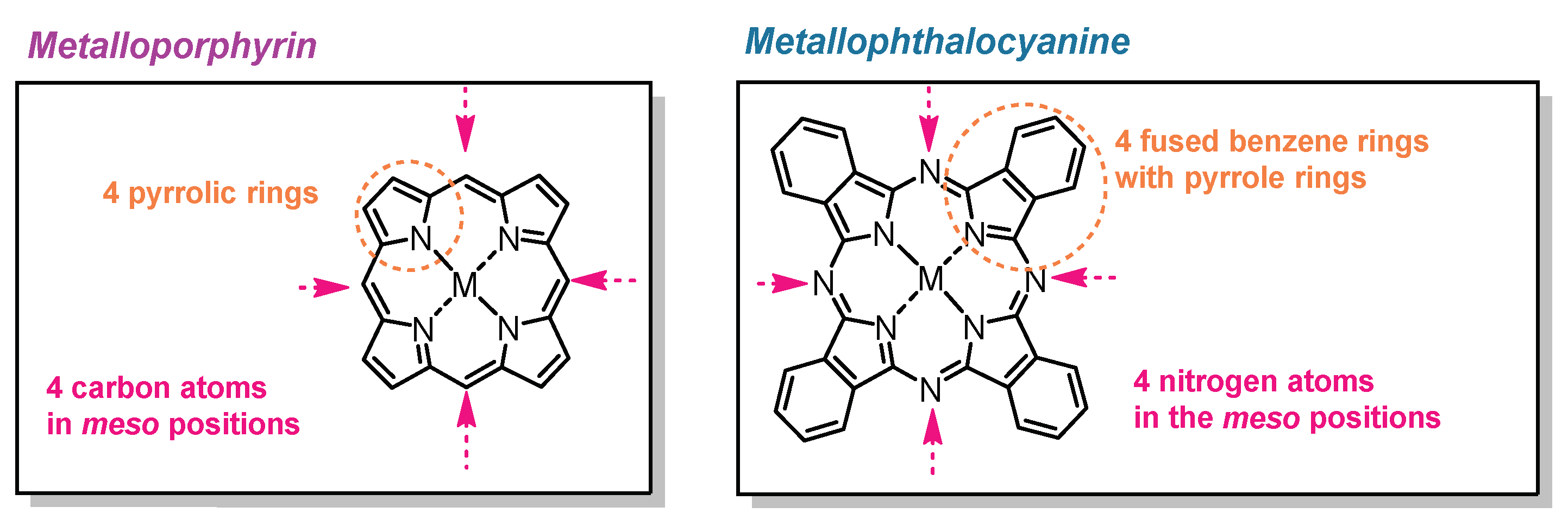
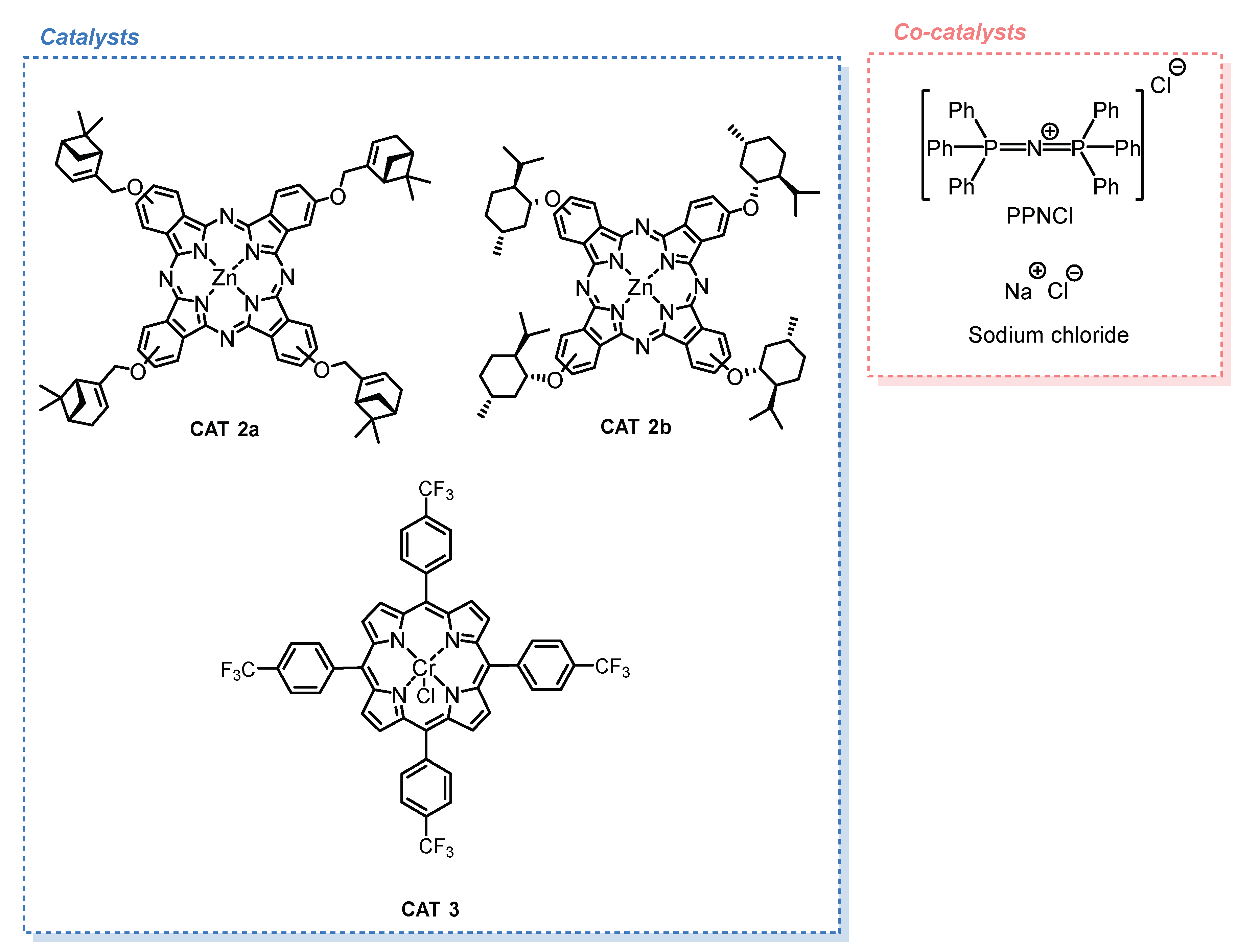
2.1. Synthesis of the Catalysts
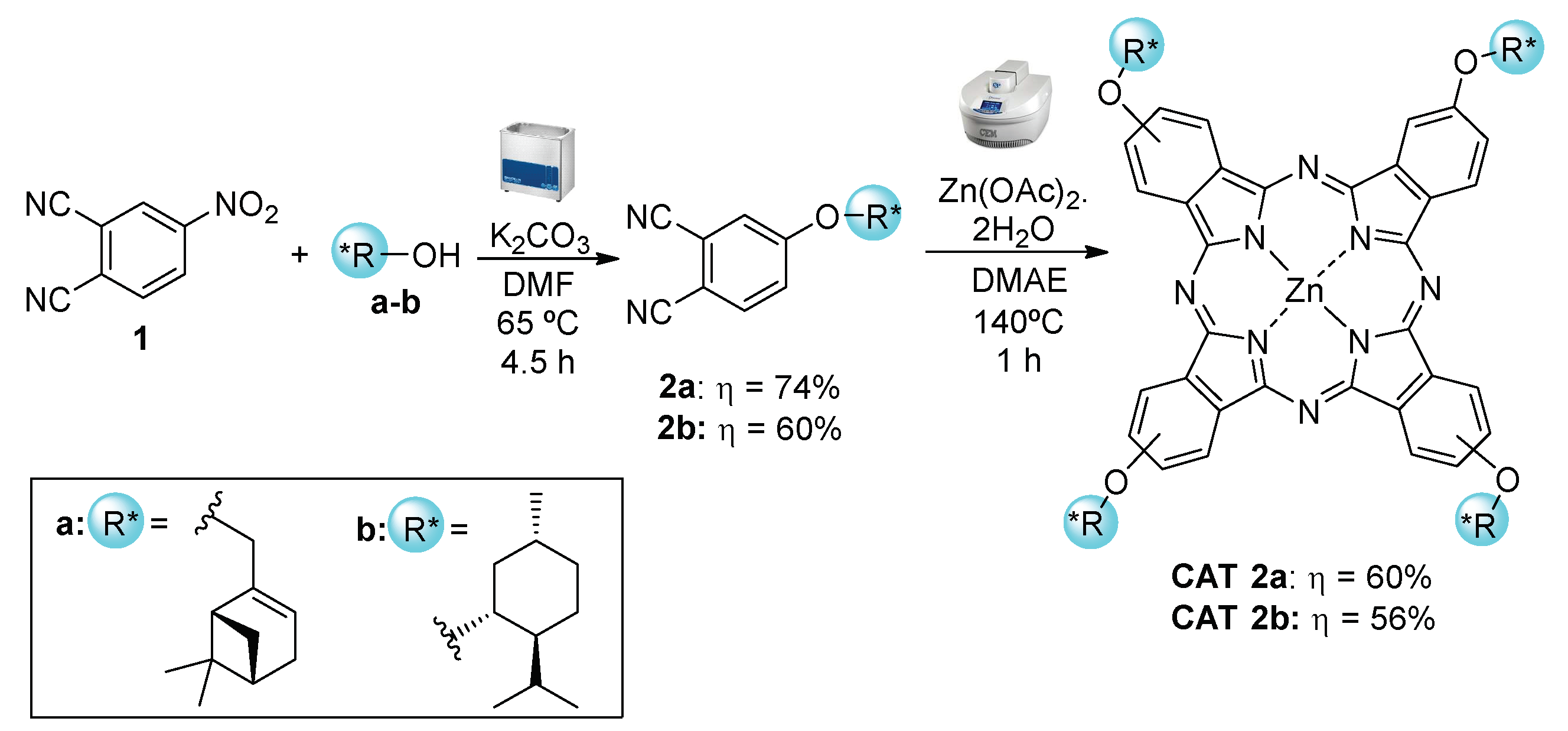
2.1.1. Monoterpene-Based Metallophthalocyanines
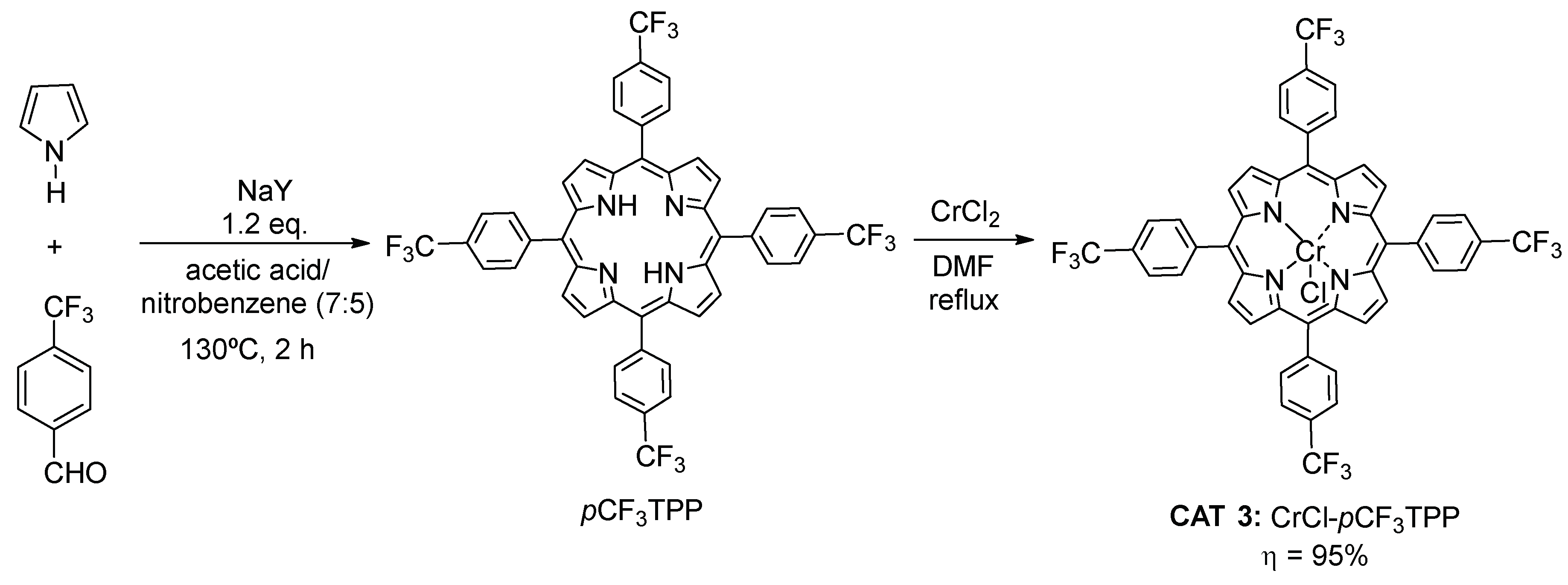
2.1.2. Fluorinated Metalloporphyrin
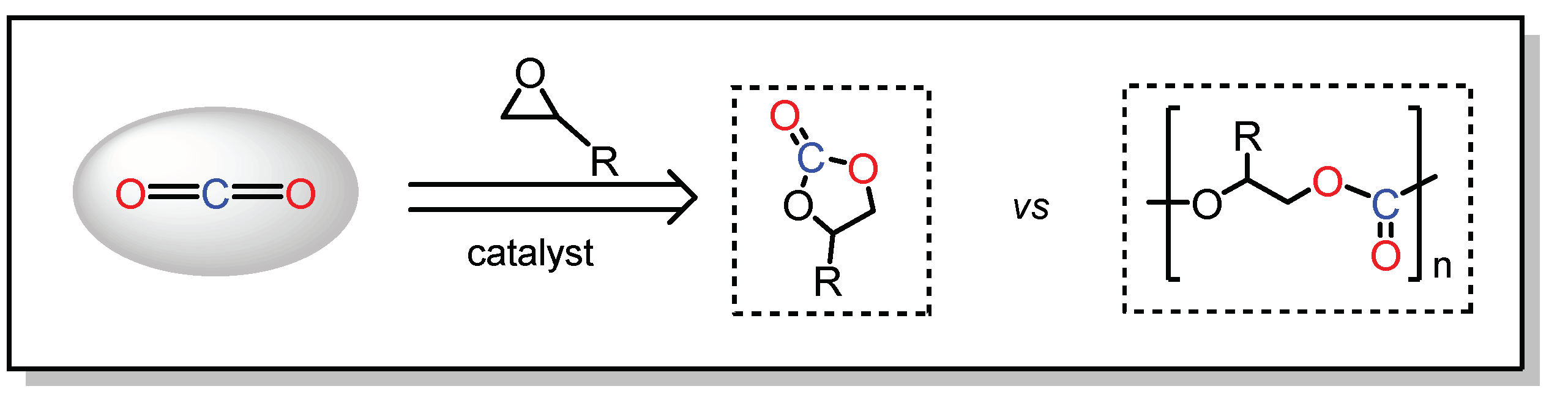
2.2. CO2 Addition Reaction to Epoxides
3. Materials and Methods
3.1. Reagents
3.2. Equipment
3.3. Synthesis of Metallophthalocyanine Catalysts CAT 2a and CAT 2b
3.4. Synthesis of Metalloporphyrin Catalyst CAT 3
3.5. CO2 Addition to Epoxides
3.6. Styrene Carbonate (SC) and Styrene Polycarbonate (SPC)
3.7. Poly(cyclohexene carbonate) (PCHC)
4. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Z.E.; Pan, S.Y.; Li, H.; Cai, J.C.; Olabi, A.G.; Anthony, E.J.; Manovic, V. Recent advances in carbon dioxide utilization. Renew. Sustain. Energy Rev. 2020, 125, 1–17. [Google Scholar] [CrossRef]
- Thonemann, N. Environmental impacts of CO2-based chemical production: A systematic literature review and meta-analysis. Appl. Energy 2020, 263, 1–21. [Google Scholar] [CrossRef]
- Aresta, M.; Dibenedetto, A.; Angelini, A. Catalysis for the Valorization of Exhaust Carbon: From CO2 to Chemicals, Materials, and Fuels. Technological Use of CO2. Chem. Rev. 2014, 114, 1709–1742. [Google Scholar] [CrossRef]
- Huh, S. Direct Catalytic Conversion of CO2 to Cyclic Organic Carbonates under Mild Reaction Conditions by Metal-Organic Frameworks. Catalysts 2019, 9, 34. [Google Scholar] [CrossRef]
- Blazek, K.; Datta, J. Renewable natural resources as green alternative substrates to obtain bio-based non-isocyanate polyurethanes-review. Crit. Rev. Environ. Sci. Technol. 2019, 49, 173–211. [Google Scholar] [CrossRef]
- Dias, L.D.; Carrilho, R.M.B.; Henriques, C.A.; Calvete, M.J.F.; Masdeu-Bultó, A.M.; Claver, C.; Rossi, L.M.; Pereira, M.M. Hybrid Metalloporphyrin Magnetic Nanoparticles as Catalysts for Sequential Transformation of Alkenes and CO2 into Cyclic Carbonates. ChemCatChem 2018, 10, 2792. [Google Scholar] [CrossRef]
- Zhao, X.; Yang, S.H.; Ebrahimiasl, S.; Arshadi, S.; Hosseinian, A. Synthesis of six-membered cyclic carbamates employing CO2 as building block: A review. J. CO2 Util. 2019, 33, 37–45. [Google Scholar] [CrossRef]
- Baena-Moreno, F.M.; Rodriguez-Galan, M.; Vega, F.; Alonso-Farinas, B.; Arenas, L.F.V.; Navarrete, B. Carbon capture and utilization technologies: A literature review and recent advances. Energy Sources Part A-Recovery Util. Environ. Eff. 2019, 41, 1403–1433. [Google Scholar] [CrossRef]
- Dalpozzo, R.; Della Ca’, N.; Gabriele, B.; Mancuso, R. Recent Advances in the Chemical Fixation of Carbon Dioxide: A Green Route to Carbonylated Heterocycle Synthesis. Catalysts 2019, 9, 511. [Google Scholar] [CrossRef]
- Dabral, S.; Schaub, T. The Use of Carbon Dioxide (CO2) as a Building Block in Organic Synthesis from an Industrial Perspective. Adv. Synth. Catal. 2019, 361, 223–246. [Google Scholar] [CrossRef]
- Anderson, T.S.; Kozak, C.M. Ring-opening polymerization of epoxides and ring-opening copolymerization of CO2 with epoxides by a zinc amino-bis(phenolate) catalyst. Eur. Polym. J. 2019, 120, 1–6. [Google Scholar] [CrossRef]
- Della Monica, F.; Buonerba, A.; Capacchione, C. Homogeneous Iron Catalysts in the Reaction of Epoxides with Carbon Dioxide. Adv. Synth. Catal. 2019, 361, 265–282. [Google Scholar] [CrossRef]
- Ng, C.K.; Toh, R.W.; Lin, T.T.; Luo, H.K.; Hor, T.S.A.; Wu, J. Metal-salen molecular cages as efficient and recyclable heterogeneous catalysts for cycloaddition of CO2 with epoxides under ambient conditions. Chem. Sci. 2019, 10, 1549–1554. [Google Scholar] [CrossRef]
- Ambrose, K.; Murphy, J.N.; Kozak, C.M. Chromium Amino-bis(phenolate) Complexes as Catalysts for Ring-Opening Polymerization of Cyclohexene Oxide. Macromolecules 2019, 52, 7403–7412. [Google Scholar] [CrossRef]
- Huiling, S.; Yernaidu, R.; Christopher, J.C. Modeling the Mechanism of CO2/Cyclohexene Oxide Copolymerization Catalyzed by Chiral Zinc β-Diiminates: Factors Affecting Reactivity and Isotacticity. ACS Catal. 2020, 10, 8870–8879. [Google Scholar] [CrossRef]
- Yi, J.J.; Sun, S.; Li, Z.L.; Gao, X.Y.; Sun, X.Z.; Wang, N.; Li, J. Pyridinium-functionalized metalloporphyrins as bifunctional catalysts for cycloaddition of epoxides and carbon dioxide. Appl. Organomet. Chem. 2020, 34, 1–7. [Google Scholar] [CrossRef]
- Carrilho, R.M.B.; Dias, L.D.; Rivas, R.; Pereira, M.M.; Claver, C.; Masdeu-Bulto, A.M. Solventless Coupling of Epoxides and CO2 in Compressed Medium Catalysed by Fluorinated Metalloporphyrins. Catalysts 2017, 7, 210. [Google Scholar] [CrossRef]
- Kasuga, K.; Kato, T.; Kabata, N.; Handa, M. Cycloaddition of carbon dioxide to 1,2-epoxypropane catalyzed by tetra-t-butylphthalocyaninatoaluminium(III) hydroxide. Bull. Chem. Soc. Jpn. 1996, 69, 2885–2888. [Google Scholar] [CrossRef]
- Srivastava, R.; Srinivas, D.; Ratnasamy, P. Synthesis of cyclic carbonates from olefins and CO2 over zeolite-based catalysts. Catal. Lett. 2003, 89, 81–85. [Google Scholar] [CrossRef]
- Gonzalez, A.C.S.; Damas, L.; Aroso, R.T.; Tomé, V.A.; Dias, L.D.; Pina, J.; Carrilho, R.M.B.; Pereira, M.M. Monoterpene-based metallophthalocyanines: Sustainable synthetic approaches and photophysical studies. J. Porphyr. Phthalocyanines 2020, 24, 947–958. [Google Scholar] [CrossRef]
- Silva, M.; Fernandes, A.; Bebiano, S.S.; Calvete, M.J.F.; Ribeiro, M.F.; Burrows, H.D.; Pereira, M.M. Size and ability do matter! Influence of acidity and pore size on the synthesis of hindered halogenated meso-phenyl porphyrins catalysed by porous solid oxides. Chem. Commun. 2014, 50, 6571–6573. [Google Scholar] [CrossRef] [PubMed]
- Taherimehr, M.; Pescarmona, P.P. Green Polycarbonates Prepared by the Copolymerization of CO2 with Epoxides. J. Appl. Polym. Sci. 2014, 131, 1–17. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Catalyst | Co-Catalyst | Conversion (%) b | TON c | Selectivity (%) d | Polymers | |||
|---|---|---|---|---|---|---|---|---|---|
| SC | PSC | % CO2 f | Mng | PDg | |||||
| 1 | CAT 2a | - | 0 | - | - | - | - | - | - |
| 2 | CAT 2a | PPNCl | 63 | 900 | 100 | - | - | - | - |
| 3 | CAT 2a | NaCl | 0 | - | - | - | - | - | - |
| 4 | CAT2b | PPNCl | 59 | 843 | 100 | - | - | - | - |
| 5 | CAT 3 | - | 85 | 1224 | 36 | 64 | 91 | 4200 | 1.20 |
| 6 e | CAT 3 | PPNCl | 100 | 1425 | 100 | 0 | - | - | - |
| 7 | - | PPNCl | 30 | 429 | 100 | - | - | - | - |

| Entry | Catalyst | Co-Catalyst | Conversion (%) b | TON | Selectivity (%) c | % CO2 | Mn (g/mol) | PD | |
|---|---|---|---|---|---|---|---|---|---|
| PCHC | CHC | ||||||||
| 1 | CAT 2a | PPNCl | 0 | - | - | - | - | - | - |
| 2 | CAT 3 | - | 0 | - | - | - | - | - | - |
| 3 | CAT 3 | PPNCl | 86 | 1224 | 99 | 1 | 98 | 4800 | 1.25 |
| 4 d | CAT 3 | PPNCl | 86 | 1224 | 96 | 4 | 98 | 12,500 | 1.38 |
| 5 | - | PPNCl | 28 | 200 | 0 | 100 | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gonzalez, A.C.S.; Aroso, R.T.; Carrilho, R.M.B.; Claver, C.; Masdeu-Bultó, A.M.; Pereira, M.M. Sustainable Synthesis of Polymeric Materials versus Fine Chemicals via CO2 Addition to Epoxides. Chem. Proc. 2021, 3, 17. https://doi.org/10.3390/ecsoc-24-08357
Gonzalez ACS, Aroso RT, Carrilho RMB, Claver C, Masdeu-Bultó AM, Pereira MM. Sustainable Synthesis of Polymeric Materials versus Fine Chemicals via CO2 Addition to Epoxides. Chemistry Proceedings. 2021; 3(1):17. https://doi.org/10.3390/ecsoc-24-08357
Chicago/Turabian StyleGonzalez, Andreia C. S., Rafael T. Aroso, Rui M. B. Carrilho, Carmen Claver, Anna M. Masdeu-Bultó, and Mariette M. Pereira. 2021. "Sustainable Synthesis of Polymeric Materials versus Fine Chemicals via CO2 Addition to Epoxides" Chemistry Proceedings 3, no. 1: 17. https://doi.org/10.3390/ecsoc-24-08357