
Development and Validation of a Novel RP-HPLC Method for the Determination of Cetrimide and Chlorhexidine Gluconate in Antiseptic Solution



Abstract
:1. Introduction
2. Materials and Methods
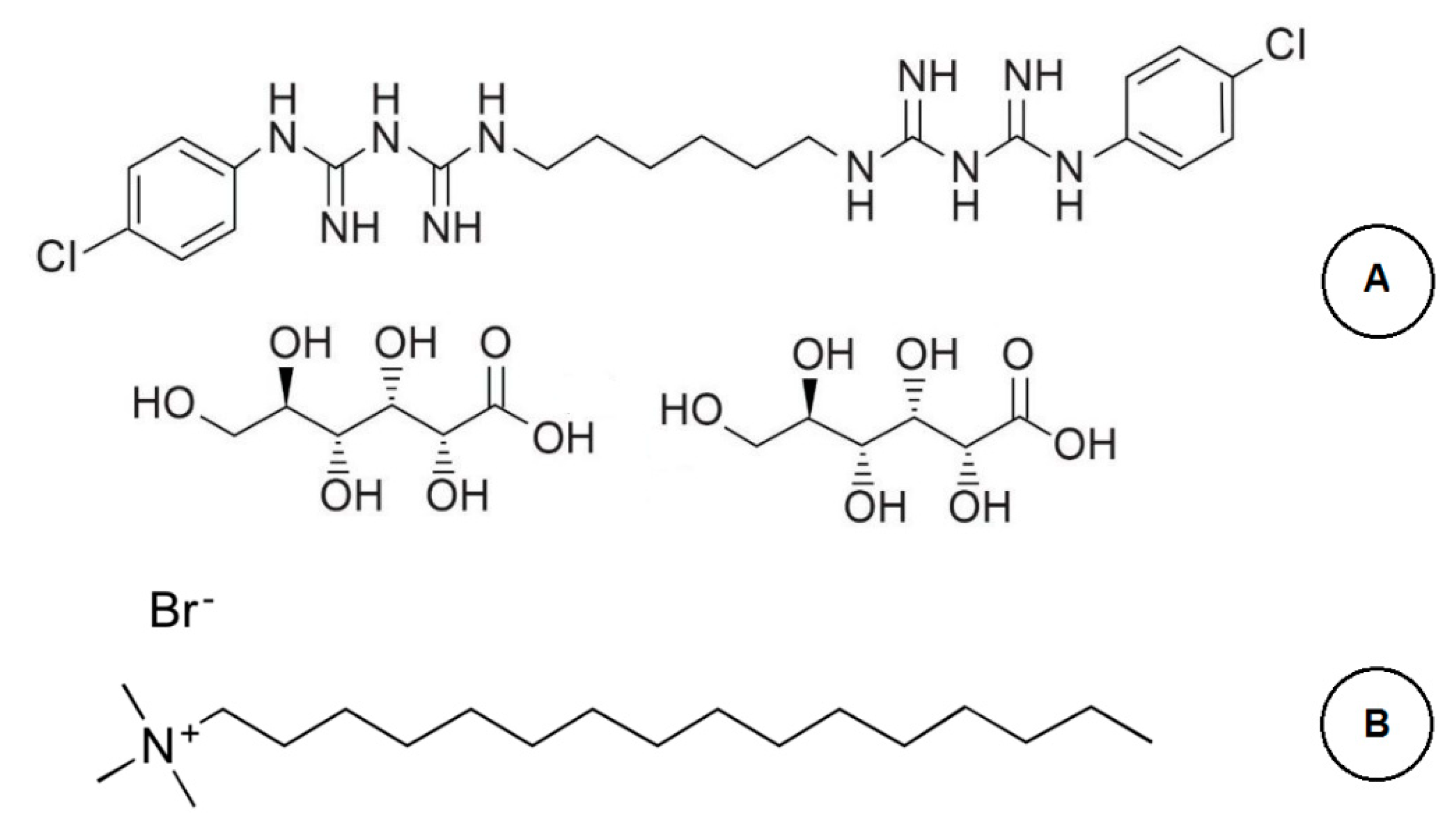
2.1. Reagents and Solvents
2.2. Instrumentation
2.3. Chromatographic Conditions
2.4. Preparation of Stock and Working Solutions
2.4.1. Preparation of Stock Standard Solutions and Working Standard Solutions
2.4.2. Preparation of Stock Solution for Spiked Samples
2.4.3. Sample Preparation
2.5. Validation Process
2.5.1. Specificity
2.5.2. Linearity, LOD and LOQ
2.5.3. Accuracy
2.5.4. Precision
2.5.5. Stability
2.6. Robustness Testing
3. Results and Discussion
3.1. Method Development
3.2. Validation Data
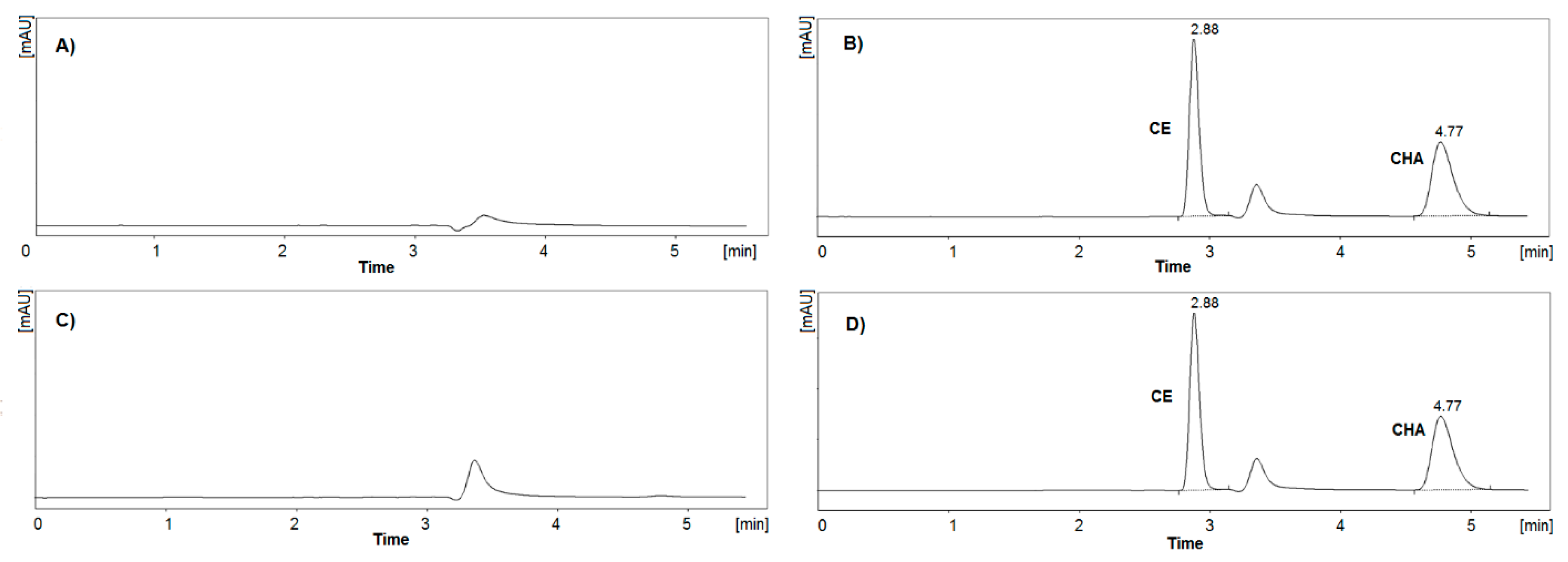
3.2.1. Specificity
3.2.2. Linearity, LOD and LOQ
3.2.3. Accuracy
3.2.4. Precision
3.2.5. Stability
3.3. Robustness Results
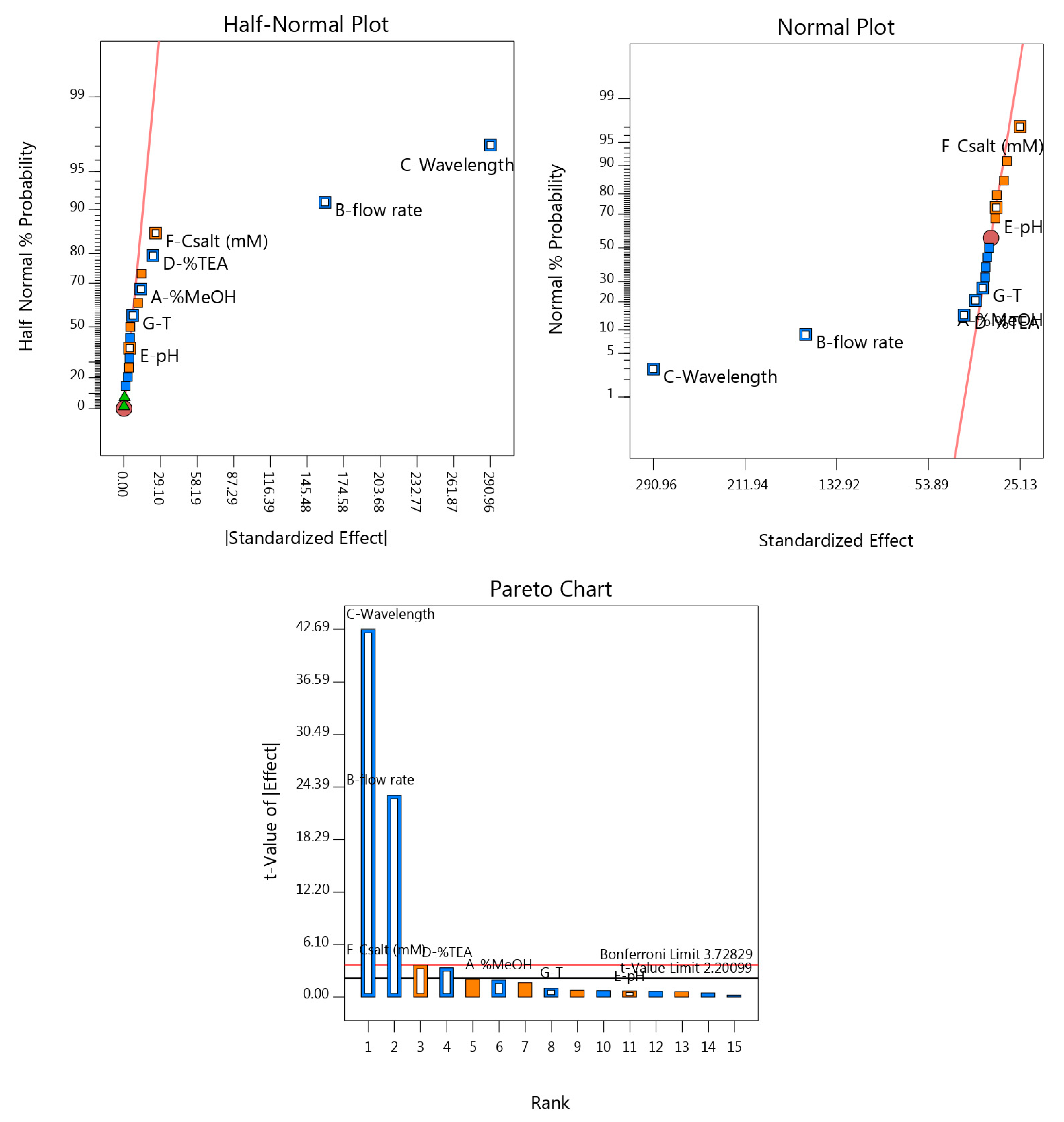
3.3.1. Statistical Approach
3.3.2. Graphical Approach
3.3.3. Comparison of the Two Approaches
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McDonnell, G.; Russell, A.D. Antiseptics and Disinfectants: Activity, Action, and Resistance. Clin. Microbiol. Rev. 1999, 12, 147–179. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boothe, H.W. Antiseptics and Disinfectants. Vet. Clin. Small Anim. Pract. 1998, 28, 233–248. [Google Scholar] [CrossRef]
- Cross, J.; Singer, E.J. Cationic Surfactants: Analytical and Biological Evaluation, 1st ed.; CRC Press: Boca Raton, FL, USA, 1994; pp. 1–26. [Google Scholar]
- Havlíková, L.; Matysová, L.; Nováková, L.; Hájková, R.; Solich, P. HPLC determination of chlorhexidine gluconate and p-chloroaniline in topical ointment. J. Pharm. Biomed. Anal. 2007, 43, 1169–1173. [Google Scholar] [CrossRef] [PubMed]
- Nicolay, A.; Wolff, E.; Vergnes, M.F.; Kaloustian, J.; Portugal, H. Rapid HPLC Method for Determination of Parachlo-roaniline in Chlorhexidine Antiseptic Agent in Mouthrinses, Ophthalmic and Skin Solution. Am. J. Anal. Chem. 2011, 2, 422–428. [Google Scholar] [CrossRef] [Green Version]
- Zeng, P.; Rao, A.; Wiedmann, T.S.; Bowles, W. Solubility Properties of Chlorhexidine Salts. Drug Dev. Ind. Pharm. 2009, 35, 172–176. [Google Scholar] [CrossRef]
- Galinos. Available online: https://www.galinos.gr/web/drugs/main/substances/cetrimide/usage (accessed on 26 November 2021).
- Galinos. Available online: https://www.galinos.gr/web/drugs/main/substances/chlorhexidine/usage (accessed on 26 November 2021).
- Galinos. Available online: https://www.galinos.gr/web/drugs/main/packages/12750/usage (accessed on 21 December 2021).
- Council of Europe; European Pharmacopoeia Commission; European Directorate for the Quality of Medicines & Healthcare. European Pharmacopoeia, 10th ed.; Council of Europe: Strasbourg, France, 2019; pp. 2159–2177. [Google Scholar]
- Rasmussen, C.D.; Nielsen, H.B.; Andersen, J.E.T. Analysis of the purity of cetrimide by titrations. PDA J. Pharm. Sci. Technol. 2006, 60, 104–110. [Google Scholar]
- Andraws, G.; Trefi, S. Ionisable substances chromatography: A new approach for the determination of Ketoprofen, Etoricoxib, and Diclofenac sodium in pharmaceuticals using ion—pair HPLC. Heliyon 2020, 6, 4613. [Google Scholar] [CrossRef]
- Trefi, S.; Bitar, Y.; Gilard, V. Separation and Quantification of Sacubitril-Valsartan Combination in Tablets by a New Ion-pair HPLC. Res. J. Pharm. Technol. 2019, 12, 1017–1022. [Google Scholar] [CrossRef]
- Hammash, L.; Bitar, Y.; Trefi, S. Novel Ion Pair HPLC Methods for the Assessment of Sitagliptin and Pioglitazone in Tablets. Res. J. Pharm. Technol. 2019, 12, 3713–3719. [Google Scholar] [CrossRef]
- Trefi, S. Simultaneous assessment of atorvastatin-ezetimibe combination in tablets by an ion-pair rp-hplc. Res. J. Pharm. Technol. 2020, 13, 4195–4199. [Google Scholar] [CrossRef]
- USP Monographs: Chlorhexidine Gluconate Solution. Available online: http://www.pharmacopeia.cn/v29240/usp29nf24s0_m15620.html (accessed on 30 November 2021).
- Colombo, M. Validation of a Simple Hplc Method for the Determination of Chlorhexidine Digluconate in Solution. Bachelor’s Thesis, Federal University of Rio Grande do Sul (UFRGS), Porto Alegre, Brazil, November 2013. [Google Scholar]
- Rajput, S.J.; Sathe, M.A. Application of doe and statistical analysis for development and validation of analytical method for chlrohexidine gluconate and cetrimide in its bulk and pharmaceutical dosage forms. Int. J. Pharm. Sci. Res. 2018, 9, 2800–2806. [Google Scholar] [CrossRef]
- Lee, T.D. Introduction to Modern Liquid Chromatography, Third Edition. J. Am. Soc. Mass Spectrom. 2011, 22, 196. [Google Scholar] [CrossRef] [Green Version]
- Vander Heyden, Y.; Nijhuis, A.; Smeyers-Verbeke, J.; Vandeginste, B.G.; Massart, D.L. Guidance for robustness/ruggedness tests in method validation. J. Pharm. Biomed. Anal. 2001, 24, 723–753. [Google Scholar] [CrossRef]
- Agilent|Chemical Analysis, Life Sciences, and Diagnostics. Available online: https://www.agilent.com/cs/library/ (accessed on 24 December 2021).
- HypersilTM SAS C1 HPLC Columns. Available online: https://www.thermofisher.com/order/catalog/product/30505-014001 (accessed on 22 December 2021).
- Wirth, M.J.; Fatunmbi, H.O. Horizontal polymerization of mixed trifunctional silanes on silica: A potential chroma-to-graphic stationary phase. Anal. Chem. 1992, 64, 2783–2786. [Google Scholar] [CrossRef]
- Neofotistos, A.D.; Gkountanas, K.; Boutsikaris, H.; Dotsikas, Y. A Validated RP-HPLC Method for the Determination of Bu-tamirate Citrate and Benzoic Acid in Syrup, Based on an Experimental Design Assessment of Robustness. Separations 2021, 8, 163. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Experimental Parameters | Levels | ||
|---|---|---|---|
| −1 | 0 (Nominal Value) | +1 | |
| Flow Rate | 0.90 | 1.00 | 1.10 |
| % MeOH | 83 | 85 | 87 |
| T | 23 | 25 | 27 |
| λ | 204 | 205 | 206 |
| pH | 2.8 | 3.0 | 3.2 |
| Csalt | 48 | 50 | 52 |
| % Et3N | 0.15 | 0.20 | 0.25 |
| Run | Experimental Factor | ||||||
|---|---|---|---|---|---|---|---|
| Flow | pH | % MeOH | Csalt | T | λ | % Et3N | |
| 1 | 0.9 | 2.8 | 87 | 52 | 23 | 206 | 0.15 |
| 2 | 1.1 | 3.2 | 83 | 52 | 23 | 204 | 0.15 |
| 3 | 1.1 | 2.8 | 83 | 48 | 27 | 206 | 0.15 |
| 4 | 1.1 | 3.2 | 87 | 48 | 23 | 206 | 0.15 |
| 5 | 1.1 | 2.8 | 87 | 52 | 27 | 204 | 0.15 |
| 6 | 1.1 | 2.8 | 83 | 52 | 23 | 206 | 0.25 |
| 7 | 1.0 | 3.0 | 85 | 50 | 25 | 205 | 0.20 |
| 8 | 1.0 | 3.0 | 85 | 50 | 25 | 205 | 0.20 |
| 9 | 0.9 | 2.8 | 83 | 48 | 23 | 204 | 0.15 |
| 10 | 0.9 | 2.8 | 87 | 48 | 27 | 206 | 0.25 |
| 11 | 0.9 | 3.2 | 83 | 52 | 27 | 206 | 0.15 |
| 12 | 0.9 | 3.2 | 87 | 48 | 27 | 204 | 0.15 |
| 13 | 0.9 | 3.2 | 83 | 48 | 23 | 206 | 0.25 |
| 14 | 1.1 | 3.2 | 83 | 48 | 27 | 204 | 0.25 |
| 15 | 1.1 | 2.8 | 87 | 48 | 23 | 204 | 0.25 |
| 16 | 0.9 | 2.8 | 83 | 52 | 27 | 204 | 0.25 |
| 17 | 0.9 | 3.2 | 87 | 52 | 23 | 204 | 0.25 |
| 18 | 1.1 | 3.2 | 87 | 52 | 27 | 206 | 0.25 |
| 19 | 1.0 | 3.0 | 85 | 50 | 25 | 205 | 0.20 |
| Compound | Regression Equation | r2 | LOD (μg/mL) | LOQ (μg/mL) |
|---|---|---|---|---|
| Cetrimide | y = 5.3 × (±0.06) − 28.5 (±9.0) | 0.9997 | 4.14 | 12.53 |
| Chlorhexidine | y = 53.7 × (±0.7) − 78.1 (±11.8) | 0.9994 | 0.54 | 1.64 |
| Compound | Concentration (μg/mL) | Mean % Recovery | %RSD |
|---|---|---|---|
| Cetrimide | 112.50 | 100.98 | 0.37 |
| 150.00 | 101.73 | ||
| 187.50 | 101.22 | ||
| Chlorhexidine | 15.38 | 98.65 | 0.73 |
| 20.50 | 100.11 | ||
| 25.63 | 99.38 |
| Compound | Day 1 | Day 2 | Day 3 |
|---|---|---|---|
| Cetrimide | 100.52 | 99.32 | 101.23 |
| 101.47 | 101.47 | 100.55 | |
| 101.64 | 100.09 | 101.59 | |
| 101.35 | 101.48 | 101.27 | |
| 100.42 | 100.05 | 102.04 | |
| 99.19 | 101.88 | ||
| %RSDr | 0.56 | 1.00 | 0.53 |
| Chlorhexidine | 99.00 | 99.86 | 99.11 |
| 99.42 | 100.89 | 98.97 | |
| 100.11 | 99.65 | 99.32 | |
| 101.74 | 100.74 | 99.42 | |
| 101.67 | 99.75 | 99.79 | |
| 96.90 | 99.32 | 100.02 | |
| %RSDr | 1.82 | 0.63 | 0.40 |
| Run | Response | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| tR CE (min) | tR CHA (min) | Area CE (mAU × min) | Area CHA (mAU × min) | Rs | NCE | NCHA | TF CE | TF CHA | |
| 1 | 3.39 | 4.36 | 744.28 | 753.55 | 4.32 | 7294 | 3505 | 1.46 | 1.82 |
| 2 | 2.80 | 3.36 | 901.50 | 650.48 | 3.09 | 5783 | 3898 | 1.18 | 1.43 |
| 3 | 2.71 | 3.62 | 562.53 | 608.63 | 5.10 | 6373 | 4084 | 1.10 | 1.53 |
| 4 | 2.67 | 3.96 | 557.72 | 600.73 | 6.07 | 5271 | 3394 | 1.33 | 1.88 |
| 5 | 2.59 | 4.65 | 836.80 | 639.52 | 8.90 | 4935 | 3443 | 1.33 | 1.79 |
| 6 | 2.75 | 3.50 | 555.00 | 606.63 | 4.20 | 6562 | 3817 | 1.10 | 1.47 |
| 7 | 2.87 | 4.69 | 797.55 | 694.35 | 8.23 | 6089 | 4050 | 1.40 | 1.63 |
| 8 | 2.95 | 4.61 | 782.14 | 691.46 | 7.81 | 6433 | 3830 | 1.30 | 1.65 |
| 9 | 3.33 | 4.41 | 1013.38 | 793.58 | 5.31 | 7066 | 4590 | 1.27 | 1.59 |
| 10 | 3.31 | 4.74 | 685.68 | 727.07 | 6.04 | 8065 | 3330 | 1.27 | 1.85 |
| 11 | 3.42 | 4.17 | 734.79 | 765.96 | 3.67 | 7439 | 4471 | 1.17 | 1.63 |
| 12 | 3.31 | 4.71 | 999.12 | 766.10 | 5.87 | 6982 | 3283 | 1.27 | 1.75 |
| 13 | 3.33 | 4.45 | 693.60 | 744.76 | 5.29 | 7038 | 4279 | 1.17 | 1.59 |
| 14 | 2.71 | 3.65 | 827.25 | 658.65 | 5.28 | 6342 | 4144 | 1.20 | 1.47 |
| 15 | 2.71 | 3.84 | 818.13 | 645.80 | 5.32 | 6373 | 2941 | 1.20 | 1.82 |
| 16 | 3.33 | 4.41 | 1023.55 | 808.28 | 5.34 | 7038 | 4590 | 1.18 | 1.59 |
| 17 | 3.27 | 4.93 | 1002.07 | 791.00 | 6.97 | 6814 | 3598 | 1.27 | 1.80 |
| 18 | 2.70 | 3.81 | 560.49 | 607.20 | 5.44 | 6310 | 3136 | 1.33 | 1.71 |
| 19 | 2.94 | 4.57 | 799.21 | 697.61 | 7.67 | 6404 | 3952 | 1.30 | 1.71 |
| Experimental Parameter | Ex Values | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| tR CE | tR CHA | Area CE | Area CHA | Rs | NCE | NCHA | TF CE | TF CHA | |
| Flow Rate | 0.63 | −0.72 | −159.63 | −141.58 | 0.07 | −1223.38 | −348.63 | −0.04 | −0.07 |
| pH | 0.01 | −0.06 | 4.65 | 0.23 | −0.35 | −215.88 | −12.13 | 0.002 | −0.03 |
| % MeOH | −0.05 | 0.43 | −13.41 | −13.25 | 1.46 | −199.63 | −905.38 | 0.14 | 0.27 |
| Csalt | 0.02 | −0.02 | 25.13 | 9.66 | −0.29 | −166.88 | 51.63 | 0.03 | −0.03 |
| T | −0.02 | 0.12 | −6.94 | −0.64 | 0.64 | 160.37 | 57.38 | −0.02 | −0.01 |
| λ | 0.03 | −0.17 | −290.97 | 42.36 | −0.74 | 377.38 | −58.88 | 0.002 | 0.03 |
| % Et3N | −0.01 | 0.01 | −23.04 | 1.36 | 0.19 | 424.88 | −104.13 | −0.05 | −0.02 |
| Response | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| tR CE | tR CHA | Area CE | Area CHA | Rs | NCE | NCHA | TF CE | TF CHA | |
| (SE)e | 0.02 | 0.03 | 4.70 | 1.54 | 0.15 | 95.39 | 55.11 | 0.03 | 0.02 |
| Ecritical | 0.10 | 0.13 | 20.25 | 6.62 | 0.63 | 410.48 | 237.13 | 0.12 | 0.08 |
| Factors | tR CE | tR CHA | Area CE | Area CHA | Rs | NCE | NCHA | TF CE | TF CHA |
|---|---|---|---|---|---|---|---|---|---|
| Flow | √√ | √√ | √√ | √√ | √√ | √√ | |||
| pH | |||||||||
| %MeOH | √√ | √√ | √√ | √√ | √√ | √√ | |||
| Csalt | √√ | √ | |||||||
| T | √ | ||||||||
| Λ | √ | √√ | √√ | √ | |||||
| % Et3N | √√ | √ |
| Factors | Limits |
|---|---|
| Flow Rate | (0.995, 1.005) |
| pH | (2.8, 3.2) |
| %MeOH | (84.5, 85.5) |
| Csalt | (48.6, 51.4) |
| T | (23, 27) |
| λ | (204.93, 205.07) |
| % Et3N | (0.16, 0.24) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vrachas, A.; Gkountanas, K.; Boutsikaris, H.; Dotsikas, Y. Development and Validation of a Novel RP-HPLC Method for the Determination of Cetrimide and Chlorhexidine Gluconate in Antiseptic Solution. Analytica 2022, 3, 79-91. https://doi.org/10.3390/analytica3010006
Vrachas A, Gkountanas K, Boutsikaris H, Dotsikas Y. Development and Validation of a Novel RP-HPLC Method for the Determination of Cetrimide and Chlorhexidine Gluconate in Antiseptic Solution. Analytica. 2022; 3(1):79-91. https://doi.org/10.3390/analytica3010006
Chicago/Turabian StyleVrachas, Andreas, Kostas Gkountanas, Haris Boutsikaris, and Yannis Dotsikas. 2022. "Development and Validation of a Novel RP-HPLC Method for the Determination of Cetrimide and Chlorhexidine Gluconate in Antiseptic Solution" Analytica 3, no. 1: 79-91. https://doi.org/10.3390/analytica3010006