
Impact of Sugars on Hypothalamic Satiety Pathways and Its Contribution to Dysmetabolic States



{kind=link}
Abstract
:1. Introduction
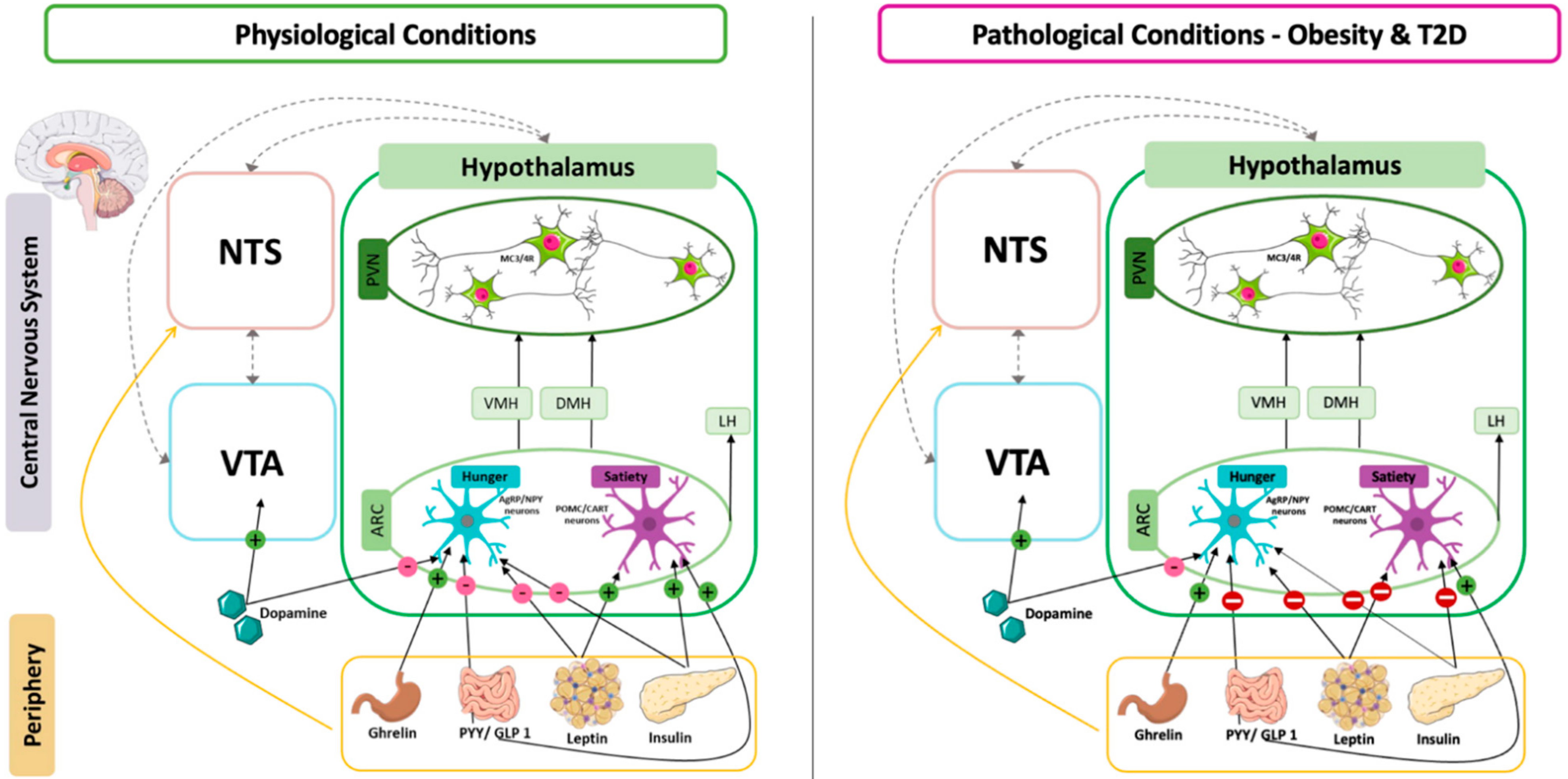
2. Control of Food Intake by the Hypothalamus
3. Deregulation of Satiety Pathways in Type 2 Diabetes and Obesity
4. Impact of Sugar Consumption on Food Behaviour
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Egger, G.; Dixon, J. Beyond obesity and lifestyle: A review of 21st century chronic disease determinants. Biomed. Res. Int. 2014, 2014, 731685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golay, A.; Ybarra, J. Link between obesity and type 2 diabetes. Best Pract. Res. Clin. Endocrinol. Metab. 2005, 19, 649–663. [Google Scholar] [CrossRef] [PubMed]
- International Diabetes Federation. IDF Diabetes; International Diabetes Federation: Brussels, Belgium, 2019. [Google Scholar]
- DeFronzo, R.A. Pathogenesis of type 2 diabetes mellitus. Med. Clin. 2004, 88, 787–835. [Google Scholar] [CrossRef] [PubMed]
- ADA. American Diabetes Association. 2021. Available online: https://www.diabetes.org/diabetes (accessed on 19 November 2022).
- Wysham, C.; Shubrook, J. Beta-cell failure in type 2 diabetes: Mechanisms, markers, and clinical implications. Postgrad. Med. 2020, 132, 676–686. [Google Scholar] [CrossRef] [PubMed]
- Timper, K.; Brüning, J.C. Hypothalamic circuits regulating appetite and energy homeostasis: Pathways to obesity. DMM Dis. Model. Mech. 2017, 10, 679–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Billes, S.K.; Simonds, S.E.; Cowley, M.A. Leptin reduces food intake via a dopamine D2 receptor-dependent mechanism. Mol. Metab. 2012, 1, 86–93. [Google Scholar] [CrossRef]
- Khodai, T.; Luckman, S.M. Ventromedial Nucleus of the Hypothalamus Neurons under the Magnifying Glass. Endocrinology 2021, 162, bqab141. [Google Scholar] [CrossRef]
- Hetherington, A.W.; Ranson, S.W. The relation of various hypothalamic lesions to adiposity in the rat. J. Comp. Neurol. 1942, 76, 475–499. [Google Scholar] [CrossRef]
- Delgado, J.M.; Anand, B.K. Increase of food intake induced by electrical stimulation of the lateral hypothalamus. Am. J. Physiol. 1953, 172, 162–168. [Google Scholar] [CrossRef]
- Al-Dossary, A.F.; Al-Majed, A.; Hossain, M.E.; Rahman, M.K.; Jennings, S.; Bargawi, R. The Need to Feed: Homeostatic and Hedonic Control of Eating. In Proceedings of the 17th Middle East Oil & Gas Show and Conference 2011 (MEOS 2011), Manama, Bahrain, 25–28 September 2011; Volume 3, pp. 1660–1672. [Google Scholar]
- Anand, B.K.; Brobeck, J.R. Localization of a ‘Feeding Center’ in the Hypothalamus of the Rat. Proc. Soc. Exp. Biol. Med. 1946, 6–7. [Google Scholar] [CrossRef]
- Stuber, G.D.; Wise, R.A. Lateral hypothalamic circuits for feeding and reward. Nat. Neurosci. 2016, 19, 198–205. [Google Scholar] [CrossRef] [Green Version]
- Ono, T.; Nakamura, K.; Nishijo, H.; Fukuda, M. Hypothalamic neuron involvement in integration of reward, aversion, and cue signals. J. Neurophysiol. 1986, 56, 63–79. [Google Scholar] [CrossRef]
- Valassi, E.; Scacchi, M.; Cavagnini, F. Neuroendocrine control of food intake. Nutr. Metab. Cardiovasc. Dis. 2008, 18, 158–168. [Google Scholar] [CrossRef]
- Hruby, V.J.; Cone, R.D. Role of melanocortinergic neurons in feeding and the agouti obesity syndrome. Nature 1997, 385, 165–168. [Google Scholar]
- Marie, L.S.; Miura, G.I.; Marsh, D.J.; Yagaloff, K.; Palmiter, R.D. A metabolic defect promotes obesity in mice lacking melanocortin-4 receptors. Proc. Natl. Acad. Sci. USA 2000, 97, 12339–12344. [Google Scholar] [CrossRef] [Green Version]
- Voss-Andreae, A.; Murphy, J.G.; Ellacott, K.L.J.; Stuart, R.C.; Nillni, E.A.; Cone, R.D.; Fan, W. Role of the central melanocortin circuitry in adaptive thermogenesis of brown adipose tissue. Endocrinology 2007, 148, 1550–1560. [Google Scholar] [CrossRef] [Green Version]
- Vaisse, C.; Clement, K.; Guy-Grand, B.; Froguel, P. A frameshift mutation in human MC4R is associated with a dominant form of obesity. Nat. Genet. 1998, 20, 113–114. [Google Scholar] [CrossRef]
- Markham, A. Setmelanotide: First Approval. Drugs 2021, 81, 397–403. [Google Scholar] [CrossRef]
- Ahima, R.S.; Antwi, D.A. Brain Regulation of Appetite and Satiety. Endocrinol. Metab. Clin. North Am. 2008, 37, 811–823. [Google Scholar] [CrossRef] [Green Version]
- Tong, J.; D’Alessio, D. Ghrelin and hypothalamic development: Too little and too much of a good thing. J. Clin. Investig. 2015, 125, 490–492. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Lin, X.; Lin, S. Neuropeptide Y and Metabolism Syndrome: An Update on Perspectives of Clinical Therapeutic Intervention Strategies. Front. Cell Dev. Biol. 2021, 9, 695623. [Google Scholar] [CrossRef] [PubMed]
- Waterson, M.J.; Horvath, T.L. Neuronal Regulation of Energy Homeostasis: Beyond the Hypothalamus and Feeding. Cell Metab. 2015, 22, 962–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, G.J. Integrative capacity of the caudal brainstem in the control of food intake. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1275–1280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tyree, S.M.; de Lecea, L. Lateral hypothalamic control of the ventral tegmental area: Reward evaluation and the driving of motivated behavior. Front. Syst. Neurosci. 2017, 11, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowe, M.R.; Butryn, M.L. Hedonic hunger: A new dimension of appetite? Physiol. Behav. 2007, 91, 432–439. [Google Scholar] [CrossRef]
- Castro, D.C.; Cole, S.L.; Berridge, K.C. Lateral hypothalamus, nucleus accumbens, and ventral pallidum roles in eating and hunger: Interactions between homeostatic and reward circuitry. Front. Syst. Neurosci. 2015, 9, 90. [Google Scholar] [CrossRef] [Green Version]
- Capucho, A.; Chegão, A.; Martins, F.; Miranda, H.V.; Conde, S. Dysmetabolism and Neurodegeneration: Trick or Treat? Nutrients 2022, 14, 1425. [Google Scholar] [CrossRef]
- Ruth, M. Obesity is associated with hypothalamic injury in rodents and humans. Yearb. Endocrinol. 2012, 2012, 119–120. [Google Scholar] [CrossRef]
- Schneeberger, M.; Dietrich, M.O.; Sebastián, D.; Imbernón, M.; Castaño, C.; Garcia, A.; Esteban, Y.; Gonzalez-Franquesa, A.; Rodríguez, I.C.; Bortolozzi, A.; et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell 2013, 155, 172–187. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKβ/NF-kB and ER stress Link Overnutrition. Cell 2009, 135, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Farr, O.M.; Gavirieli, A. What have we learned about leptin and obesity. Physiol. Behav. 2017, 176, 139–148. [Google Scholar]
- Ambwani, R.K.S.; Misra, A.K. Evaluation of Leptin as a Marker of Insulin Resistance in Type 2 Diabetes Mellitus. Int. J. Appl. Basic Med. Res. 2017, 2019, 193–195. [Google Scholar]
- Vaisse, C.; Halaas, J.L.; Horvath, C.M.; Darnell, J.E.; Stoffel, M.; Friedman, J.M. Leptin activation of Stat3 in the hypothalamus of wild–type and ob/ob mice but not db/db mice. Nat. Genet. 1996, 14, 95–97. [Google Scholar] [CrossRef]
- Ernst, M.B.; Wunderlich, C.M.; Hess, S.; Paehler, M.; Mesaros, A.; Koralov, S.; Kleinridders, A.; Husch, A.; Münzberg, H.; Hampel, B.; et al. Enhanced Stat3 activation in POMC neurons provokes negative feedback inhibition of leptin and insulin signaling in obesity. J. Neurosci. 2009, 29, 11582–11593. [Google Scholar] [CrossRef] [Green Version]
- Könner, A.C.; Brüning, J.C. Selective insulin and leptin resistance in metabolic disorders. Cell Metab. 2012, 16, 144–152. [Google Scholar] [CrossRef] [Green Version]
- Suriano, F.; Vieira-Silva, S.; Falony, G.; Roumain, M.; Paquot, A.; Pelicaen, R.; Régnier, M.; Delzenne, N.M.; Raes, J.; Muccioli, G.G.; et al. Novel insights into the genetically obese (ob/ob) and diabetic (db/db) mice: Two sides of the same coin. Microbiome 2021, 9, 147. [Google Scholar] [CrossRef]
- Ahima, R.S.; Saper, C.B.; Flier, J.S.; Elmquist, J.K. Leptin regulation of neuroendocrine systems. Front. Neuroendocrinol. 2000, 21, 263–307. [Google Scholar] [CrossRef]
- Loh, K.; Zhang, L.; Brandon, A.; Wang, Q.; Begg, D.; Qi, Y.; Fu, M.; Kulkarni, R.; Teo, J.; Baldock, P.; et al. Insulin controls food intake and energy balance via NPY neurons. Mol. Metab. 2017, 6, 574–584. [Google Scholar] [CrossRef]
- Gelling, R.W.; Morton, G.J.; Morrison, C.; Niswender, K.D.; Myers, M.G.; Rhodes, C.J.; Schwartz, M.W. Insulin action in the brain contributes to glucose lowering during insulin treatment of diabetes. Cell Metab. 2006, 3, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Cassaglia, P.A.; Shi, Z.; Brooks, V.L. Insulin increases sympathetic nerve activity in part by suppression of tonic inhibitory neuropeptide y inputs into the paraventricular nucleus in female rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 311, R97–R103. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.; López, M.; Rahmouni, K. The cellular and molecular bases of leptin and ghrelin resistance in obesity. Nat. Rev. Endocrinol. 2017, 13, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Briggs, D.I.; Enriori, P.J.; Lemus, M.B.; Cowley, M.; Andrews, Z.B. Diet-induced obesity causes ghrelin resistance in arcuate NPY/AgRP neurons. Endocrinology 2010, 151, 4745–4755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrestha, Y.B.; Wickwire, K.; Giraudo, S. Effect of reducing hypothalamic ghrelin receptor gene expression on energy balance. Peptides 2009, 30, 1336–1341. [Google Scholar] [CrossRef] [PubMed]
- Decarie-Spain, L.; Kanoski, S. Ghrelin and glucagon-like peptide-1: A gut-brain axis battle for food reward. Nutrients 2021, 13, 977. [Google Scholar] [CrossRef] [PubMed]
- González-Garciá, I.; Fernø, J.; Diéguez, C.; Nogueiras, R.; López, M. Hypothalamic Lipids: Key Regulators of Whole Body Energy Balance. Neuroendocrinology 2017, 104, 398–411. [Google Scholar] [CrossRef]
- Mayer, C.M.; Belsham, D.D. Palmitate attenuates insulin signaling and induces endoplasmic reticulum stress and apoptosis in hypothalamic neurons: Rescue of resistance and apoptosis through adenosine 5′ monophosphate-activated protein kinase activation. Endocrinology 2010, 151, 576–585. [Google Scholar] [CrossRef]
- Posey, K.A.; Clegg, D.J.; Printz, R.L.; Byun, J.; Morton, G.J.; Vivekanandan-Giri, A.; Pennathur, S.; Baskin, D.G.; Heinecke, J.W.; Woods, S.C.; et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1003–E1012. [Google Scholar] [CrossRef]
- Contreras, C.; García, I.G.; Martinez-Sanchez, N.; Seoane-Collazo, P.; Jacas, J.; Morgan, D.A.; Serra, D.; Gallego, R.; Gonzalez, F.; Casals, N.; et al. Central ceramide-induced hypothalamic lipotoxicity and ER stress regulate energy balance. Cell Rep. 2014, 9, 366–377. [Google Scholar] [CrossRef] [Green Version]
- Gaysinskaya, V.; Karatayev, O.; Shuluk, J.; Leibowitz, S. Hyperphagia induced by sucrose: Relation to circulating and CSF glucose and corticosterone and orexigenic peptides in the arcuate nucleus. Pharmacol. Biochem. Behav. 2011, 97, 521–530. [Google Scholar] [CrossRef] [Green Version]
- European Food Safety Authority (EFSA). Scientific Opinion on establishing Food-Based Dietary Guidelines. EFSA J. 2016, 8, 1460. [Google Scholar]
- Martins, F.O.; Conde, S.V. Impact of Diet Composition on Insulin Resistance. Nutrients 2022, 14, 3716. [Google Scholar] [CrossRef]
- Monteiro-Alfredo, T.; Caramelo, B.; Arbeláez, D.; Amaro, A.; Barra, C.; Silva, D.; Oliveira, S.; Seiça, R.; Matafome, P. Distinct impact of natural sugars from fruit juices and added sugars on caloric intake, body weight, glycaemia, oxidative stress and glycation in diabetic rats. Nutrients 2021, 13, 2956. [Google Scholar] [CrossRef]
- Seung, H.C.; Wolfgang, M.; Tokutake, Y.; Chohnan, S.; Lane, M.D. Differential effects of central fructose and glucose on hypothalamic malonyl-CoA and food intake. Proc. Natl. Acad. Sci. USA 2008, 105, 16871–16875. [Google Scholar]
- Colley, D.L.; Castonguay, T.W. Effects of sugar solutions on hypothalamic appetite regulation. Physiol. Behav. 2015, 139, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Lindqvist, A.; Baelemans, A.; Erlanson-Albertsson, C. Effects of sucrose, glucose and fructose on peripheral and central appetite signals. Regul. Pept. 2008, 150, 26–32. [Google Scholar] [CrossRef]
- Erlanson-Albertsson, C.; Lindqvist, A. Fructose affects enzymes involved in the synthesis and degradation of hypothalamic endocannabinoids. Regul. Pept. 2010, 161, 87–91. [Google Scholar] [CrossRef]
- Page, K.A.; Chan, O.; Arora, J.; Belfort-Deaguiar, R.; Dzuira, J.; Roehmholdt, B.; Cline, G.W.; Naik, S.; Sinha, R.; Constable, R.T.; et al. Effects of Fructose vs Glucose on Regional Cerebral Blood Flow in Brain Regions Involved with Appetite and Reward Pathways. JAMA 2013, 39, 63–70. [Google Scholar] [CrossRef] [Green Version]
- Freeman, C.; Zehra, A.; Ramirex, V.; Wiers, C.; Volkow, N.; Wang, G.-J. Impact of Sugar on the Body. Front. Biosci. 2018, 23, 2255–2266. [Google Scholar]
- Volkow, N.D.; Wang, G.-J.; Baler, R.D. Reward, dopamine and the control of food intake: Implications for obesity. Trends Cogn. Sci. 2012, 15, 37–46. [Google Scholar] [CrossRef] [Green Version]
- Small, D.M.; Jones-Gotman, M.; Dagher, A. Feeding-induced dopamine release in dorsal striatum correlates with meal pleasantness ratings in healthy human volunteers. Neuroimage 2003, 19, 1709–1715. [Google Scholar] [CrossRef]
- Range, K.D.M.; Moser, Y.A. Gustatory Reward and the Nucleus Accumbens. Physiol. Behav. 2012, 23, 531–535. [Google Scholar]
- Eiler, W.J.; Dzemidzic, M.; Soeurt, C.M.; Carron, C.R.; Oberlin, B.G.; Considine, R.V.; Harezlak, J.; Kareken, D.A. Family history of alcoholism and the human brain response to oral sucrose. NeuroImage Clin. 2018, 17, 1036–1046. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.B.; da Silva, J.A.; Almeida, J.; Cui, G.; Gerfen, C.R.; Costa, R.M.; Oliveira-Maia, A.J. Postingestive Modulation of Food Seeking Depends on Vagus-Mediated Dopamine Neuron Activity. Neuron 2020, 106, 778–788.e6. [Google Scholar] [CrossRef] [PubMed]
- Payant, M.A.; Chee, M.J. Neural mechanisms underlying the role of fructose in overfeeding. Neurosci. Biobehav. Rev. 2021, 128, 346–357. [Google Scholar] [CrossRef]
- Wang, G.J.; Volkow, N.D.; Thanos, P.K.; Fowler, J.S. Imaging of brain dopamine pathways: Implications for understanding obesity. J. Addict. Med. 2009, 3, 8–18. [Google Scholar] [CrossRef] [Green Version]
- Adam, T.C.; Epel, E.S. Stress, eating and the reward system. Physiol. Behav. 2007, 91, 449–458. [Google Scholar] [CrossRef]
- Palmiter, R.D. Is dopamine a physiologically relevant mediator of feeding behavior? Trends Neurosci. 2007, 30, 375–381. [Google Scholar] [CrossRef]
- Abizaid, A.; Liu, Z.-W.; Andrews, Z.B.; Shanabrough, M.; Borok, E.; Elsworth, J.D.; Roth, R.H.; Sleeman, M.W.; Picciotto, M.; Tschöp, M.H.; et al. Ghrelin modulates the activity and synaptic input organization of midbrain dopamine neurons while promoting appetite. J. Clin. Investig. 2006, 116, 3229–3239. [Google Scholar] [CrossRef]
- Missale, C.; Nash, S.R.; Robinson, S.W.; Jaber, M.; Caron, M.G. Dopamine receptors: From structure to function. Physiol. Rev. 1998, 78, 189–225. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capucho, A.M.; Conde, S.V. Impact of Sugars on Hypothalamic Satiety Pathways and Its Contribution to Dysmetabolic States. Diabetology 2023, 4, 1-10. https://doi.org/10.3390/diabetology4010001
Capucho AM, Conde SV. Impact of Sugars on Hypothalamic Satiety Pathways and Its Contribution to Dysmetabolic States. Diabetology. 2023; 4(1):1-10. https://doi.org/10.3390/diabetology4010001
Chicago/Turabian StyleCapucho, Adriana M., and Silvia V. Conde. 2023. "Impact of Sugars on Hypothalamic Satiety Pathways and Its Contribution to Dysmetabolic States" Diabetology 4, no. 1: 1-10. https://doi.org/10.3390/diabetology4010001