
Improved Photocatalytic H2 Evolution by Cobaloxime-Tethered Imidazole-Functionalized Periodic Mesoporous Organosilica
 , , and
, , and
Abstract
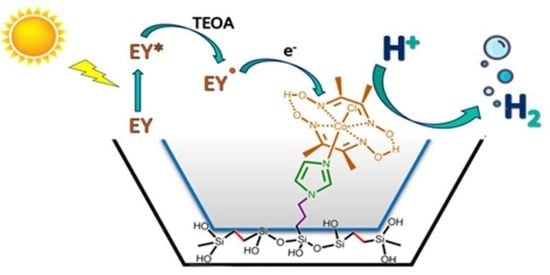
:
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Synthesis of the Materials
2.2.1. Synthesis of Cobaloxime Complexes: Co(dmgH2)(dmgH)Cl2 and Co(dmgH)2(Im)Cl
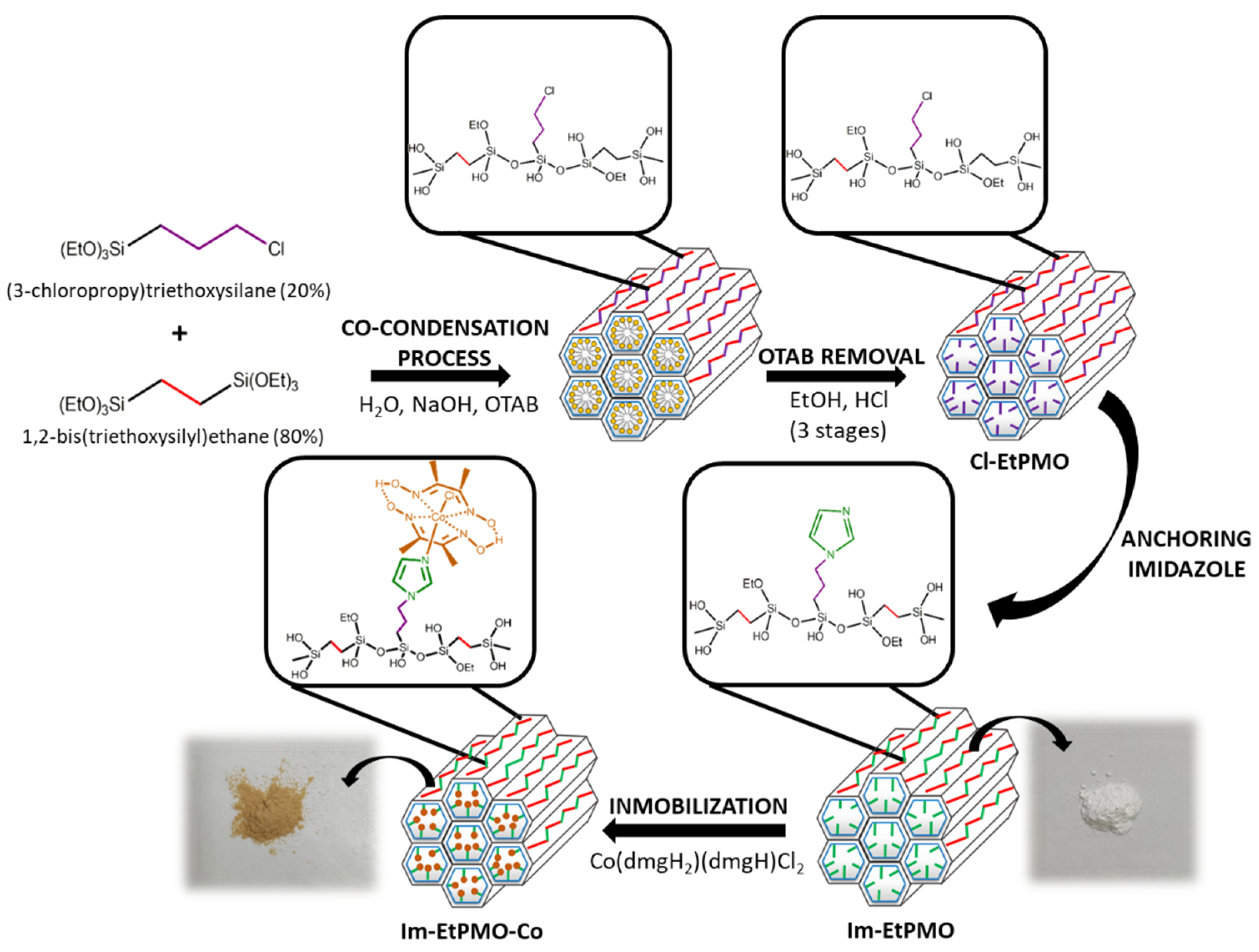
2.2.2. Synthesis of Chloropropyl Functionalized Ethylene-Bridged Periodic Mesoporous Organosilicas (Cl-EtPMO)
2.2.3. Anchoring of Imidazole on Cl-EtPMO
2.2.4. Immobilization of Dichlorocobaloxime on Im-EtPMO
2.3. Physicochemical Characterization
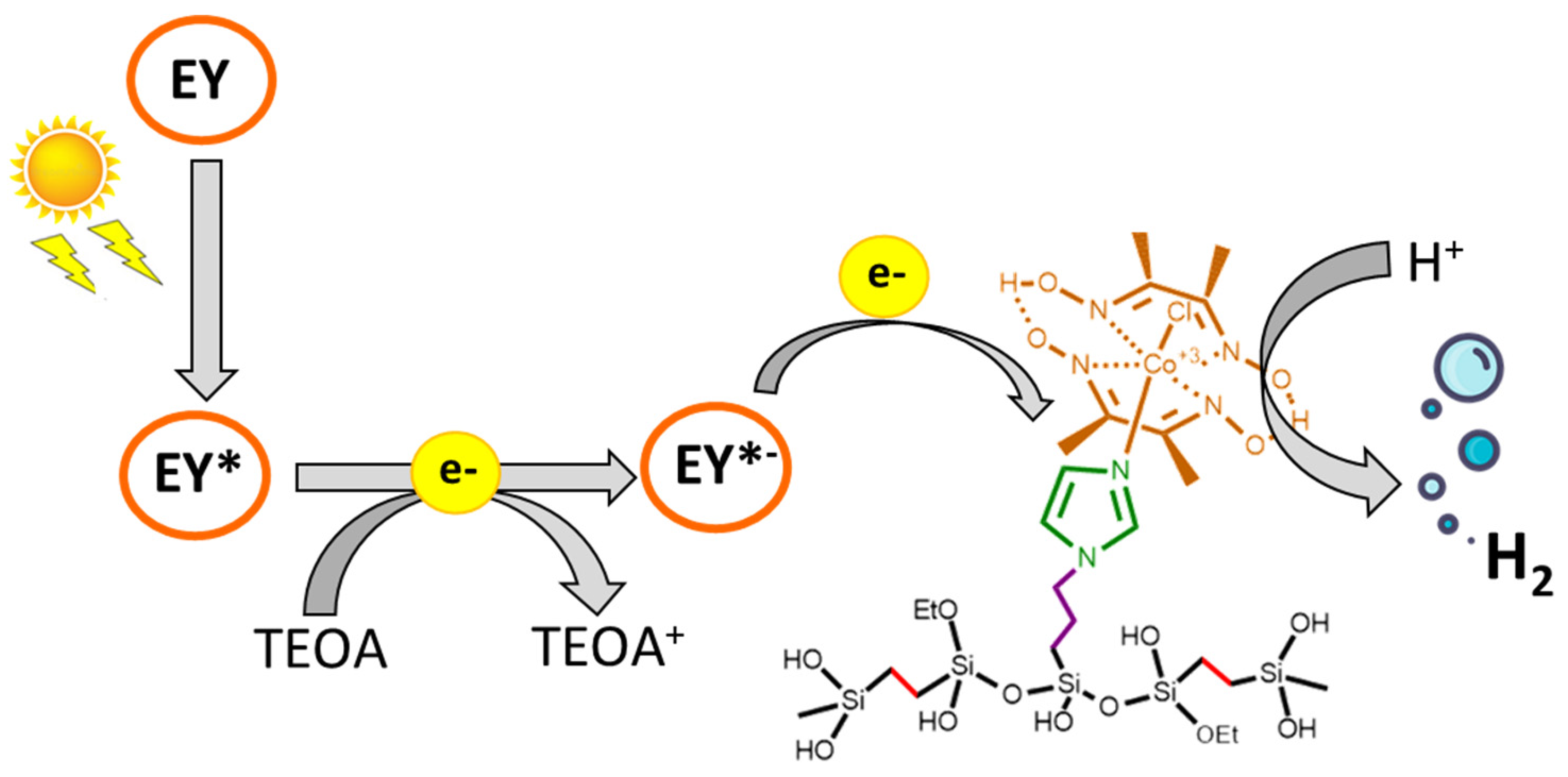
2.4. Photocatalytic Hydrogen Production Tests
3. Results and Discussion
3.1. Synthetic Strategy for Cobaloxime-Tethered Imidazole-Functionalized Ethylene-Bridged PMO Catalyst
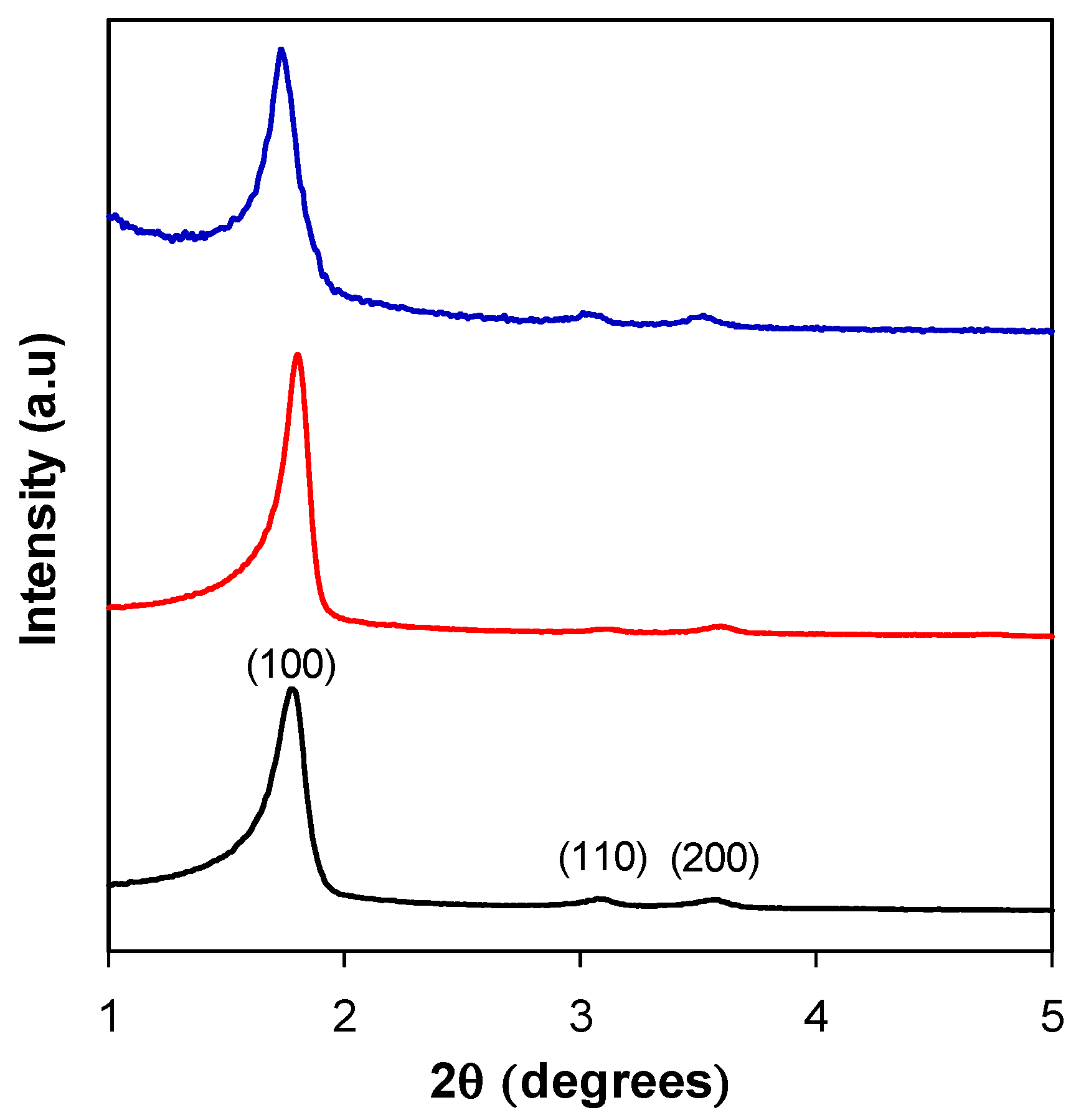
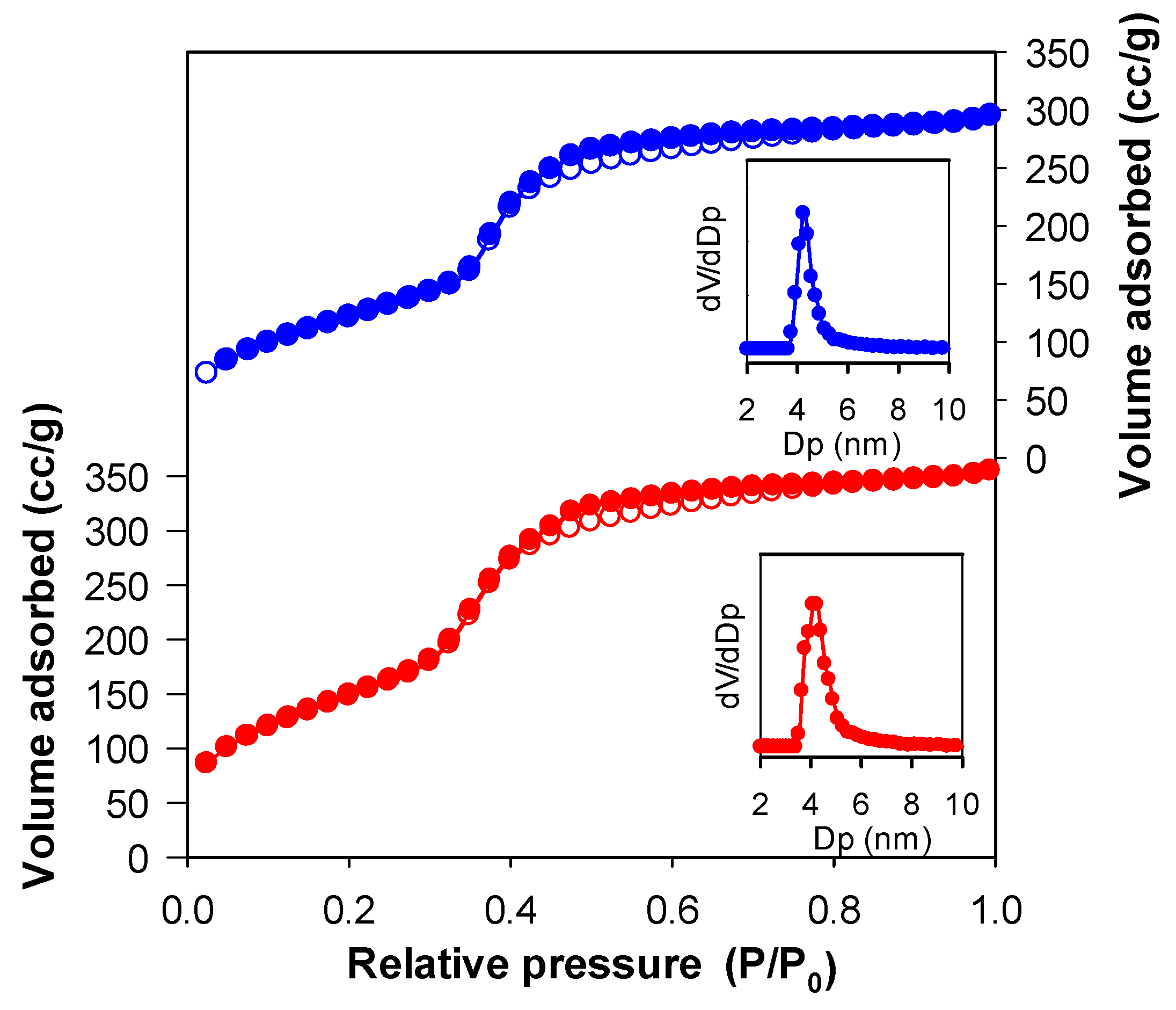
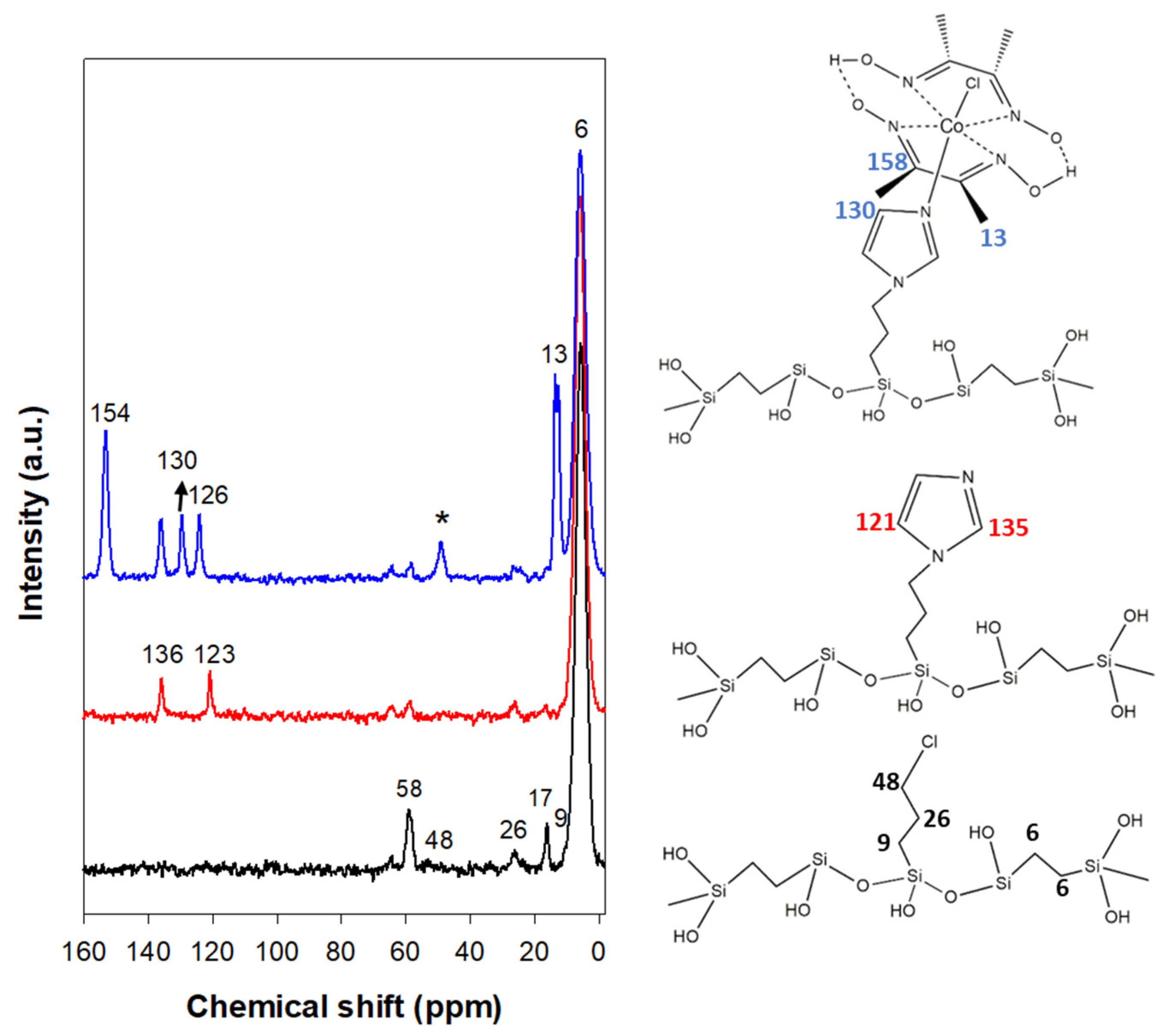
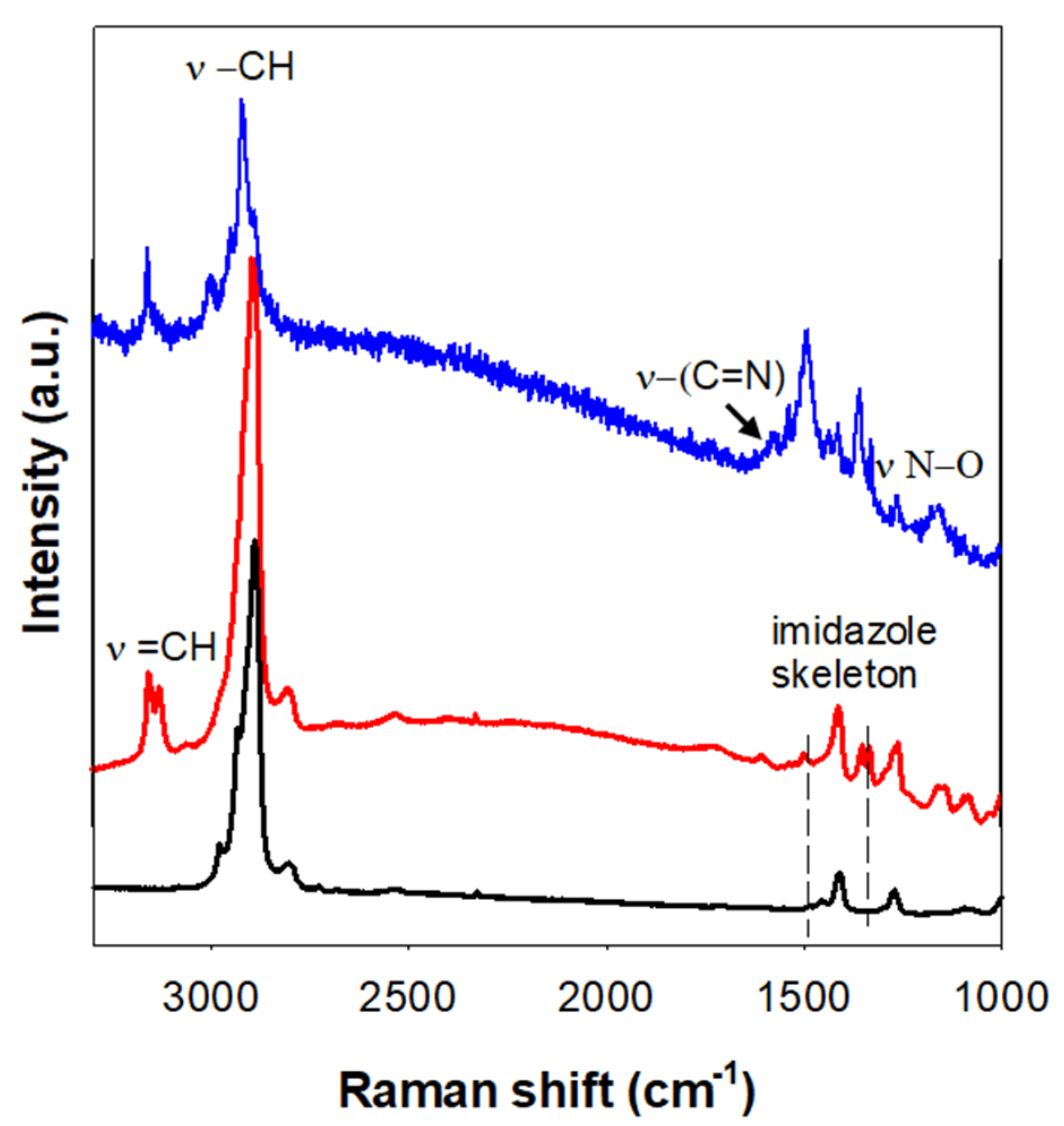
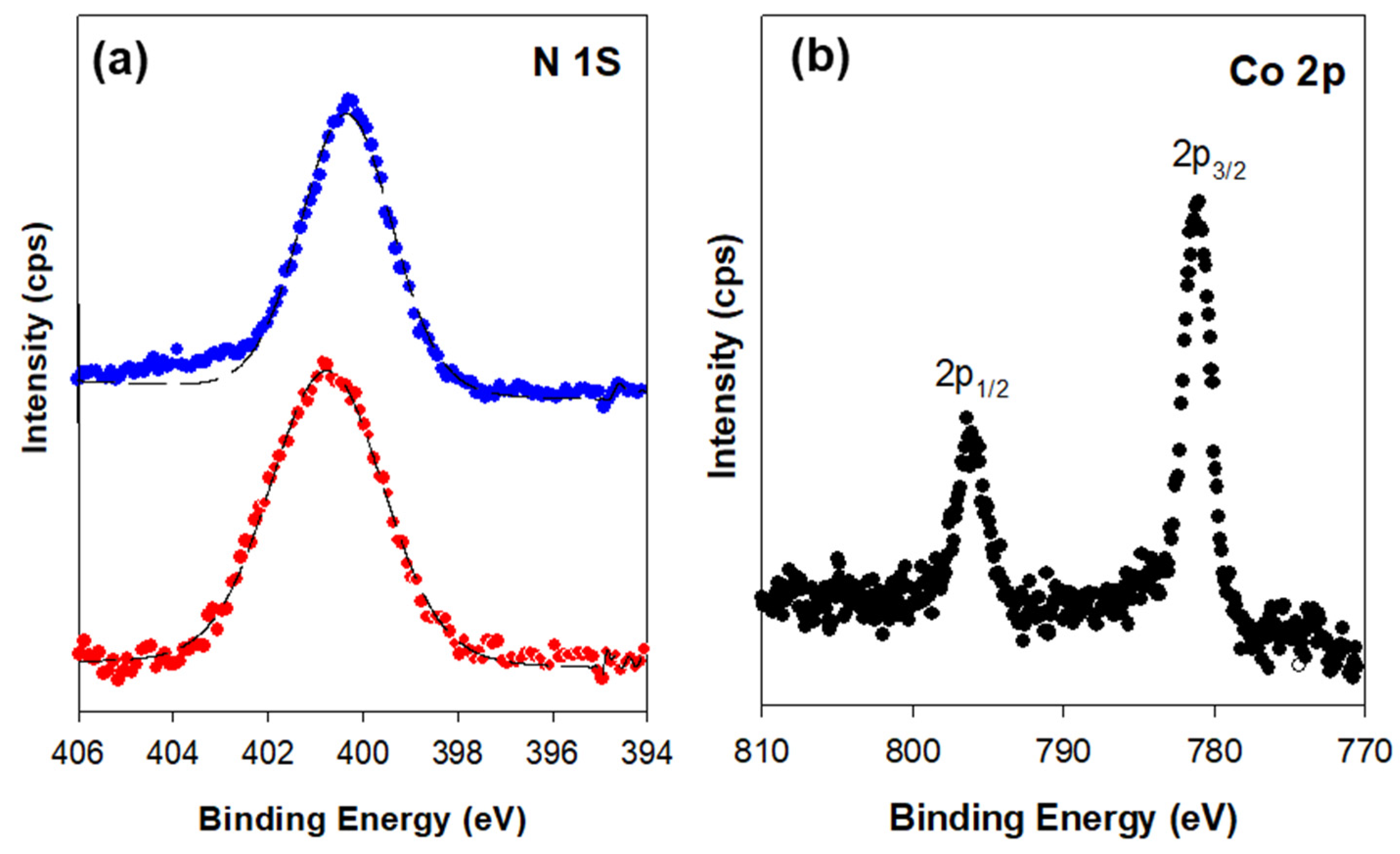
3.2. Structural Characterization
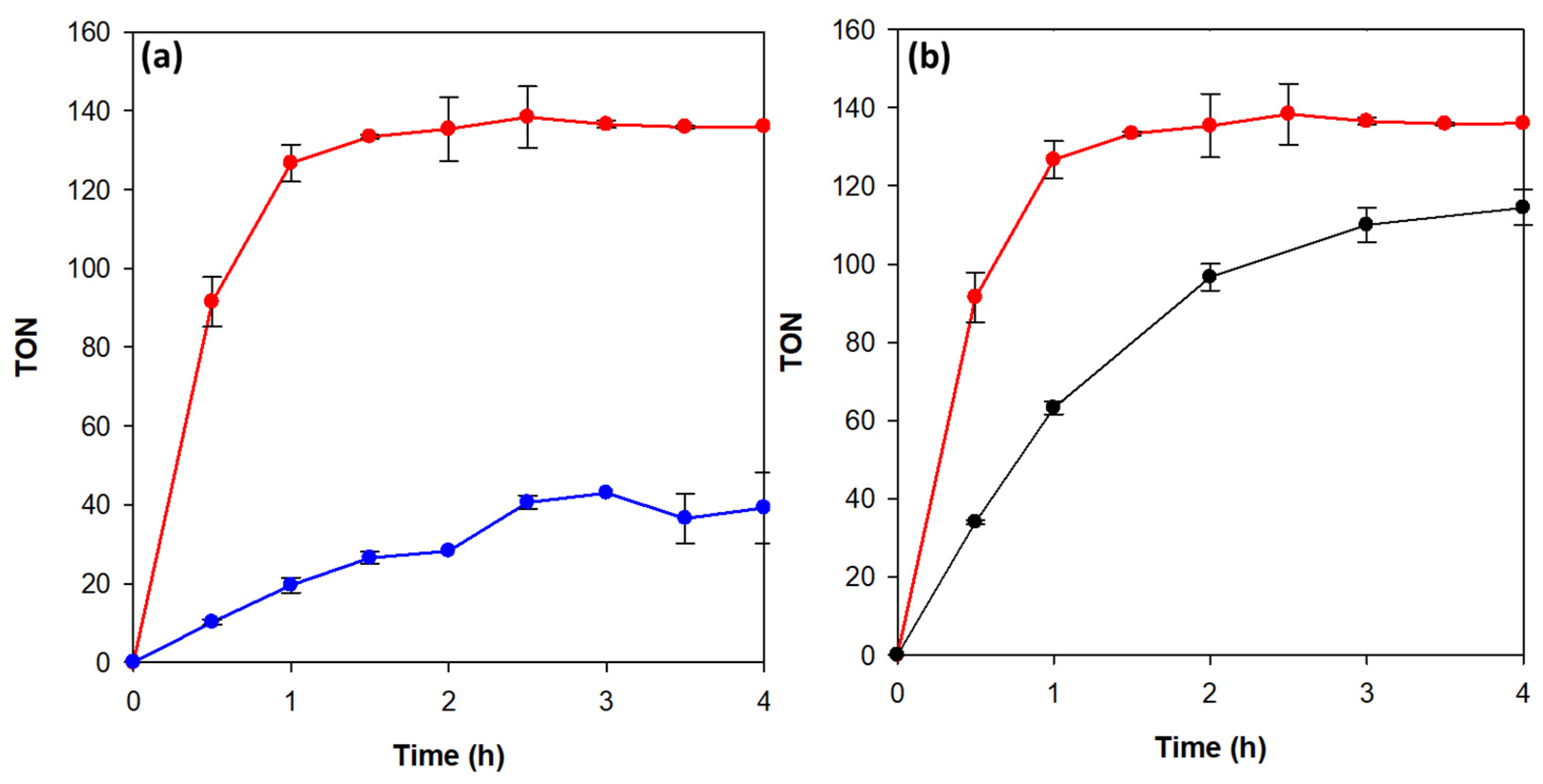
3.3. Photocatalytic Hydrogen Evolution Reaction
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nocera, D.G. The Artificial Leaf. Acc. Chem. Res. 2012, 45, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Koroneos, C.; Dompros, A.; Roumbas, G.; Moussiopoulos, N. Life Cycle Assessment of Hydrogen Fuel Production Processes. Int. J. Hydrog. Energy 2004, 29, 1443–1450. [Google Scholar] [CrossRef]
- Le Goff, A.; Artero, V.; Jousselme, B.; Tran, P.D.; Guillet, N.; Métayé, R.; Fihri, A.; Palacin, S.; Fontecave, M. From Hydrogenases to Noble Metal–Free Catalytic Nanomaterials for H2 Production and Uptake. Science 2009, 326, 1384–1387. [Google Scholar] [CrossRef]
- Gao, S.; Fan, W.; Liu, Y.; Jiang, D.; Duan, Q. Artificial Water-Soluble Systems Inspired by [FeFe]-Hydrogenases for Electro- and Photocatalytic Hydrogen Production. Int. J. Hydrog. Energy 2020, 19, 4305–4327. [Google Scholar] [CrossRef]
- Lu, Y.; Koo, J. O2 Sensitivity and H2 Production Activity of Hydrogenases—A Review. Biotechnol. Bioeng. 2019, 116, 3124–3135. [Google Scholar] [CrossRef]
- Azwar, M.Y.; Hussain, M.A.; Abdul-wahab, A.K. Development of Biohydrogen Production by Photobiological, Fermentation and Electrochemical Processes: A Review. Renew. Sustain. Energy Rev. 2014, 31, 158–173. [Google Scholar] [CrossRef]
- Pullen, S.; Fei, H.; Orthaber, A.; Cohen, S.M.; Ott, S. Enhanced Photochemical Hydrogen Production by a Molecular Diiron Catalyst Incorporated into a Metal-Organic Framework. J. Am. Chem. Soc. 2013, 135, 16997–17003. [Google Scholar] [CrossRef]
- Himiyama, T.; Waki, M.; Esquivel, D.; Onoda, A.; Hayashi, T.; Van Der Voort, P.; Inagaki, S. A Heterogeneous Hydrogen-Evolution Catalyst Based on a Mesoporous Organosilica with a Diiron Catalytic Center Modelling [FeFe]-Hydrogenase. ChemCatChem 2018, 10, 4908–4913. [Google Scholar] [CrossRef]
- Li, R.-X.; Ren, X.-T.; Tang, M.-Y.; Chen, M.-X.; Huang, G.-B.; Fang, C.-H.; Liu, T.; Feng, Z.-H.; Yin, Y.-B.; Guo, Y.-M.; et al. Fabrication of Covalently Linked Graphene-Mediated [FeFe]-Hydrogenases Biomimetic Photocatalytic Hydrogen Evolution System in Aqueous Solution. Appl. Catal. B Environ. 2018, 224, 772–782. [Google Scholar] [CrossRef]
- Amaro-Gahete, J.; Pavliuk, M.V.; Tian, H.; Esquivel, D.; Romero-Salguero, F.J.; Ott, S. Catalytic Systems Mimicking the [FeFe]-Hydrogenase Active Site for Visible-Light-Driven Hydrogen Production. Coord. Chem. Rev. 2021, 448, 214172. [Google Scholar] [CrossRef]
- Gerhard, N. Schrauzer Organocobalt Chemistry of Vitamin B12 Model Compounds (Cobaloximes). Acc. Chem. Res. 1968, 1, 97–103. [Google Scholar]
- Lakadamyali, F.; Kato, M.; Muresan, N.M.; Reisner, E. Selective Reduction of Aqueous Protons to Hydrogen with a Synthetic Cobaloxime Catalyst in the Presence of Atmospheric Oxygen. Angew. Chem.-Int. Ed. 2012, 51, 9381–9384. [Google Scholar] [CrossRef]
- Wakerley, D.W.; Gross, M.A.; Reisner, E. Proton Reduction by Molecular Catalysts in Water under Demanding Atmospheres. Chem. Commun. 2014, 50, 15995–15998. [Google Scholar] [CrossRef] [PubMed]
- Willkomm, J.; Muresan, N.M.; Reisner, E. Enhancing H2 Evolution Performance of an Immobilised Cobalt Catalyst by Rational Ligand Design. Chem. Sci. 2015, 6, 2727–2736. [Google Scholar] [CrossRef] [PubMed]
- Wakerley, D.W.; Reisner, E. Development and Understanding of Cobaloxime Activity through Electrochemical Molecular Catalyst Screening. Phys. Chem. Chem. Phys. 2014, 16, 5739–5746. [Google Scholar] [CrossRef] [PubMed]
- Artero, V.; Pe, J.; Fontecave, M. Cobalt and Nickel Diimine-Dioxime Complexes as Molecular Electrocatalysts for Hydrogen Evolution with Low Overvoltages. Proc. Natl. Acad. Sci. USA 2009, 106, 20627–20632. [Google Scholar]
- Andreiadis, E.S.; Jacques, P.A.; Tran, P.D.; Leyris, A.; Chavarot-Kerlidou, M.; Jousselme, B.; Matheron, M.; Pécaut, J.; Palacin, S.; Fontecave, M.; et al. Molecular Engineering of a Cobalt-Based Electrocatalytic Nanomaterial for H2 Evolution under Fully Aqueous Conditions. Nat. Chem. 2013, 5, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Hawecker, J.; Lehn, J.M.; Ziessel, R. Efficient Homogeneous Photochemical Hydrogen Generation and Water Reduction Mediated by Cobaloxime or Macrocyclic Cobalt Complexes. Nouv. J. Chim. 1983, 7, 271–277. [Google Scholar] [CrossRef]
- Panagiotopoulos, A.; Ladomenou, K.; Sun, D.; Artero, V.; Coutsolelos, A.G. Photochemical Hydrogen Production and Cobaloximes: The Influence of the Cobalt Axial N-Ligand on the System Stability. Dalt. Trans. 2016, 45, 6732–6738. [Google Scholar] [CrossRef]
- Sideri, I.K.; Charalambidis, G.; Coutsolelos, A.G.; Arenal, R.; Tagmatarchis, N. Pyridine vs. Imidazole Axial Ligation on Cobaloxime Grafted Graphene: Hydrogen Evolution Reaction Insights. Nanomaterials 2022, 12, 3077. [Google Scholar] [CrossRef]
- Wadsworth, B.L.; Nishiori, D.; Nguyen, N.P.; Cruz, E.A.R. Electrochemistry of Polymeric Cobaloxime-Containing Assemblies in Organic and Aqueous Solvents. ECS J. Solid State Sci. Technol. 2020, 9, 061018. [Google Scholar] [CrossRef]
- Roy, S.; Huang, Z.; Bhunia, A.; Castner, A.; Gupta, A.K.; Zou, X.; Ott, S. Electrocatalytic Hydrogen Evolution from a Cobaloxime-Based Metal-Organic Framework Thin Film. J. Am. Chem. Soc. 2019, 141, 15942–15950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, S.; Bhunia, A.; Schuth, N.; Haumann, M.; Ott, S. Light-Driven Hydrogen Evolution Catalyzed by a Cobaloxime Catalyst Incorporated in a MIL-101(Cr) Metal-Organic Framework. Sustain. Energy Fuels 2018, 2, 1148–1152. [Google Scholar] [CrossRef] [PubMed]
- Gottschling, K.; Savasci, G.; Vignolo-González, H.; Schmidt, S.; Mauker, P.; Banerjee, T.; Rovó, P.; Ochsenfeld, C.; Lotsch, B.V. Rational Design of Covalent Cobaloxime-Covalent Organic Framework Hybrids for Enhanced Photocatalytic Hydrogen Evolution. J. Am. Chem. Soc. 2020, 142, 12146–12156. [Google Scholar] [CrossRef]
- Navarro, M.Á.; Cosano, D.; Bhunia, A.; Simonelli, L.; Martin-Diaconescu, V.; Romero-Salguero, F.J.; Esquivel, D. Cobaloxime Tethered Pyridine-Functionalized Ethylene-Bridged Periodic Mesoporous Organosilica as an Efficient HER Catalyst. Sustain. Energy Fuels 2022, 6, 398–407. [Google Scholar] [CrossRef]
- Mccormick, T.M.; Han, Z.; Weinberg, D.J.; Brennessel, W.W.; Holland, P.L.; Eisenberg, R. Impact of Ligand Exchange in Hydrogen Production from Cobaloxime-Containing Photocatalytic Systems. Inorg. Chem. 2011, 50, 10660–10666. [Google Scholar] [CrossRef]
- Dolui, D.; Khandelwal, S.; Majumder, P.; Dutta, A. The Odyssey of Cobaloximes for Catalytic H2 production and Their Recent Revival with Enzyme-Inspired Design. Chem. Commun. 2020, 56, 8166–8181. [Google Scholar] [CrossRef] [PubMed]
- Dolui, D.; Khandelwal, S.; Shaik, A.; Gaat, D.; Thiruvenkatam, V.; Dutta, A. Enzyme-Inspired Synthetic Proton Relays Generate Fast and Acid-Stable Cobalt-Based H2 Production Electrocatalysts. ACS Catal. 2019, 9, 10115–10125. [Google Scholar] [CrossRef]
- Dolui, D.; Das, S.; Bharti, J.; Kumar, S.; Kumar, P.; Dutta, A. Bio-Inspired Cobalt Catalyst Enables Natural-Sunlight-Driven Hydrogen Production from Aerobic Neutral Aqueous Solution. Cell Rep. Phys. Sci. 2020, 1, 100007. [Google Scholar] [CrossRef]
- Trogler, W.C.; Stewart, R.C. Cis and Trans Effects on the Proton Magnetic Resonance Spectra of Cobaloximes. Inorg. Chem. 1974, 13, 1564–1570. [Google Scholar] [CrossRef]
- Waki, M.; Mizoshita, N.; Tani, T.; Inagaki, S. Periodic Mesoporous Organosilica Derivatives Bearing a High Density of Metal Complexes on Pore Surfaces. Angew. Chem.-Int. Ed. 2011, 50, 11667–11671. [Google Scholar] [CrossRef]
- Zhang, P.; Sun, Y.; Zhang, Q.; Guo, Y.; Song, D. Upgrading of Pyrolysis Biofuel via Esterification of Acetic Acid with Benzyl Alcohol Catalyzed by BrØnsted Acidic Ionic Liquid Functionalized Ethyl-Bridged Organosilica Hollow Nanospheres. Fuel 2018, 228, 175–186. [Google Scholar] [CrossRef]
- Xia, Y.; Mokaya, R. To Stir or Not to Stir: Formation of Hierarchical Superstructures of Molecularly Ordered Ethylene-Bridged Periodic Mesoporous Organosilicas. J. Mater. Chem. 2006, 16, 395–400. [Google Scholar] [CrossRef]
- Sing, K.S.W. Reporting Physisorption Data for Gas / Solid Systems with Special Reference to the Determination of S. Pure Appl. Chem. 1982, 54, 2201–2218. [Google Scholar] [CrossRef]
- Liu, W.; Kaczmarek, A.M.; Folens, K.; Du Laing, G.; Van Der Voort, P.; Van Deun, R. Rational Design of Lanthanide Nano Periodic Mesoporous Organosilicas (Ln-Nano-PMOs) for near-Infrared Emission. Dalt. Trans. 2021, 50, 2774–2781. [Google Scholar] [CrossRef]
- Navarro, M.Á.; Amaro-Gahete, J.; Ruiz, J.R.; Jiménez-Sanchidrián, C.; Romero-Salguero, F.J.; Esquivel, D. Copper-Complexed Dipyridyl-Pyridazine Functionalized Periodic Mesoporous Organosilica as a Heterogeneous Catalyst for Styrene Epoxidation. Dalt. Trans. 2022, 51, 4884–4897. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhou, L.; Su, Y.; Han, B.; Deng, F. 13C and 15N Spectral Editing inside Histidine Imidazole Ring through Solid-State NMR Spectroscopy. Solid State Nucl. Magn. Reson. 2013, 54, 13–17. [Google Scholar] [CrossRef]
- Bisergaeva, R.; Sirieva, Y. Synthesis of Some Cobaloxime Derivatives. Earth Environ. Sci. 2020, 421, 072007. [Google Scholar] [CrossRef]
- Markham, L.M.; Mayne, L.C.; Hudson, B.S.; Zgierski, M.Z. Resonance Raman Studies of Imidazole, Imidazolium, and Their Derivatives: The Effect of Deuterium Substitution. J. Phys. Chem. 1993, 97, 10319–10325. [Google Scholar] [CrossRef]
- Bottari, C.; Rossi, B.; Mele, A.; Damin, A.; Bordiga, S.; Musso, M.; Gessini, A.; Masciovecchio, C. Synchrotron-Based UV Resonance Raman Scattering for Investigating Ionic Liquid-Water Solutions. Condens. Matter Phys. 2019, 22, 43301. [Google Scholar] [CrossRef]
- Salama, S.; Spiro, T.G. Resonance Raman Spectra of Cobalt(II)-Imidazole Complexes: Analogues of the Binding Site of Cobalt-Substituted Zinc Proteins. J. Am. Chem. Soc. 1978, 100, 1105–1111. [Google Scholar] [CrossRef]
- Xue, G.; Dai, Q.; Jiang, S. Chemical Reactions of Imidazole with Metallic Silver Studied by the Use of SERS and XPS Techniques. J. Am. Chem. Soc. 1988, 110, 2393–2395. [Google Scholar] [CrossRef]
- Grzyb, B.; Gryglewicz, S.; Śliwak, A.; Díez, N.; Machnikowski, J.; Gryglewicz, G. Guanidine, Amitrole and Imidazole as Nitrogen Dopants for the Synthesis of N-Graphenes. RSC Adv. 2016, 6, 15782–15787. [Google Scholar] [CrossRef]
- Chuang, T.J.; Brundle, C.R.; Rice, D.W. Interpretation of the X-Ray Photoemission Spectra of Cobalt Oxides and Cobalt Oxide Surfaces. Surf. Sci. 1976, 59, 413–429. [Google Scholar] [CrossRef]
- Brown, D.G.; Weser, U. XPS Spectra of Spin-Triplet Cobalt(III) Complexes. Z. Für Naturforsch. B 1979, 34, 1468–1470. [Google Scholar] [CrossRef]
- Kumar, A.; Pandey, G. A Review on the Factors Affecting the Photocatalytic Degradation of Hazardous Materials. Mater. Sci. Eng. Int. J. 2017, 1, 106–114. [Google Scholar] [CrossRef]
- Angeles Navarro, M.; Sain, S.; Wünschek, M.; Pichler, C.M.; Romero-Salguero, F.J.; Esquivel, D.; Roy, S. Solar Driven CO2 Reduction with a Molecularly Engineered Periodic Mesoporous Organosilica Containing Cobalt Phthalocyanine. Nanoscale 2023. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | a0 a (nm) | SBET (m2 g−1) | Vp (cm3 g−1) | Dp b (nm) | w c (nm) |
|---|---|---|---|---|---|
| Im-EtPMO | 5.6 | 583 | 0.52 | 4.2 | 5.6 |
| Im-EtPMO-Co | 5.7 | 455 | 0.43 | 4.2 | 5.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Navarro, M.Á.; Martín, M.A.; Ruiz, J.R.; Jiménez-Sanchidrián, C.; Romero-Salguero, F.J.; Esquivel, D. Improved Photocatalytic H2 Evolution by Cobaloxime-Tethered Imidazole-Functionalized Periodic Mesoporous Organosilica. Hydrogen 2023, 4, 120-132. https://doi.org/10.3390/hydrogen4010008
Navarro MÁ, Martín MA, Ruiz JR, Jiménez-Sanchidrián C, Romero-Salguero FJ, Esquivel D. Improved Photocatalytic H2 Evolution by Cobaloxime-Tethered Imidazole-Functionalized Periodic Mesoporous Organosilica. Hydrogen. 2023; 4(1):120-132. https://doi.org/10.3390/hydrogen4010008
Chicago/Turabian StyleNavarro, M. Ángeles, Miguel A. Martín, José Rafael Ruiz, César Jiménez-Sanchidrián, Francisco J. Romero-Salguero, and Dolores Esquivel. 2023. "Improved Photocatalytic H2 Evolution by Cobaloxime-Tethered Imidazole-Functionalized Periodic Mesoporous Organosilica" Hydrogen 4, no. 1: 120-132. https://doi.org/10.3390/hydrogen4010008