
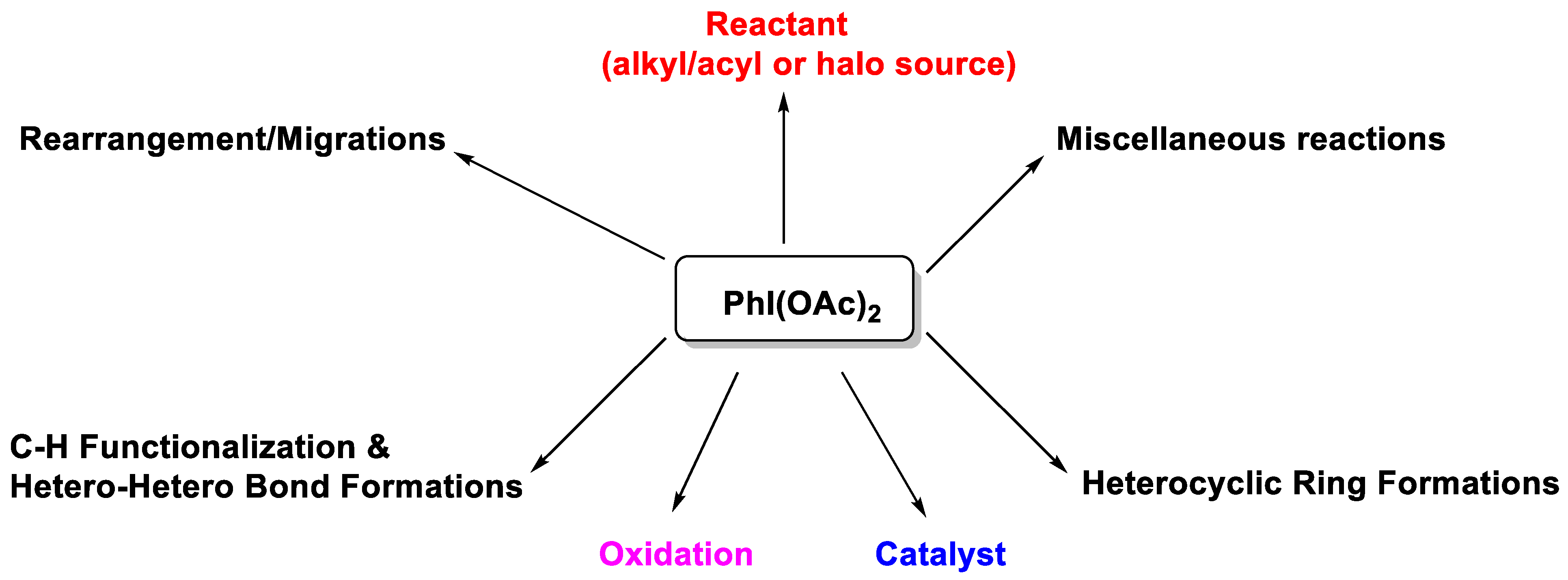
Phenyliodine(III)diacetate (PIDA): Applications in Organic Synthesis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
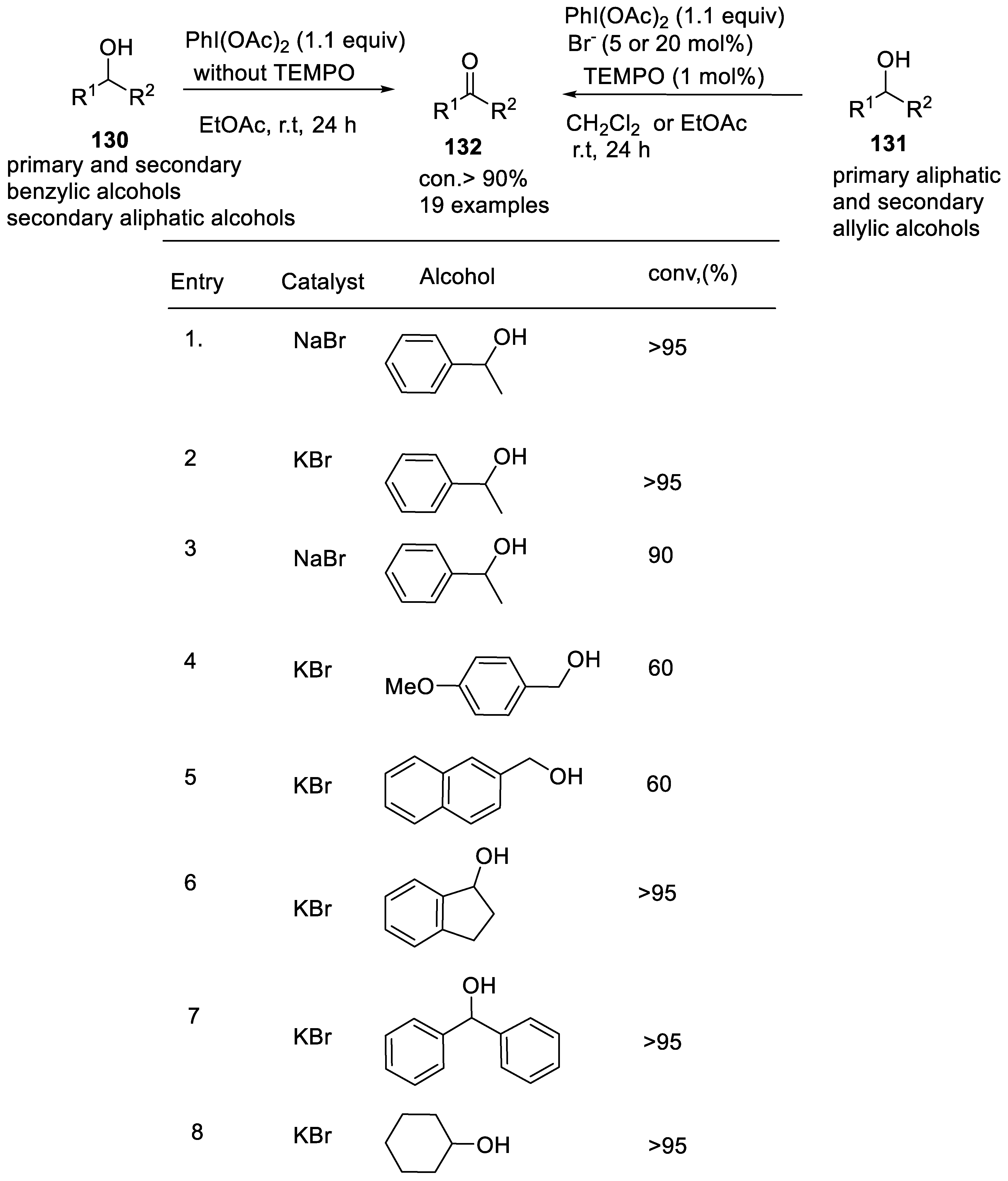{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Recent Applications of PIDA (from 2015)
2.1. C-H Functionalization and Hetero-Hetero Bond Formations
2.1.1. C-C Bond Formation
- (a)
- C-Alkylation reaction
- (b)
- C-C bond formation reaction on alkenes
2.1.2. C-O and C-X Bond Formation
2.1.3. C-N Bond Formation
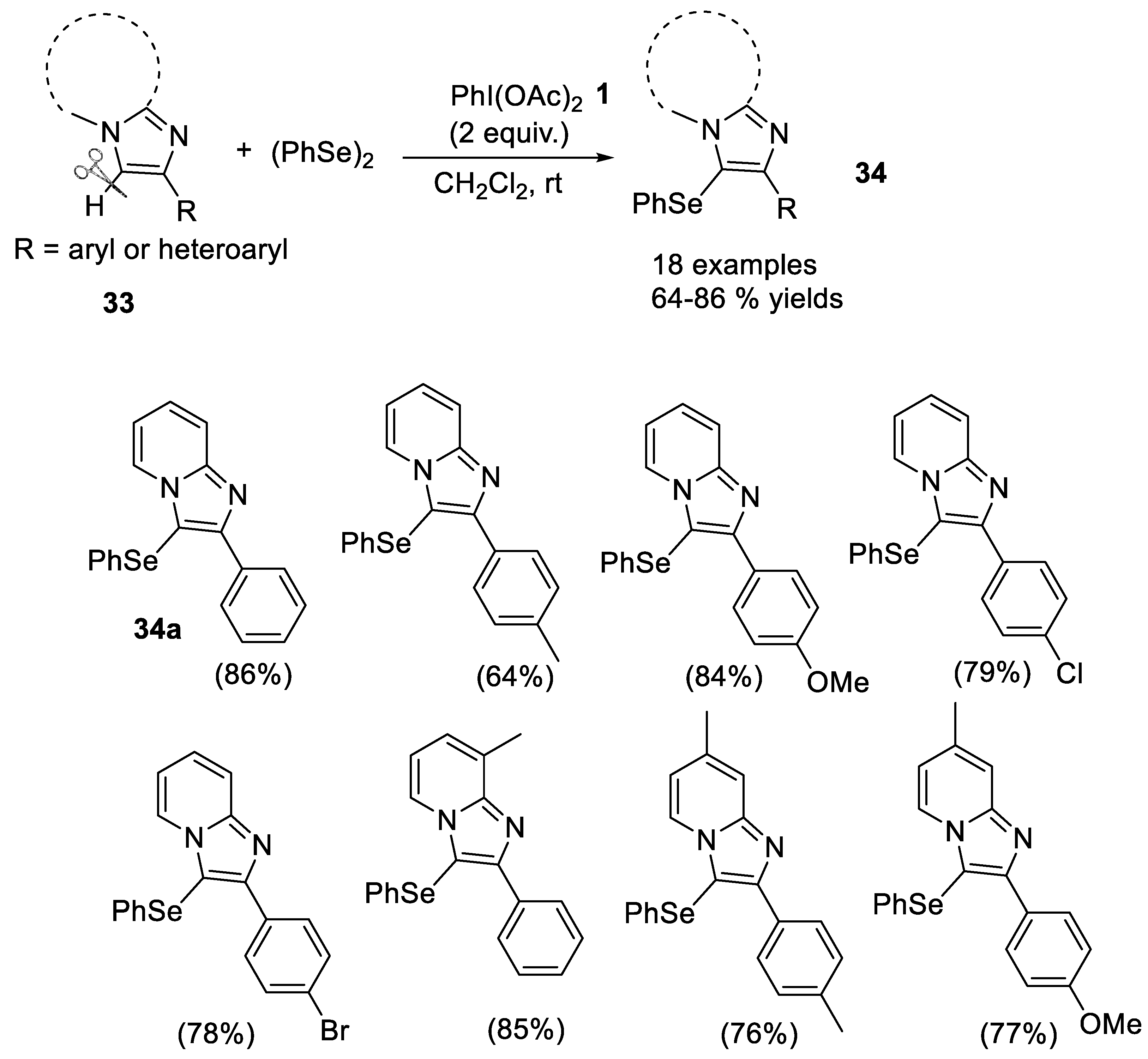
2.1.4. C-Se Bond Formation
2.1.5. Hetero-Hetero Bond Formation
2.2. Heterocyclic Ring Formations
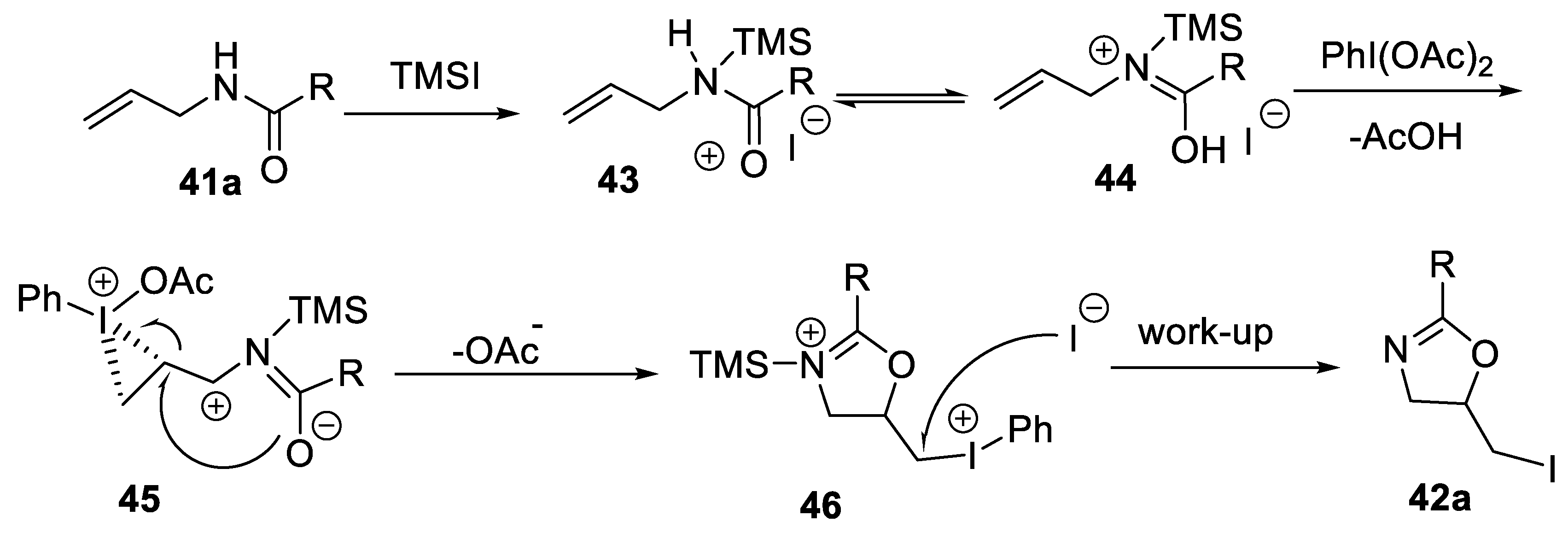
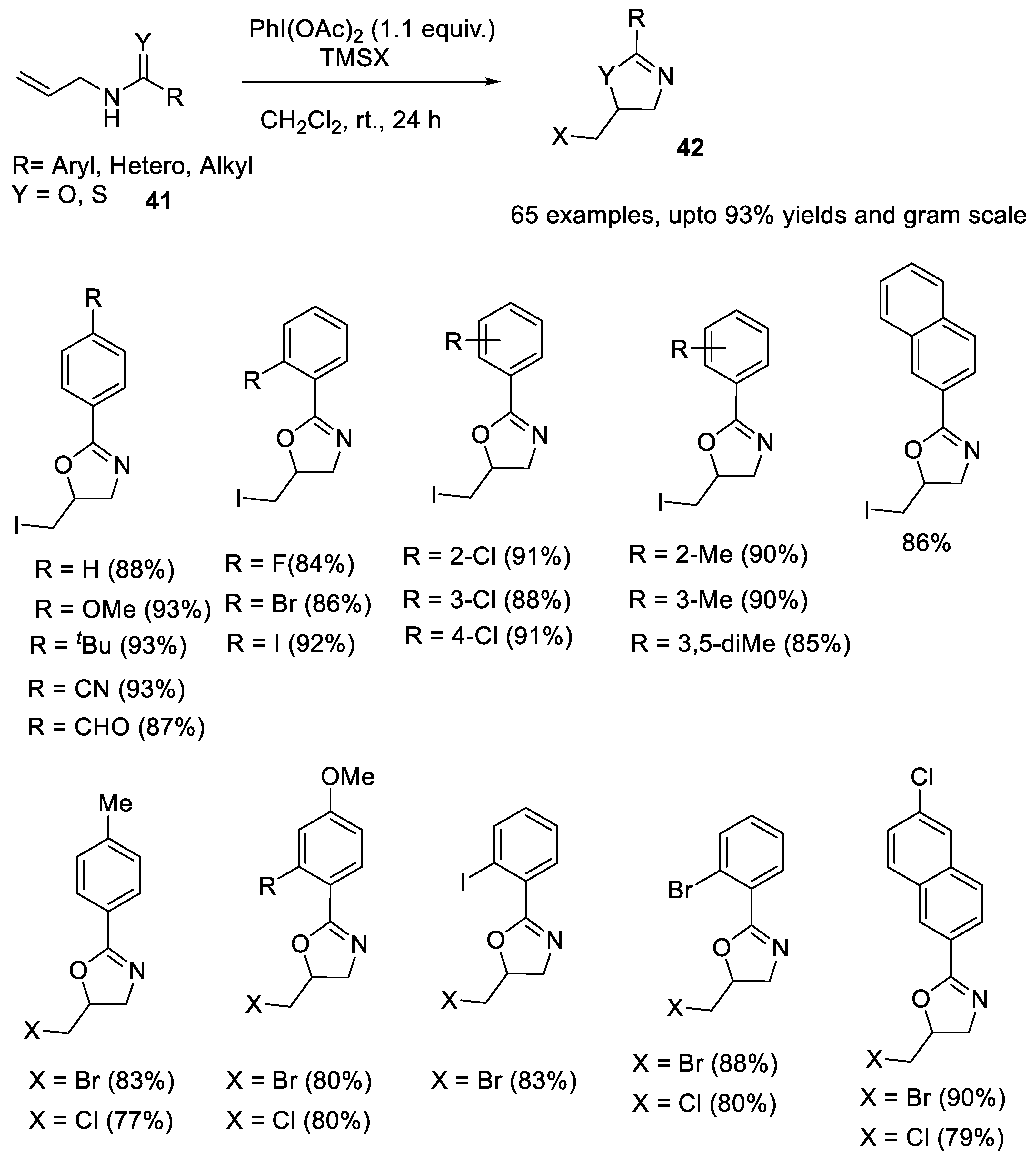
2.2.1. Synthesis of Oxazoline and Thiazoline Derivatives
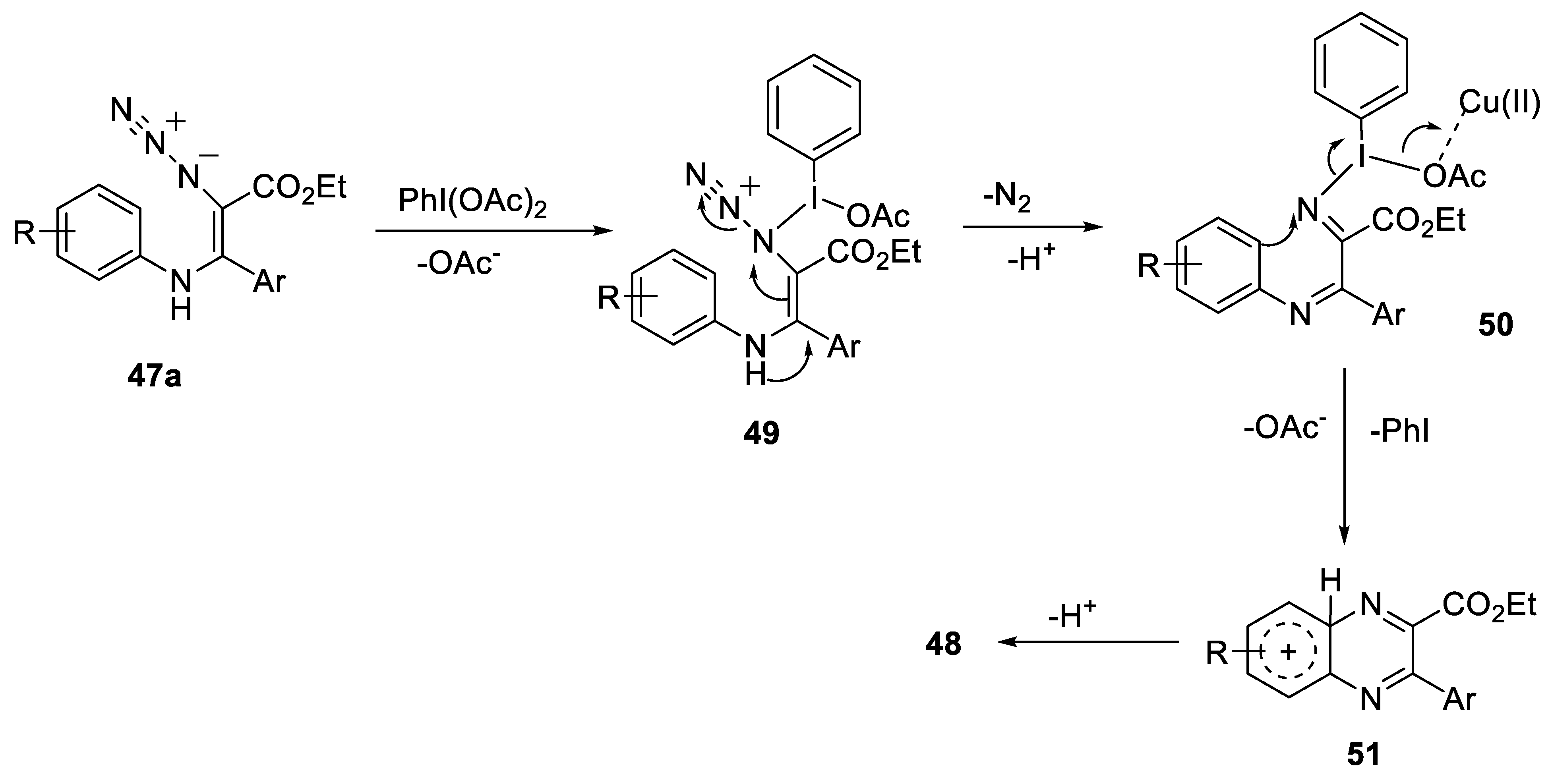
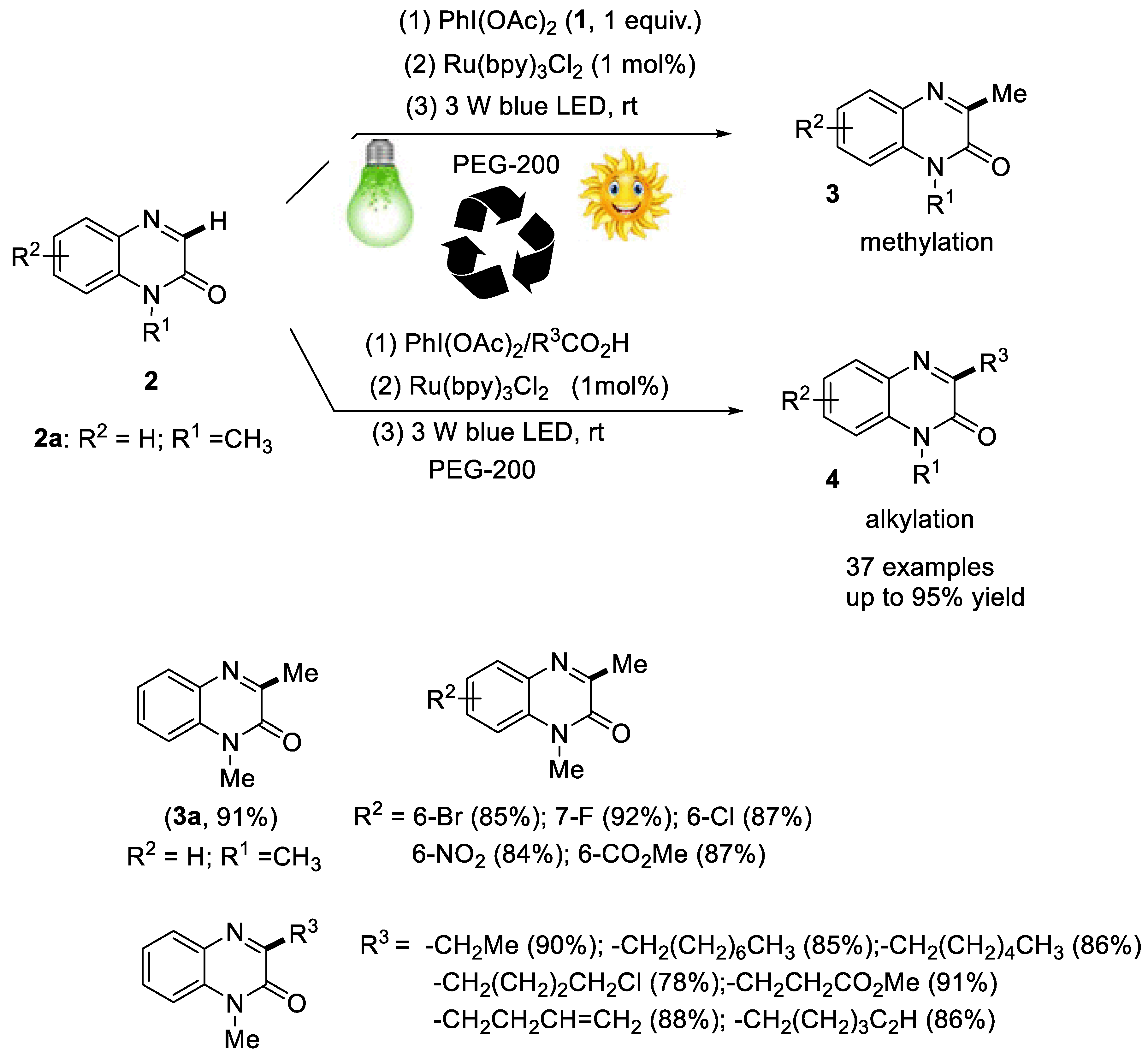
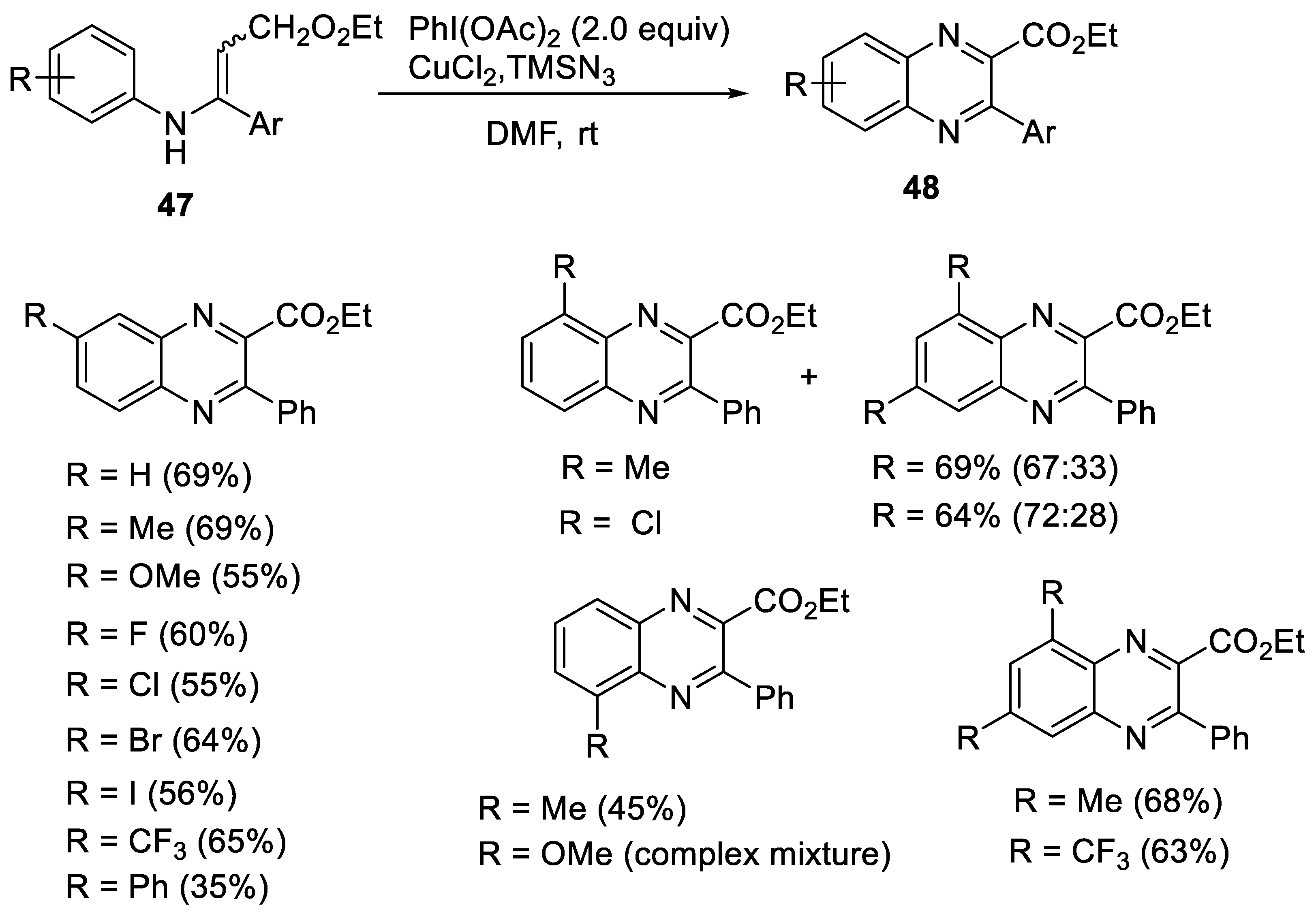
2.2.2. Synthesis of Quinoxaline Derivatives
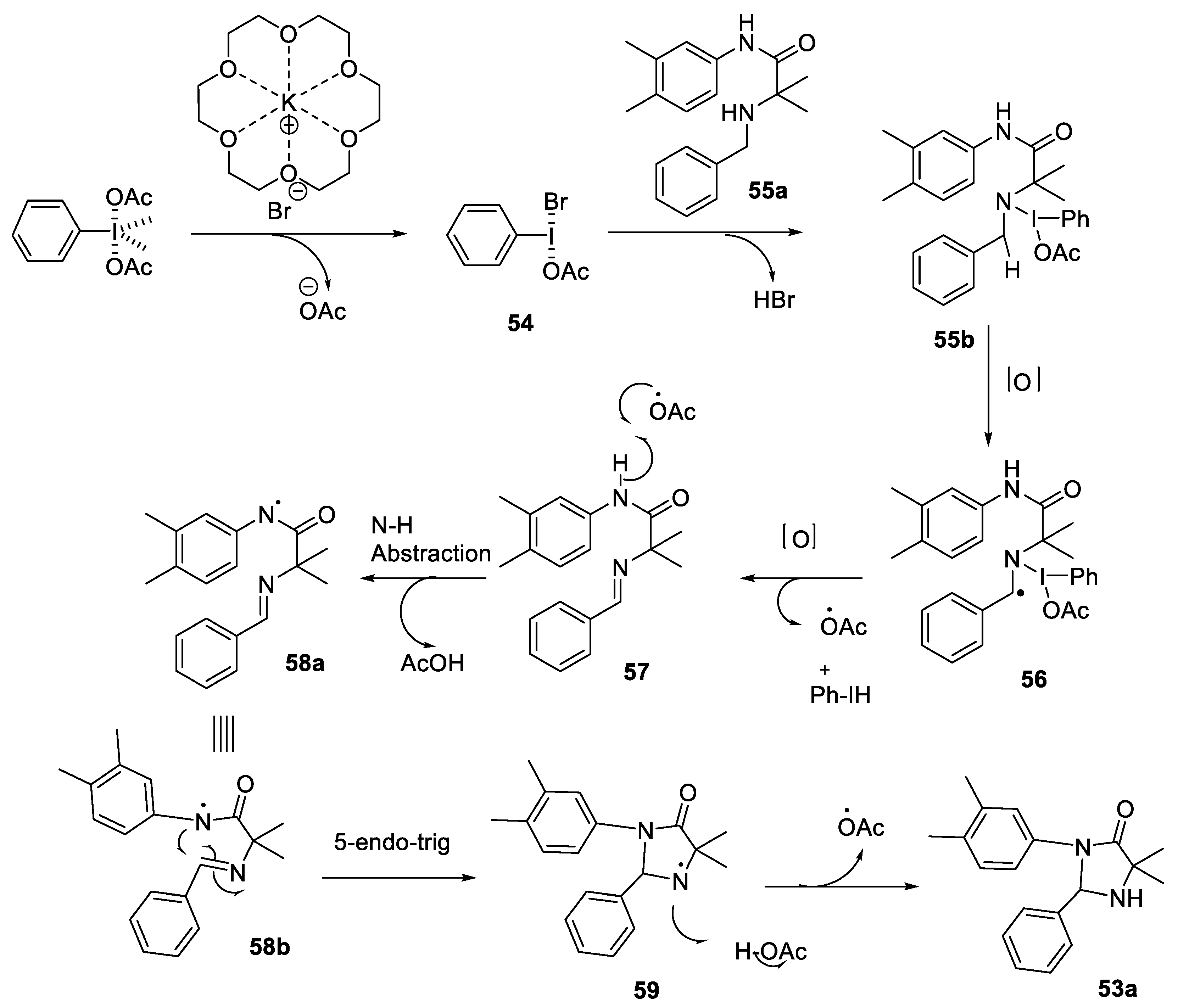
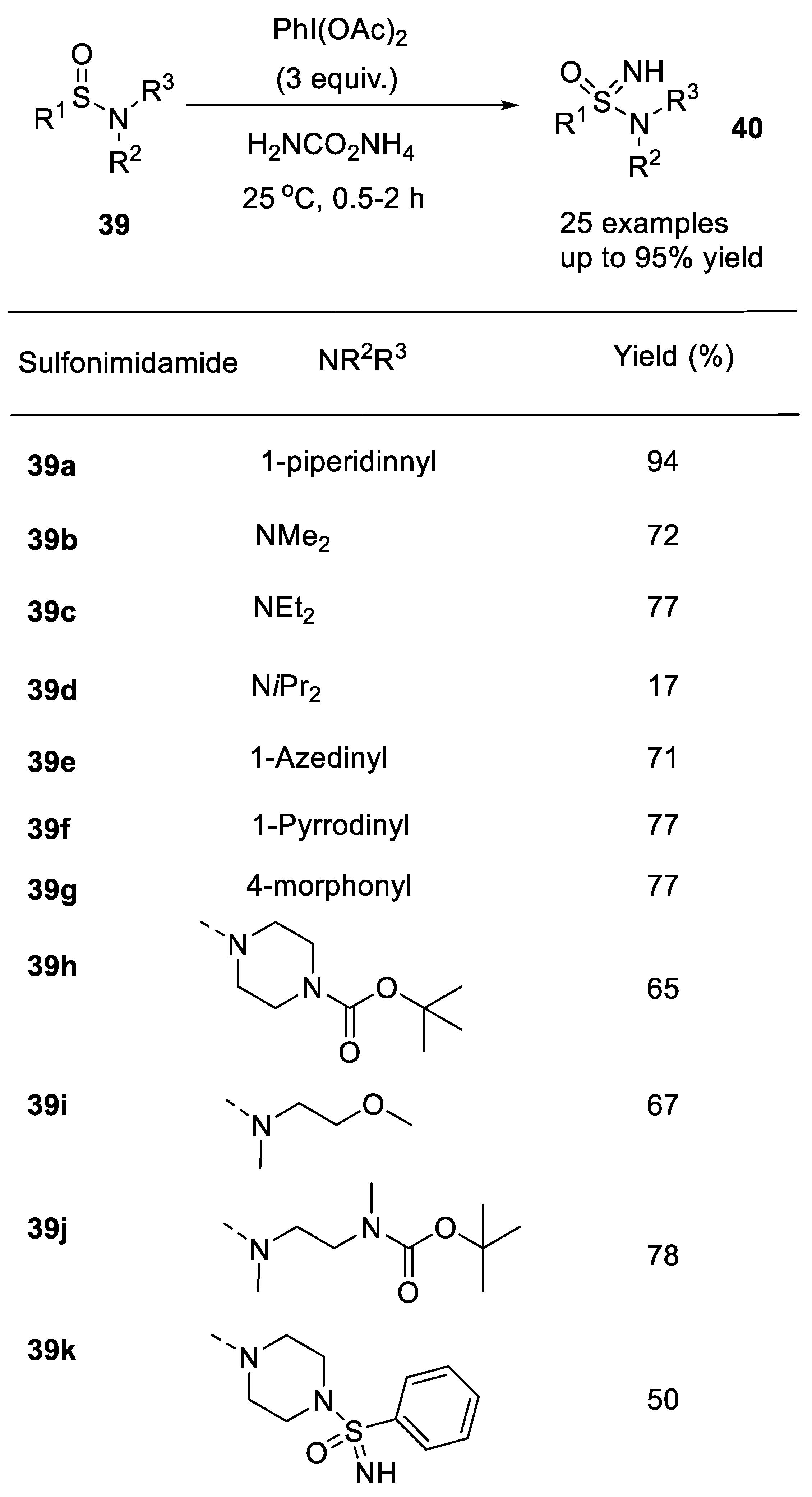
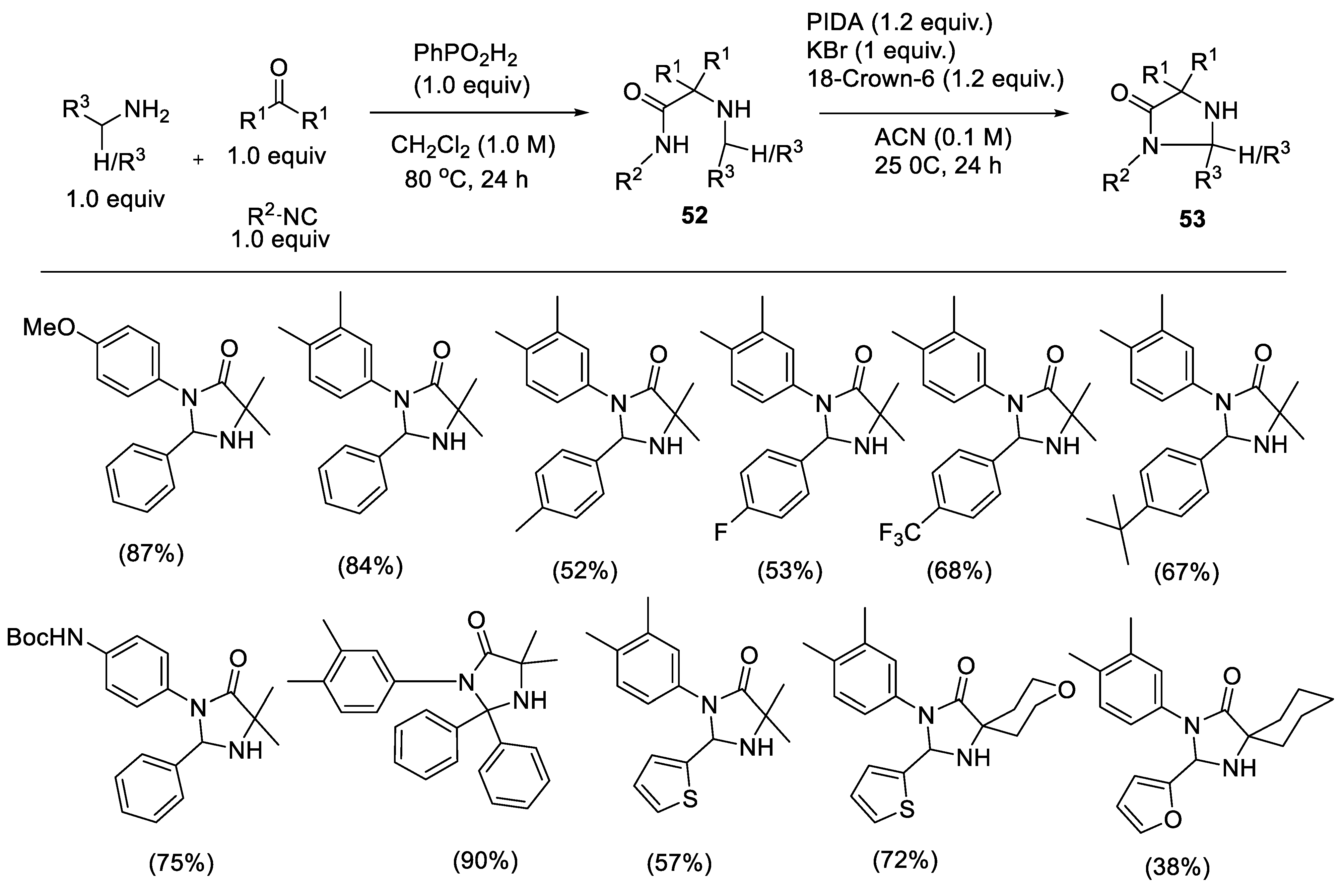
2.2.3. Synthesis of Imidazolidinones
2.3. Rearrangement/Migration Reactions
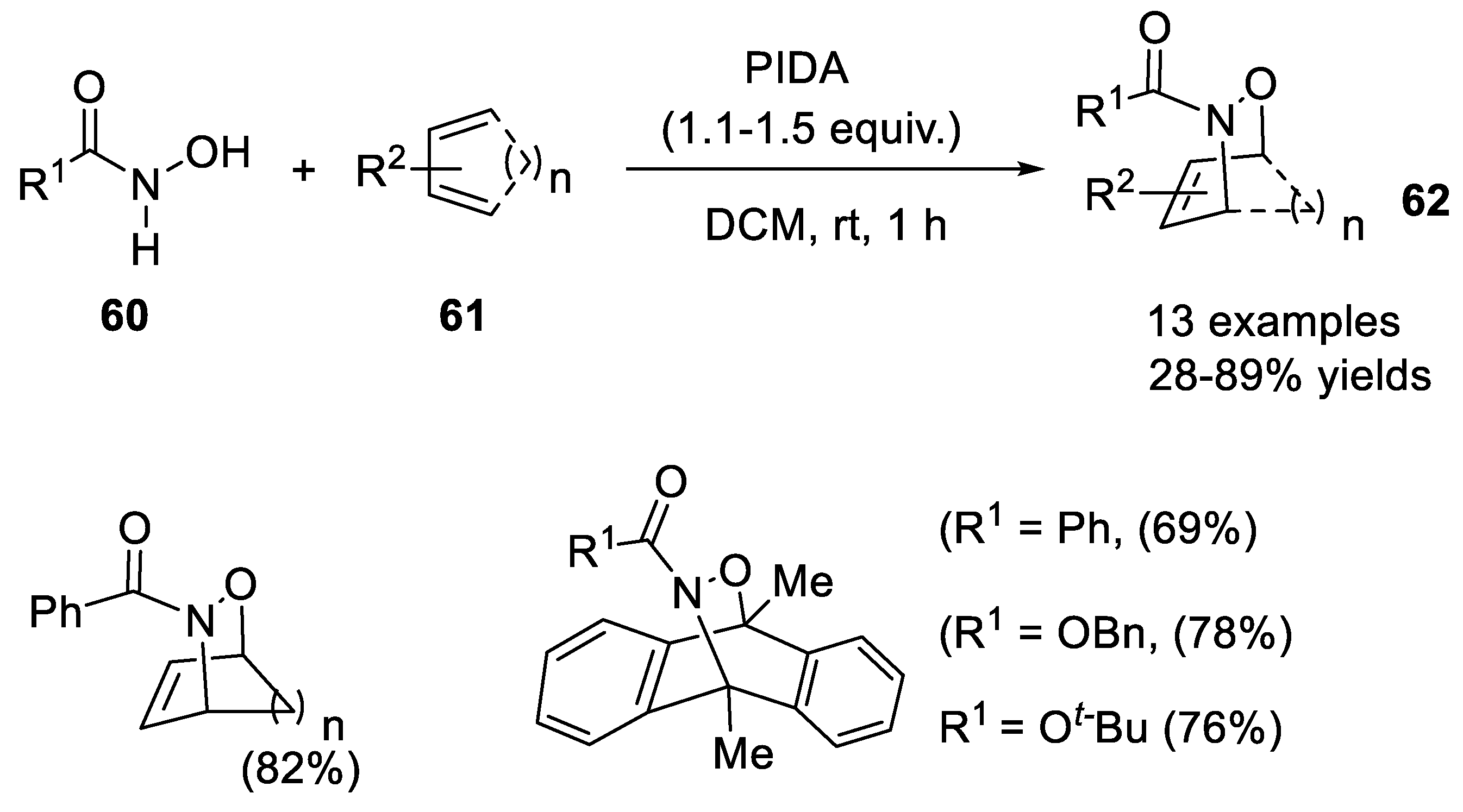
2.3.1. Hetero Diels-Alder Addition
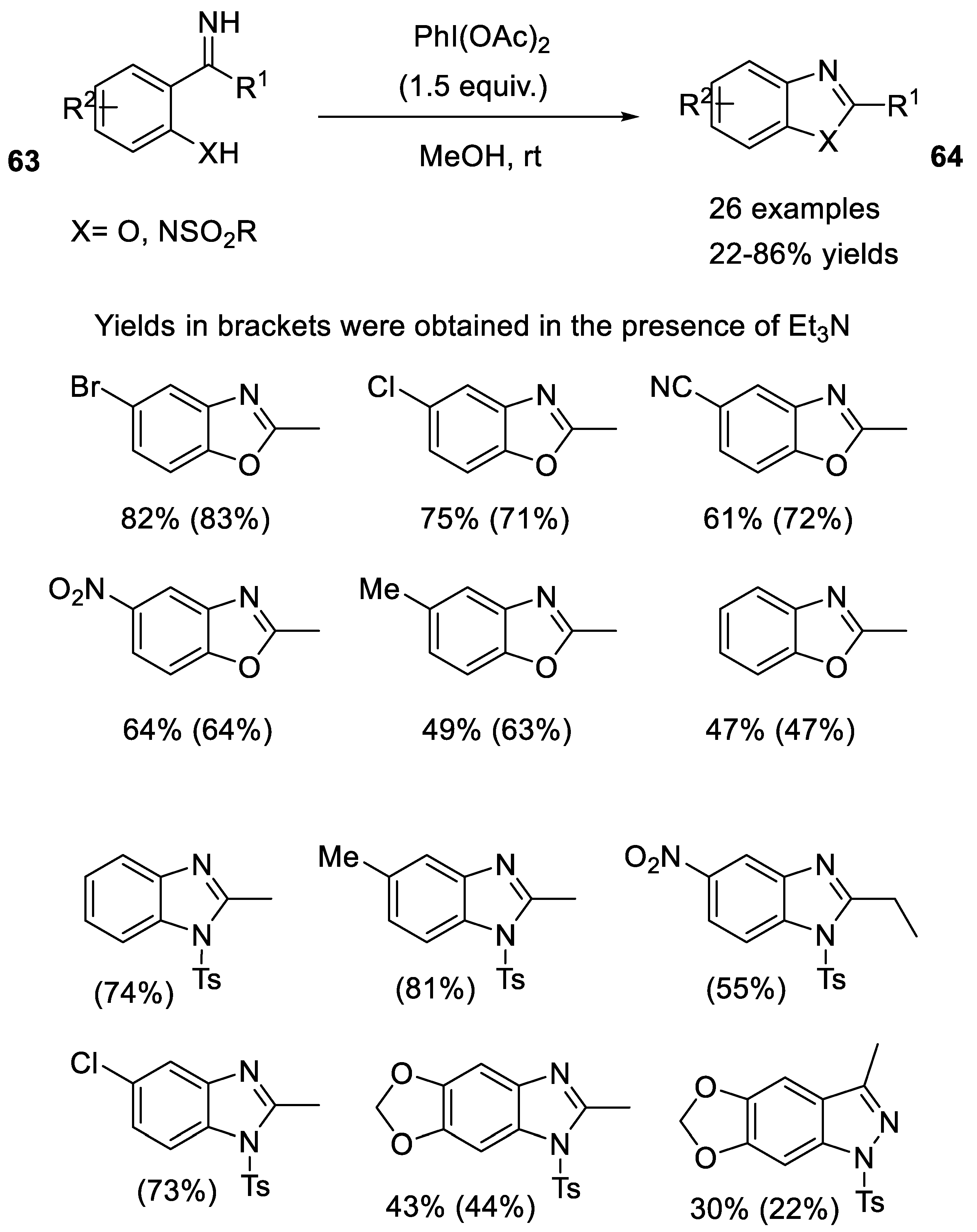
2.3.2. Beckmann-Type Rearrangement
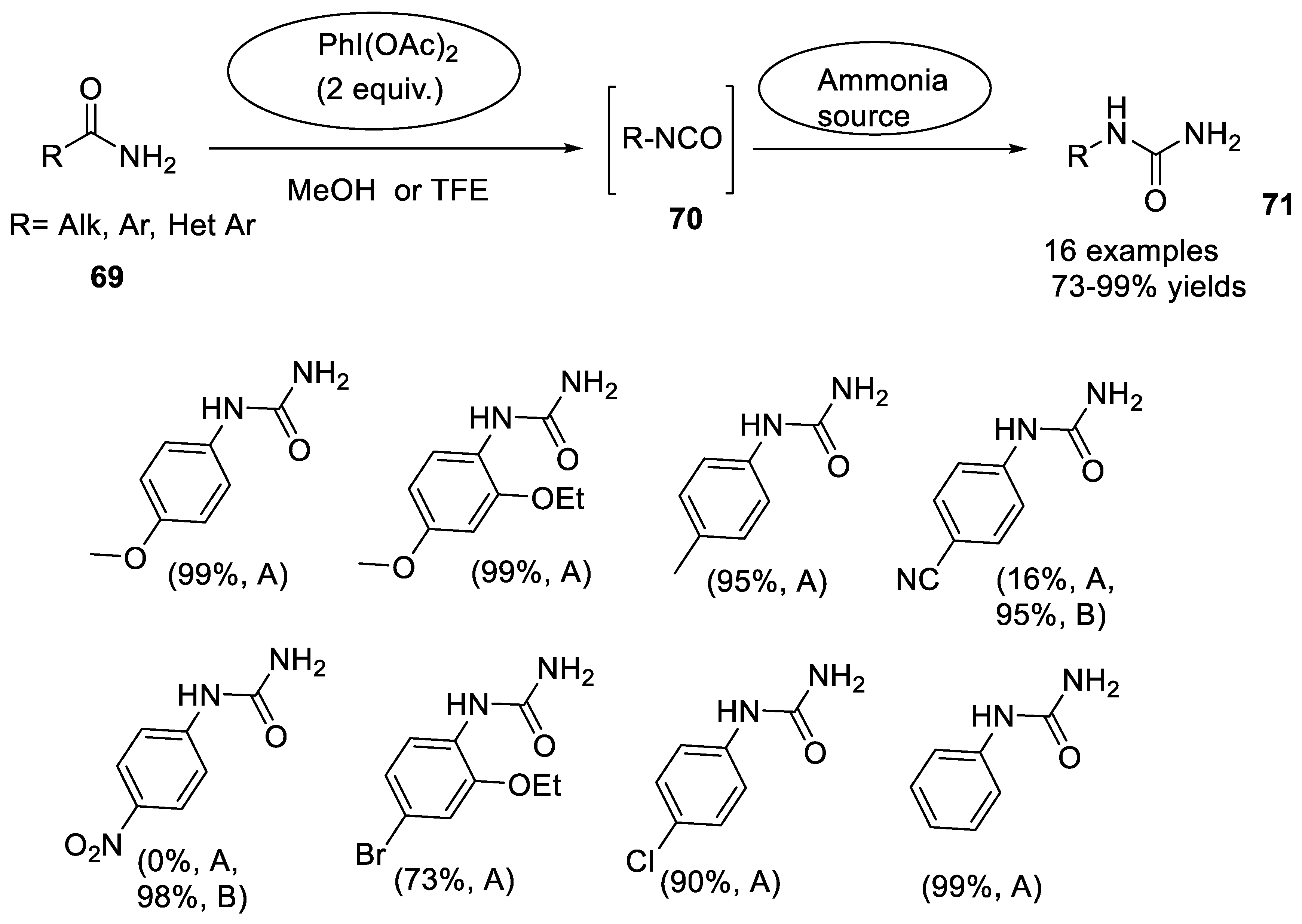
2.3.3. Hofmann Rearrangement
2.3.4. Morin Rearrangement
2.3.5. Grob-Type Fragmentation
2.3.6. 1,2-Diaza-Cope Rearrangement
2.4. Miscellaneous Reactions
2.4.1. Synthesis of Aryl(trifloxyalkenyl)iodonium Triflate Salts
2.4.2. Dearomative Spirocyclization
2.4.3. Generation of α-Aminoalkyl Radicals
2.4.4. Ring Opening Reaction
2.4.5. Oxidative Cleavage of C2–C3 Bond
2.4.6. Oxidation
2.4.7. Synthesis of Furylpyrazolino[60]fullerene Derivatives
2.4.8. Catalytic Oxidation of Styrene
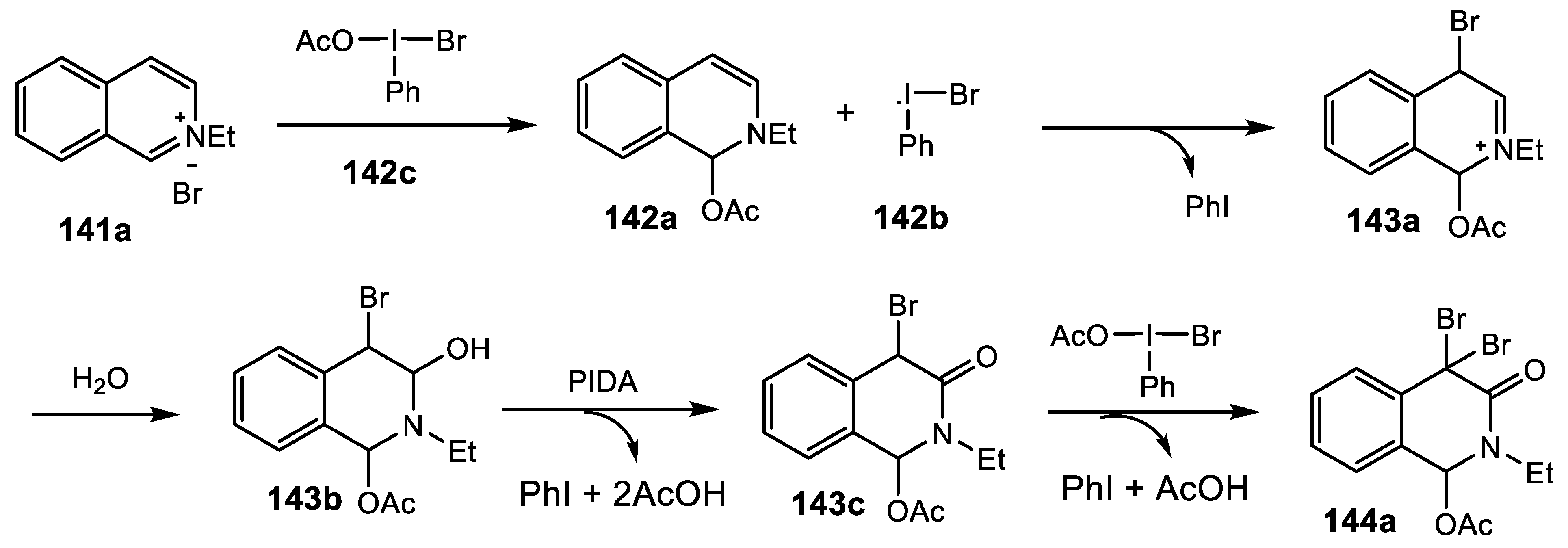
2.4.9. Synthesis of 1,4-Bridged Dihydroisoquinolin-3-Ones
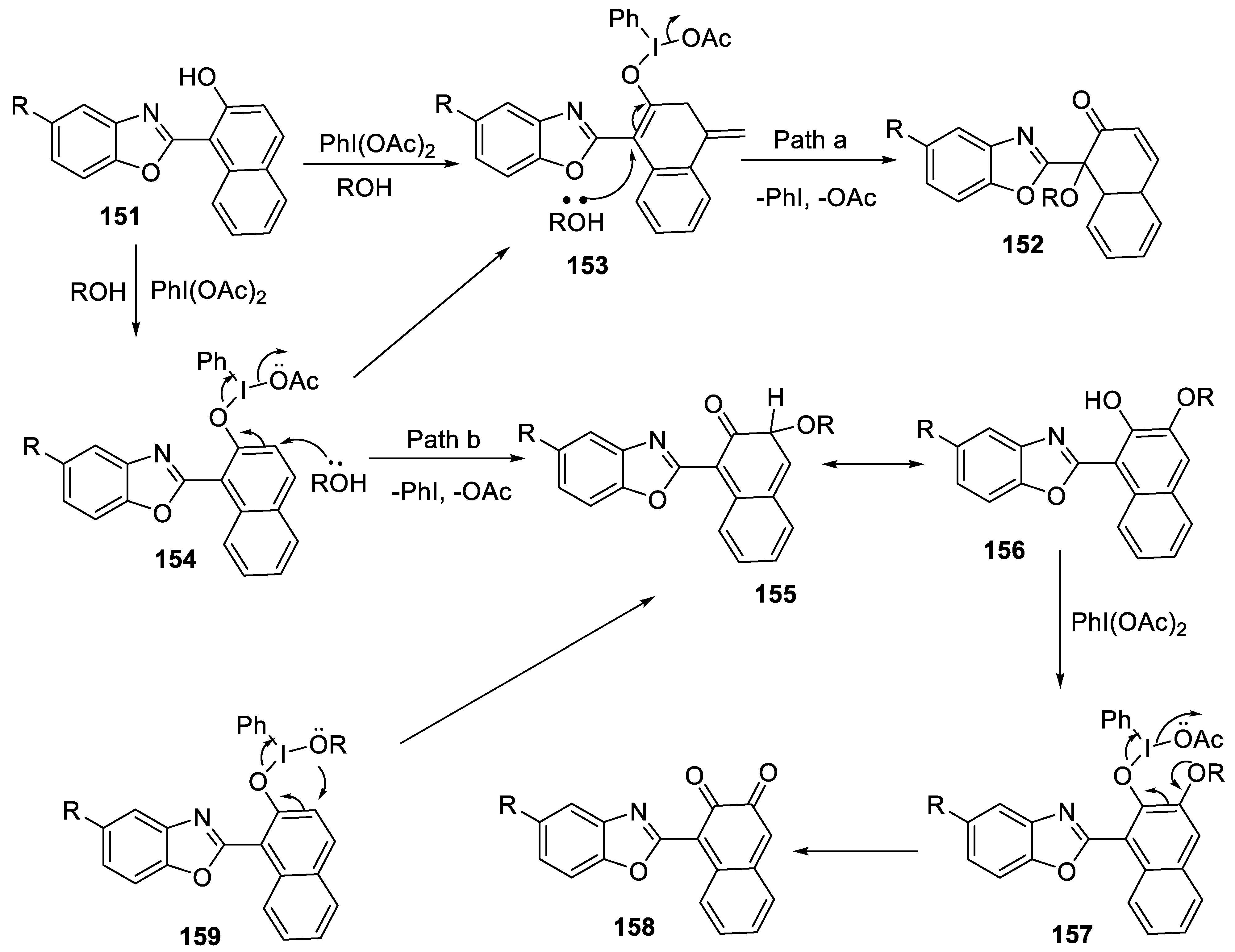
2.4.10. Regioselective Synthesis of 1-(Benzoxazol-2-yl)-1-Alkoxynaphthalen-2(1H)-Ones
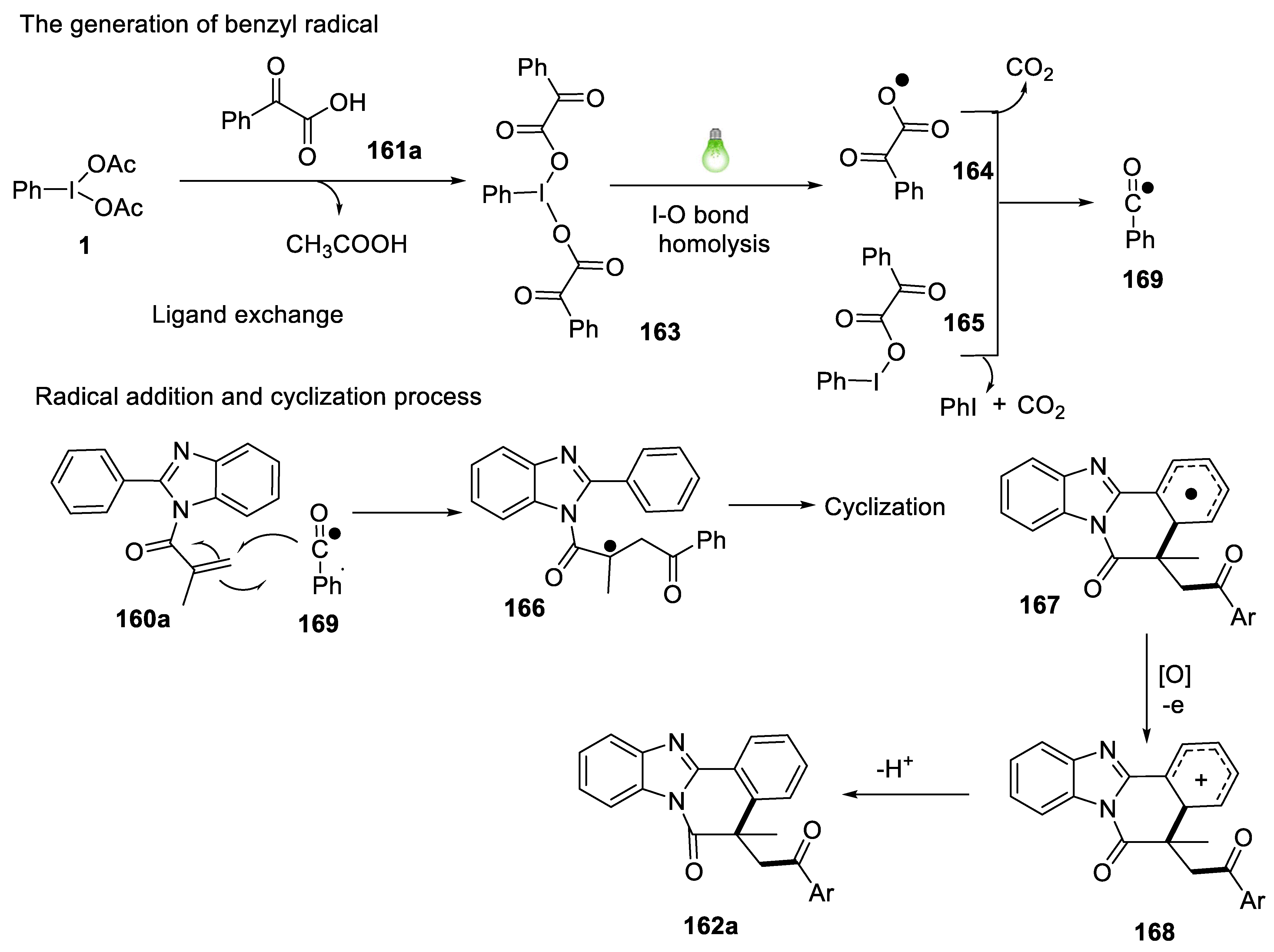
2.4.11. Synthesis of Acylated Benzimidazo/Indolo[2,1-a]isoquinolines
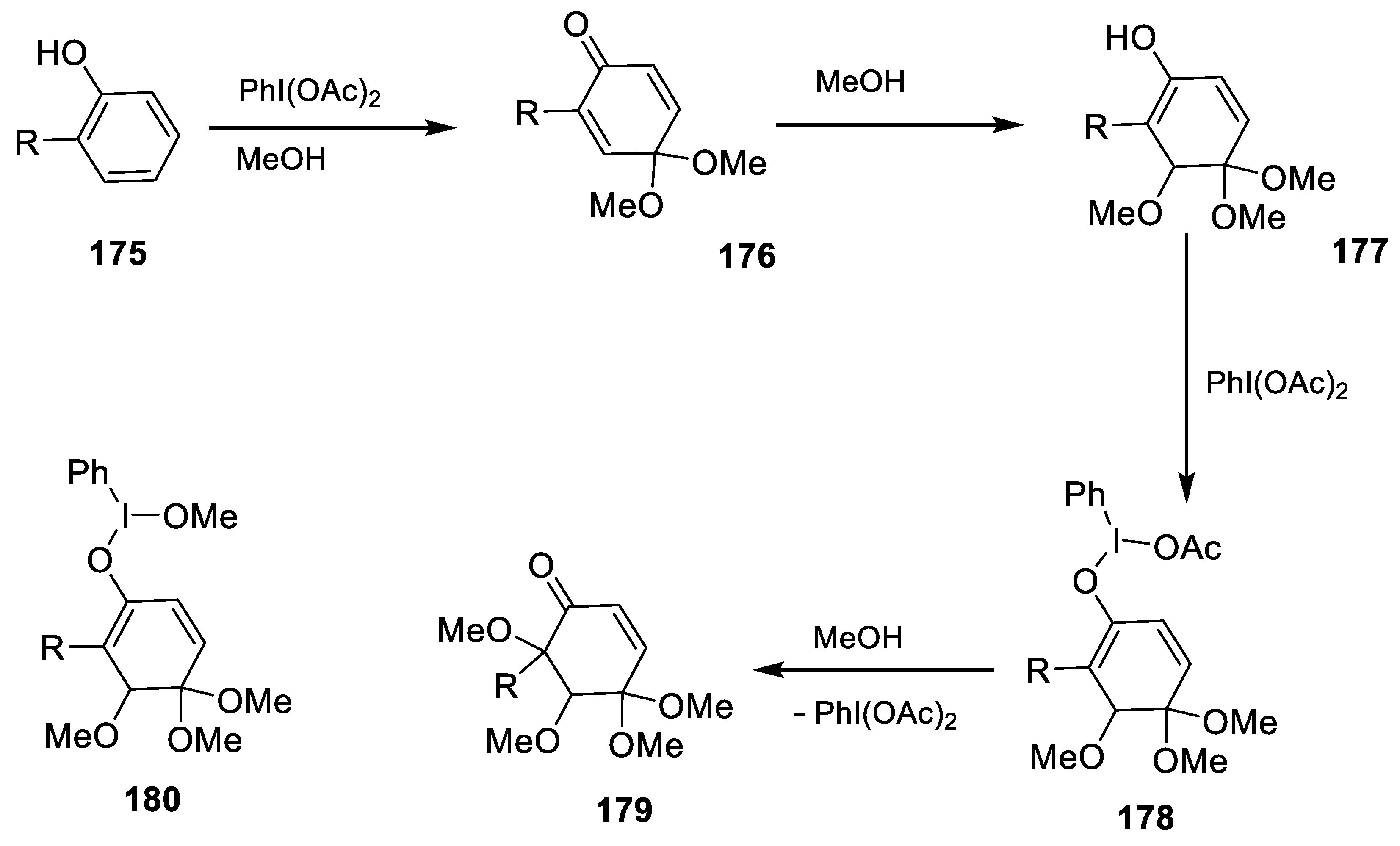
2.4.12. Oxidative Dearomatization
3. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhdankin, V.V. Application of hypervalent iodine compounds in advanced green technologies. Resour.-Effic. Technol. 2021, 1, 1–16. [Google Scholar] [CrossRef]
- Yoshimura, A.; Zhdankin, V.V. Advances in synthetic applications of hypervalent iodine compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef] [PubMed]
- Shetgaonkar, S.E.; Krishnan, M.; Singh, F.V. Hypervalent iodine reagents for oxidative rearrangements. Mini Rev. Org. Chem. 2021, 18, 138–158. [Google Scholar] [CrossRef]
- Zhang, B.; Li, X.; Guob, B.; Du, Y. Hypervalent iodine reagent-mediated reactions involving rearrangement processes. Chem. Commun. 2020, 56, 14119–14136. [Google Scholar] [CrossRef]
- Soni, R.; Sihag, M.; Rani, N.; Kinger, M.; Aneja, D.K. Aqueous mediated reactions involving hypervalent iodine reagents. Asian J. Org. Chem. 2022, 11, e202200125. [Google Scholar] [CrossRef]
- Rani, N.; Soni, R.; Sihag, M.; Kinger, M.; Aneja, D.K. Combined approach of hypervalent iodine reagents and transition metals in organic reactions. Adv. Synth. Catal. 2022, 364, 1798–1848. [Google Scholar] [CrossRef]
- Kiprof, P. The nature of iodine oxygen bonds in hypervalent 10-i-3 iodine compounds. Arkivoc 2005, iv, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Wang, T.-L.; Zhang, B.-S.; Liu, J.-J.; Liu, X.-J.; Wang, X.-C.; Quan, Z.-J. Visible light promoted polyhalomethylation of alkenes: Alkylation and cyclization. Org. Chem. Front. 2022, 9, 1004–1009. [Google Scholar] [CrossRef]
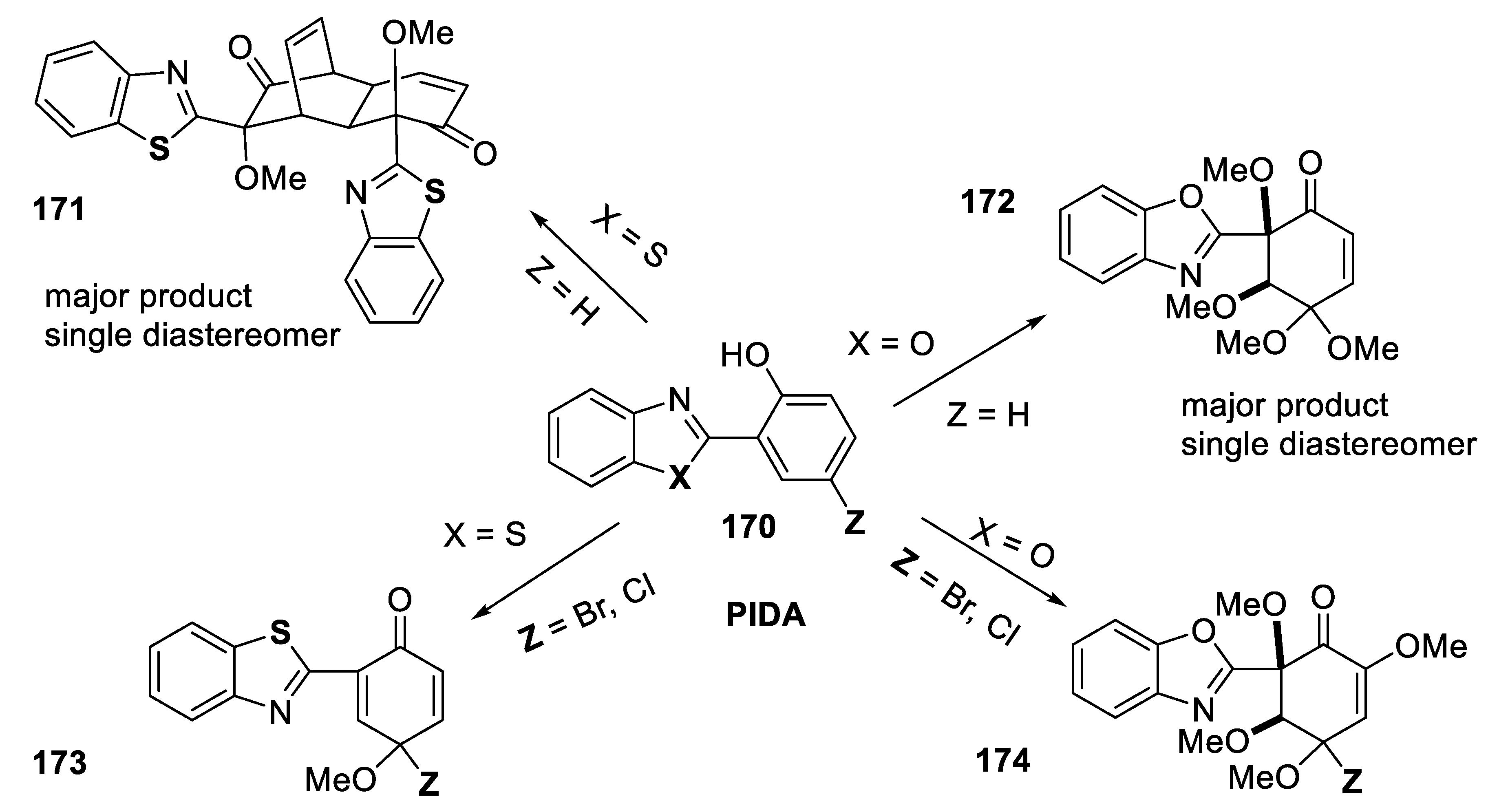
- Yadav, G.; Kumar, S.; Kumar, D.; Kataria, R.; Laroche, C.; Kerwin, S.M. Phenyliodine(III) diacetate-mediated dearomatization of 2-(2-hydroxyaryl)benzoxazoles and 2-(2-hydroxyaryl)benzothiazoles: Regio- and stereoselective synthesis of tetramethoxycyclohexenones and bicyclo[2.2.2]octenones. J. Mol. Struct. 2022, 1266, 133520. [Google Scholar] [CrossRef]
- Freitas, R.H.C.N. (Diacetoxyiodo)benzene: More than an oxidant. Aus. J. Chem. 2017, 70, 338–340. [Google Scholar] [CrossRef]
- Willgerodt, C.J. Ueber einige aromatische Jodidchloride. J. Prakt. Chem. 1886, 33, 154–160. [Google Scholar] [CrossRef] [Green Version]
- Iinuma, M.; Moriyama, K.; Togo, H. Simple and practical method for preparation of [(diacetoxy)iodo]arenes with iodoarenes and m-chloroperoxybenzoic acid. Synlett 2012, 23, 2663–2666. [Google Scholar] [CrossRef]
- Hossain, D.; Kitamura, T. Alternative, easy preparation of (diacetoxyiodo)arenes from iodoarenes using potassium peroxodisulfate as the oxidant. Synthesis 2005, 2005, 1932–1934. [Google Scholar] [CrossRef]
- Togo, H.; Nabana, T.; Yamaguchi, K. Preparation and reactivities of novel (diacetoxyiodo)arenes bearing heteroaromatics. J. Org. Chem. 2000, 65, 8391–8394. [Google Scholar] [CrossRef]
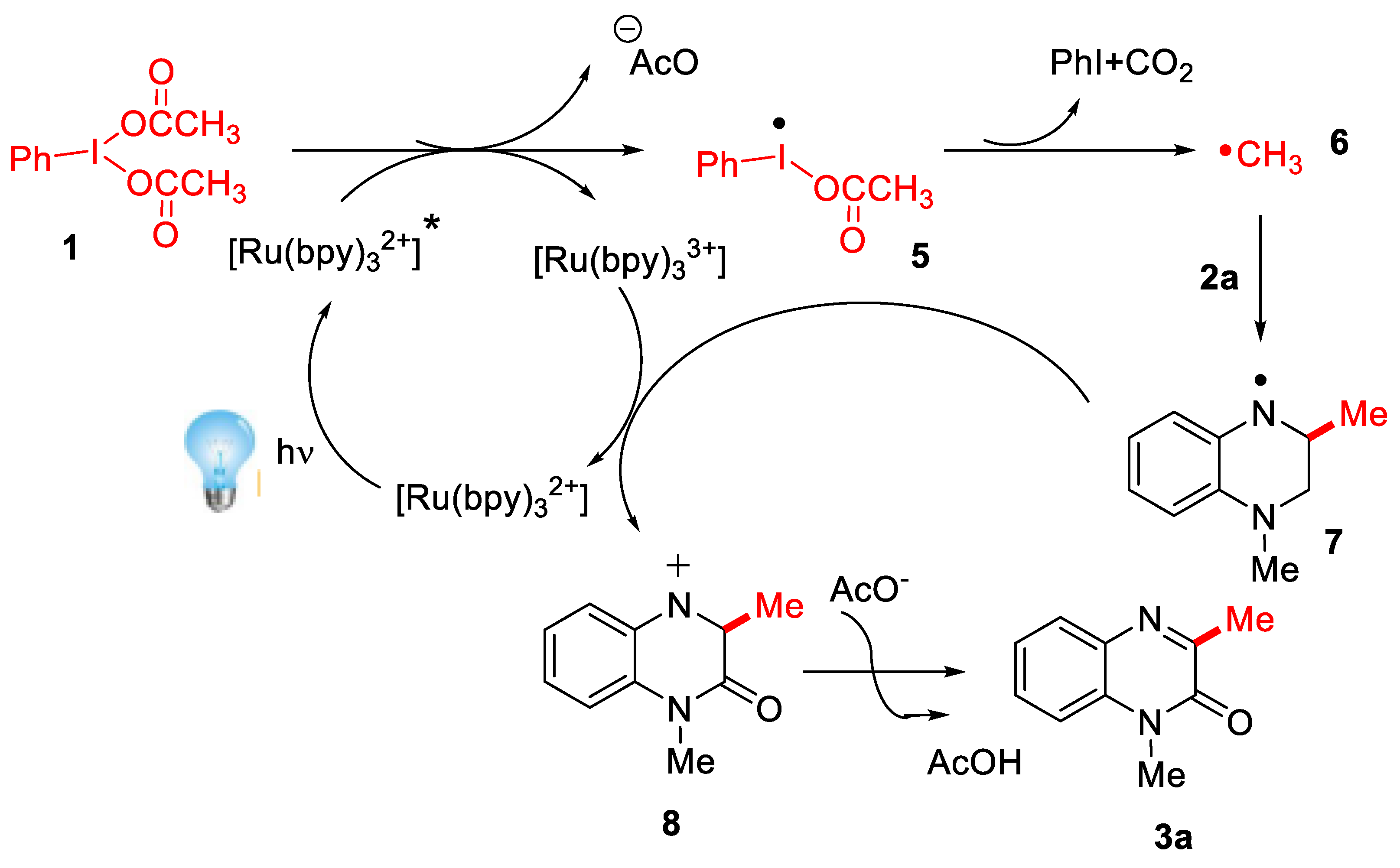
- Xie, L.-Y.; Jiang, L.-L.; Tan, J.-X.; Wang, Y.; Xu, X.-Q.; Zhang, B.; Cao, Z.; He, W.-M. Visible-light-initiated decarboxylative alkylation of quinoxalin-2(1H)-ones with phenyliodine(III) dicarboxylates in recyclable ruthenium(II) catalytic system. ACS Sustain. Chem. Eng. 2019, 7, 14153–14160. [Google Scholar] [CrossRef]
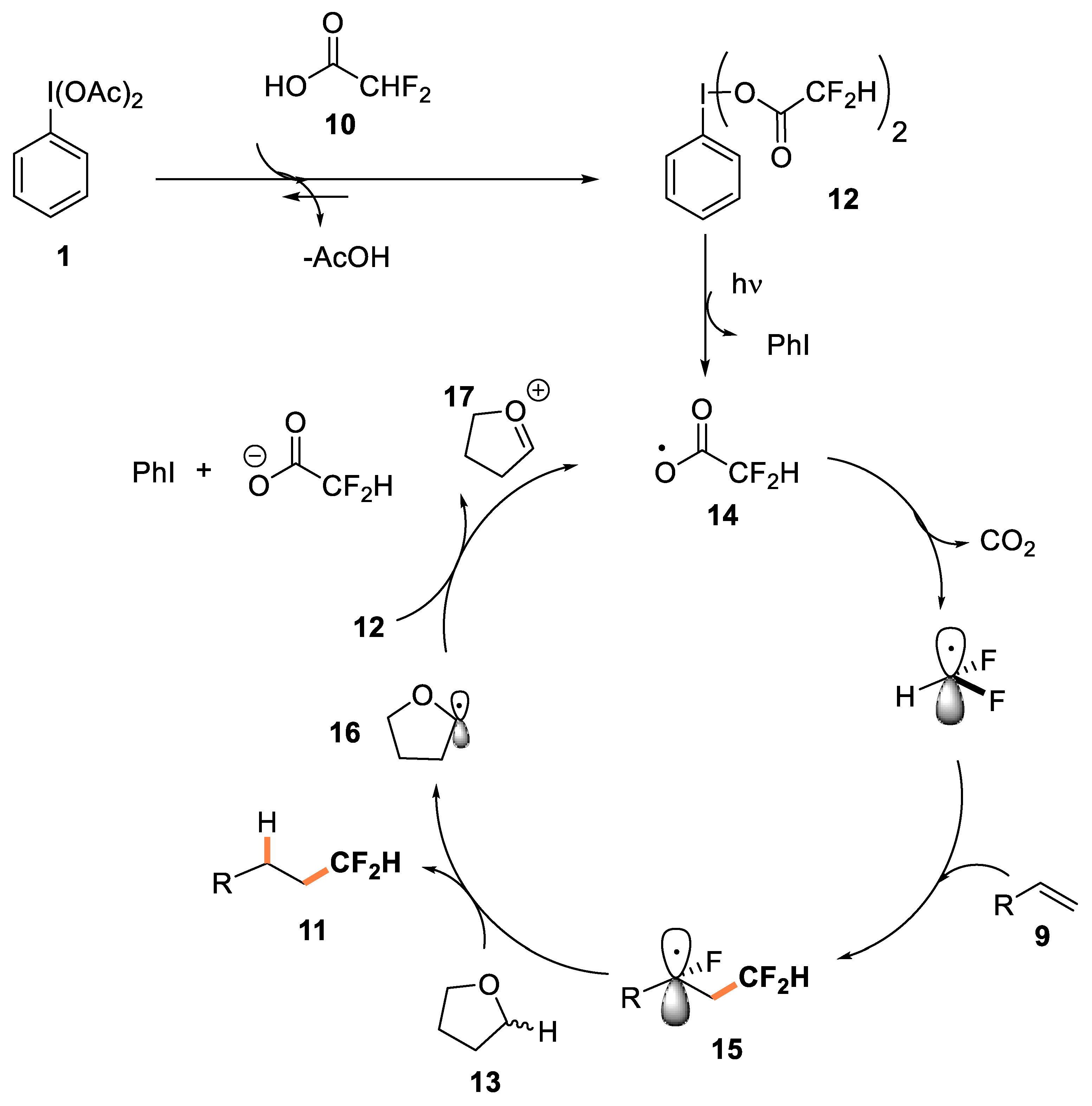
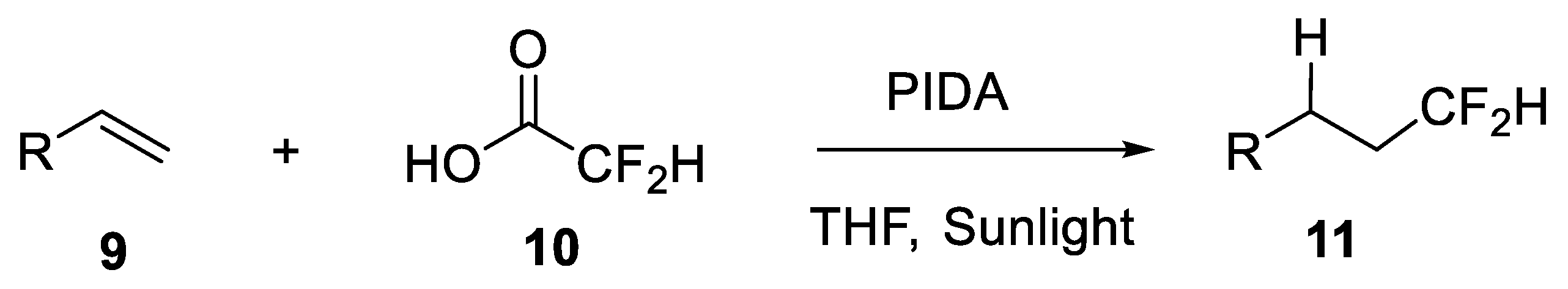
- Meyer, C.F.; Hell, S.M.; Misale, A.; Trabanco, A.; Gouverneur, V. Hydrodifluoromethylation of alkenes with difluoroacetic acid. Angew. Chem. Int. Ed. 2019, 58, 8829–8833. [Google Scholar] [CrossRef]
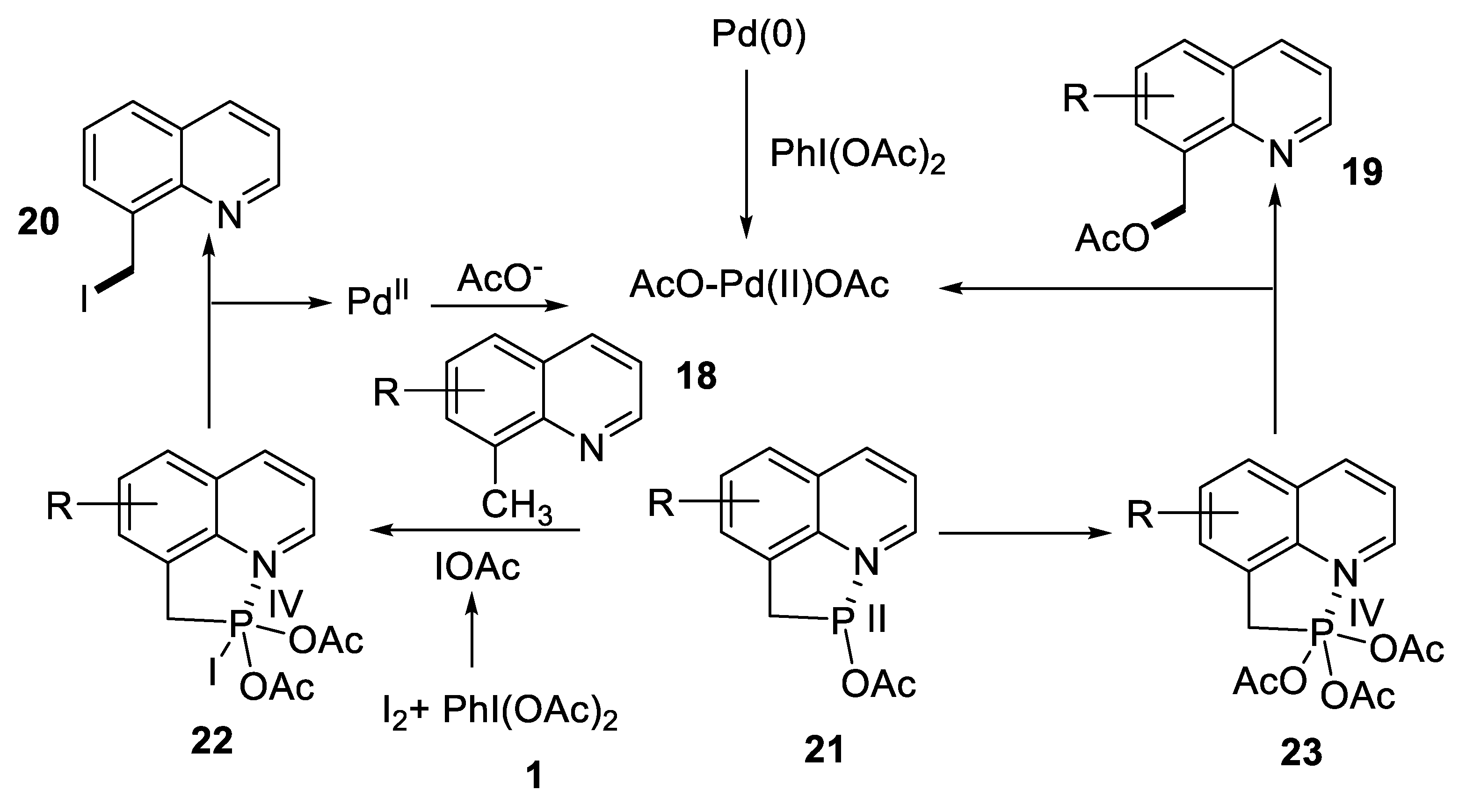
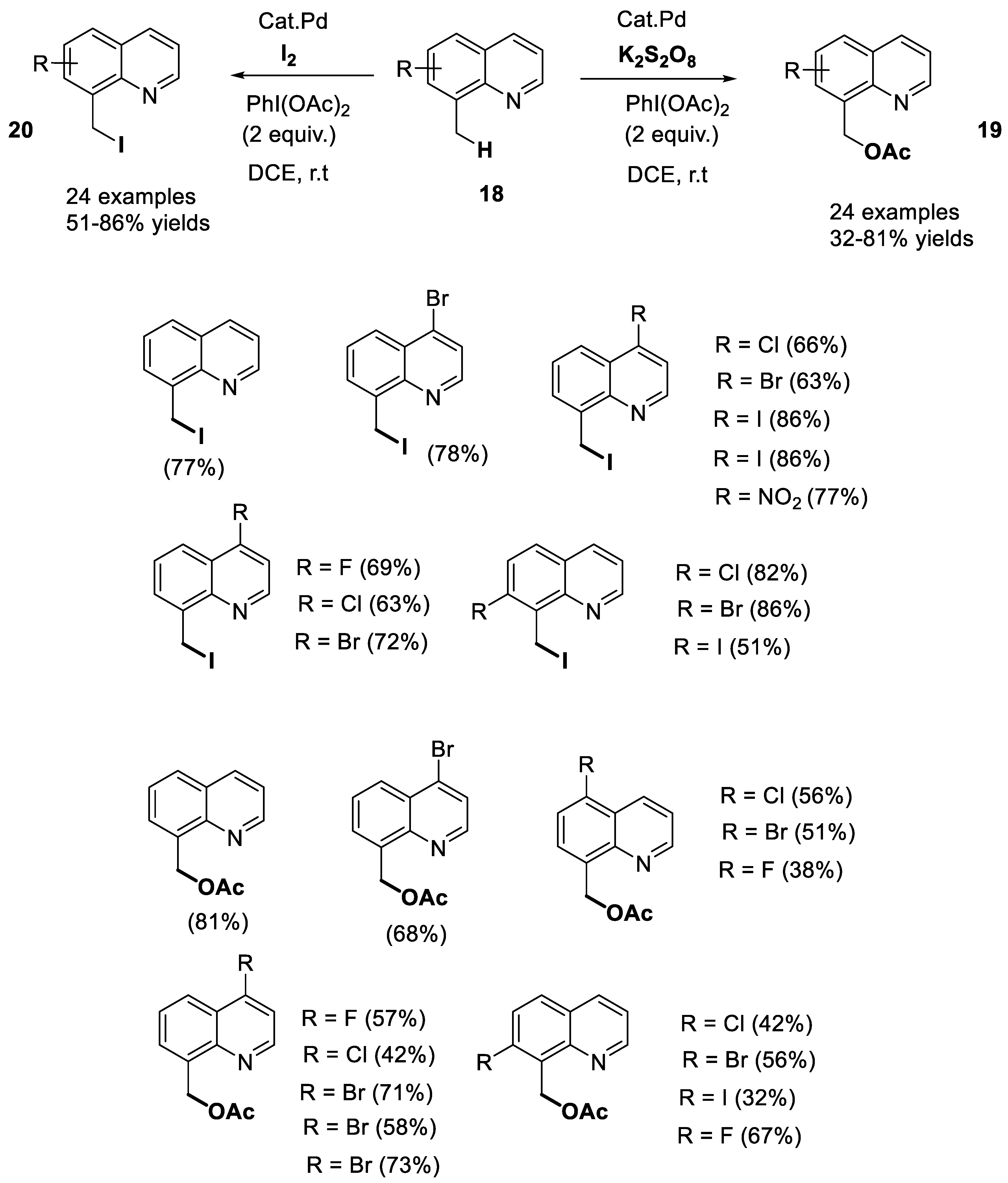
- Zhang, M.-L.; Zhang, X.-L.; Guo, R.-L.; Wang, M.-Y.; Zhao, B.-Y.; Yang, J.-H.; Jia, Q.; Wang, Y.-Q. Switchable, reagent-controlled C(sp3)-H selective iodination and acetoxylation of 8-methylquinolines. J. Org. Chem. 2022, 87, 5730–5743. [Google Scholar] [CrossRef]
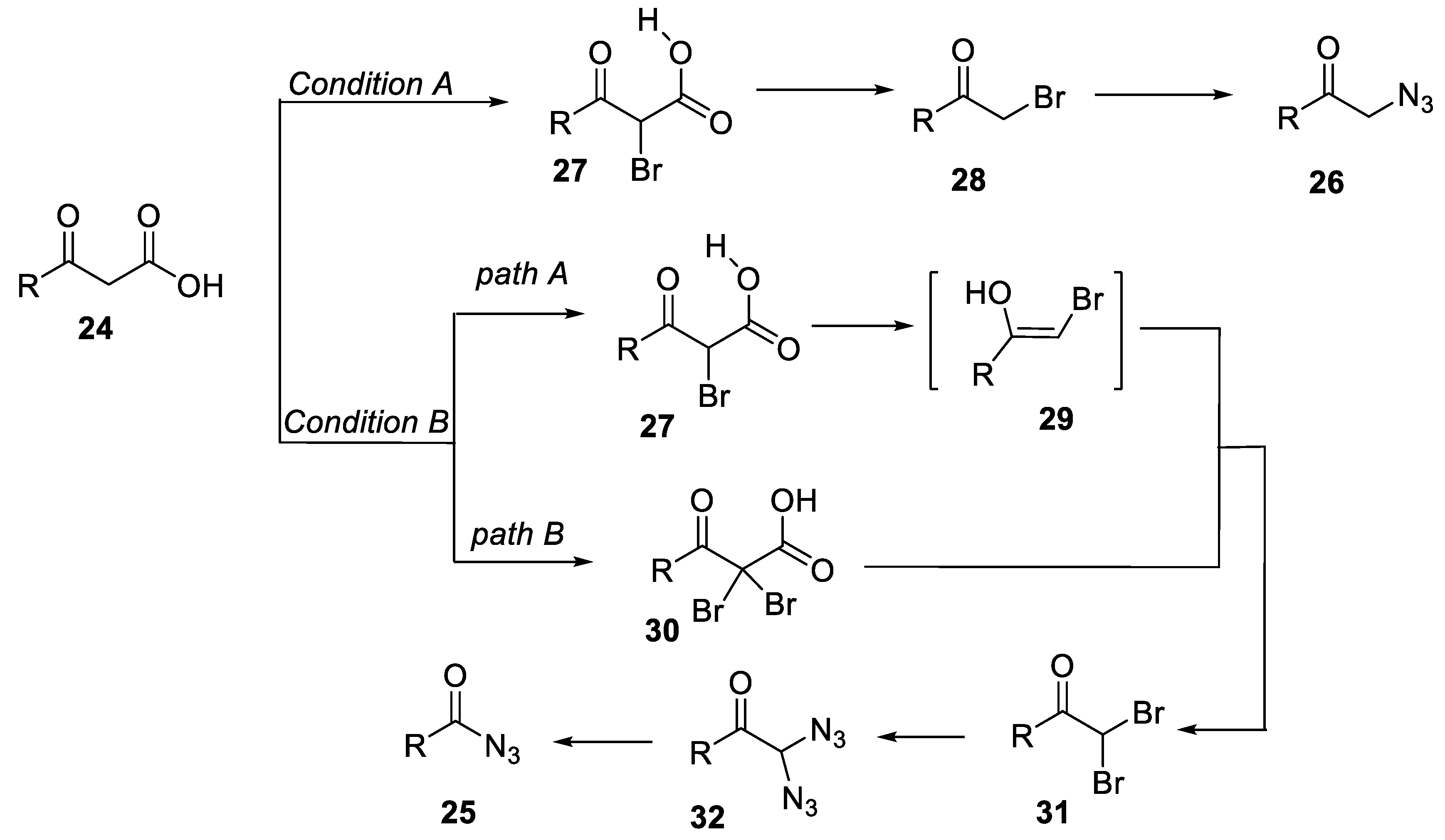
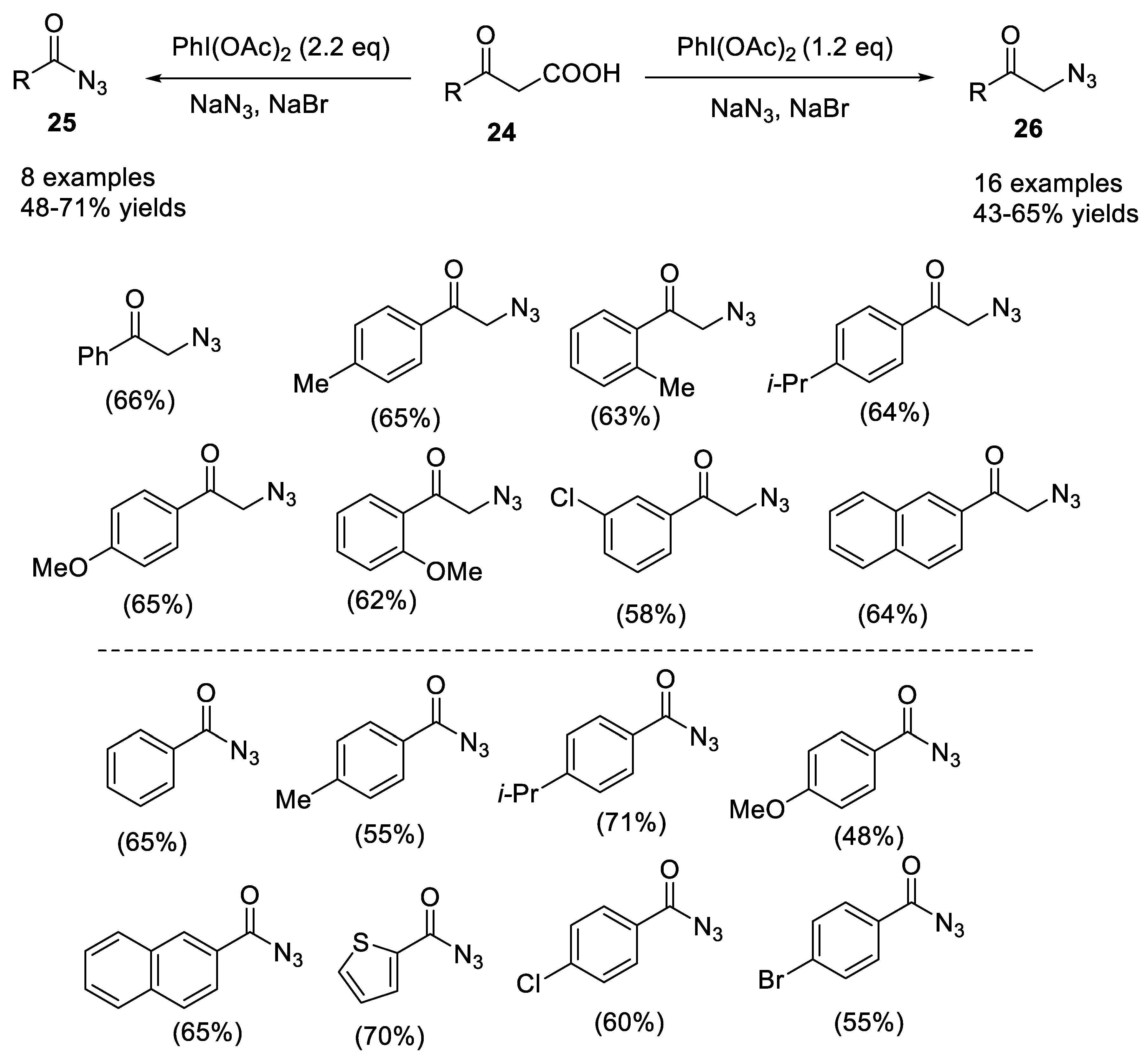
- Zheng, Z.-J.; Yu, T.-Y.; Xu, P.-F.; Wei, H. (Diacetoxyiodo)benzene-mediated selective synthesis of α-azido ketones or acyl azides from β-keto acids. Asian J. Org. Chem. 2018, 7, 1579–1582. [Google Scholar] [CrossRef]
- Opai, I.A.; Shirsath, P.D.; Kalbandhe, A.H.; Karade, N.N. (Diacetoxyiodo)benzene mediated metal-free C(sp2)-H phenylselenation of imidazo[1,2-a]pyridines and imidazo[2,1-b]thiazoles using diphenyl diselenide. Arkivoc 2021, 7, 101–111. [Google Scholar] [CrossRef]
- Izzo, F.; Schaefer, M.; Stockman, R.; Luecking, U. A new, practical one-pot synthesis of unprotected sulfonimidamides by transfer of electrophilic NH to sulfinamides. Chem. Eur. J. 2017, 23, 15189–15193. [Google Scholar] [CrossRef]
- Liu, G.-Q.; Yang, C.-H.; Li, Y.-M. Modular preparation of 5-halomethyl-2-oxazolines via PIDA-promoted intramolecular halooxygenation of N-allylcarboxamides. J. Org. Chem. 2015, 80, 11339–11350. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Li, D.; Yu, W. Synthesis of quinoxaline derivatives via tandem oxidative azidation/cyclization reaction of N-arylenamines. Org. Lett. 2016, 18, 868–871. [Google Scholar] [CrossRef]
- Schofield, K.; Foley, C.; Hulme, C. 5-endo trig oxidative radical cyclizations of Ugi-3CR products toward 1,4-imidazolidinones. Org. Lett. 2021, 23, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, H.; Yoshimura, A.; Noguchi, K.; Nemykin, V.N.; Zhdankin, V.V.; Saito, A. Oxidative cycloaddition of hydroxamic acids with dienes or guaiacols mediated by iodine(III) reagents. Beilstein J. Org. Chem. 2018, 14, 531–536. [Google Scholar] [CrossRef] [Green Version]
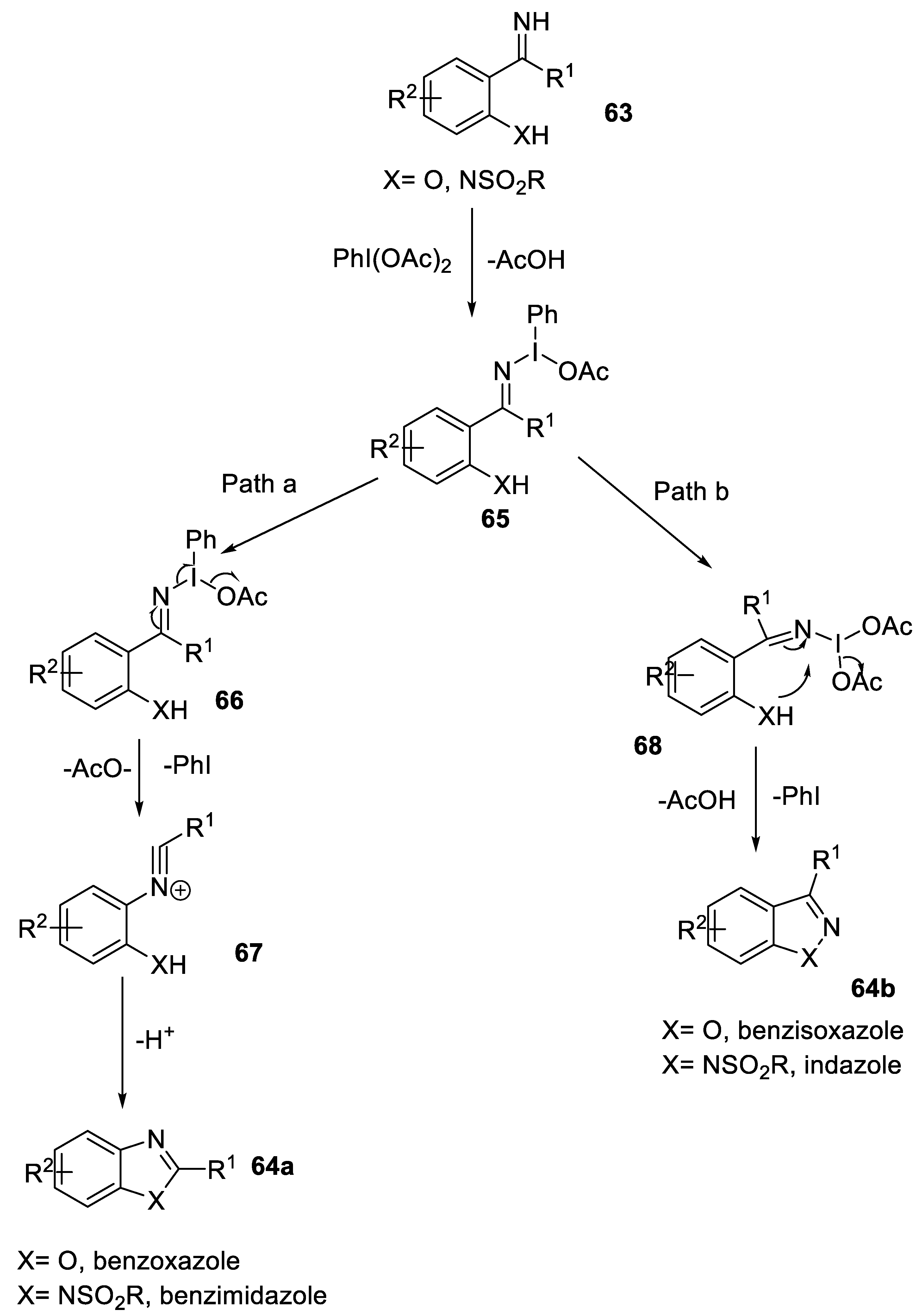
- Zhang, X.; Huang, R.; Marrot, J.; Coeffard, V.; Xiong, Y. Hypervalent iodine-mediated synthesis of benzoxazoles and benzimidazoles via an oxidative rearrangement. Tetrahedron 2015, 71, 700–708. [Google Scholar] [CrossRef]
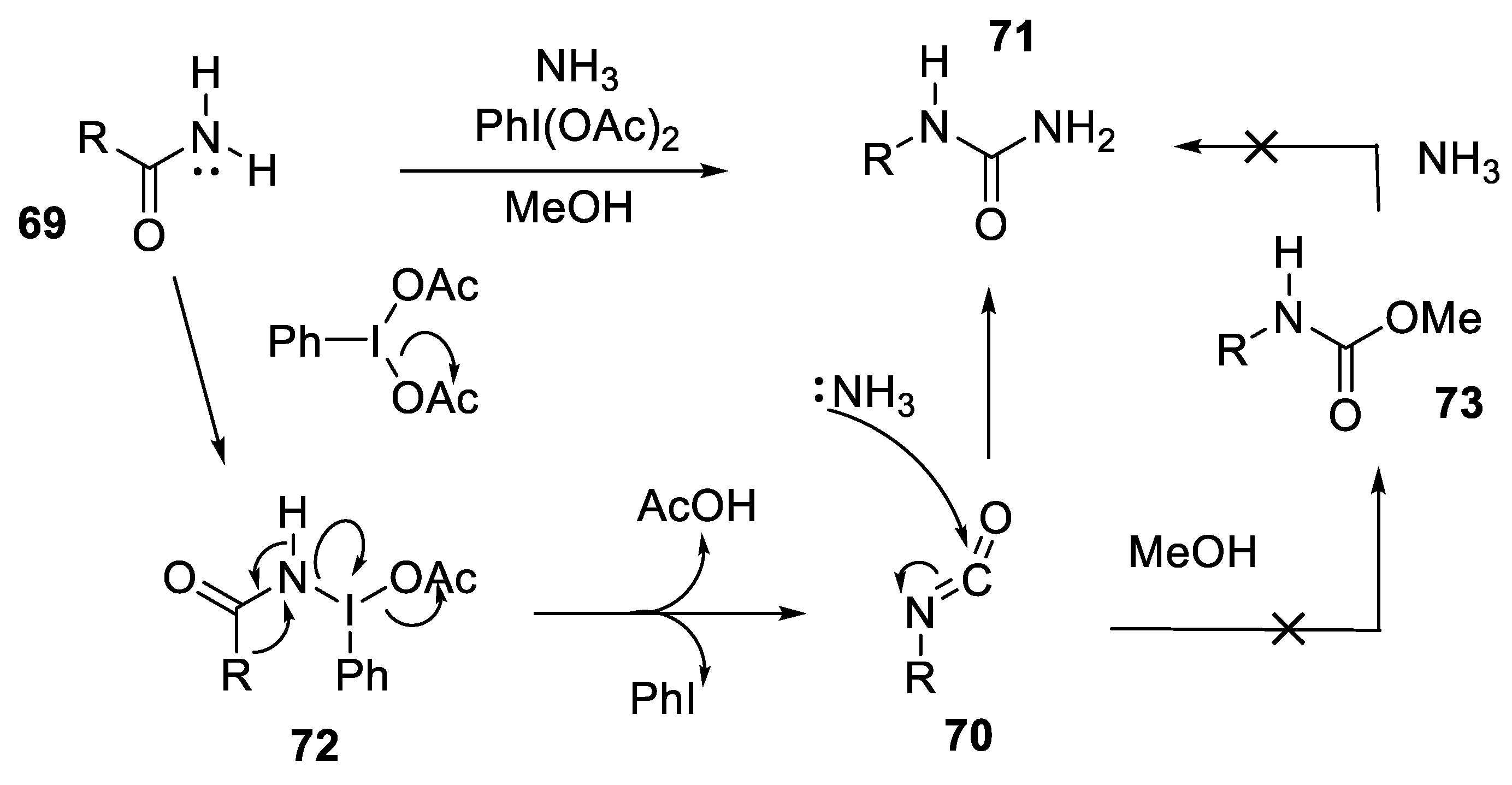
- Rosa, N.S.; Glachet, T.; Ibert, Q.; Lohier, J.-F.; Franck, X.; Reboul, V. A Straightforward Synthesis of N-Substituted Ureas from Primary Amides. Synthesis 2020, 52, 2099–2105. [Google Scholar] [CrossRef]
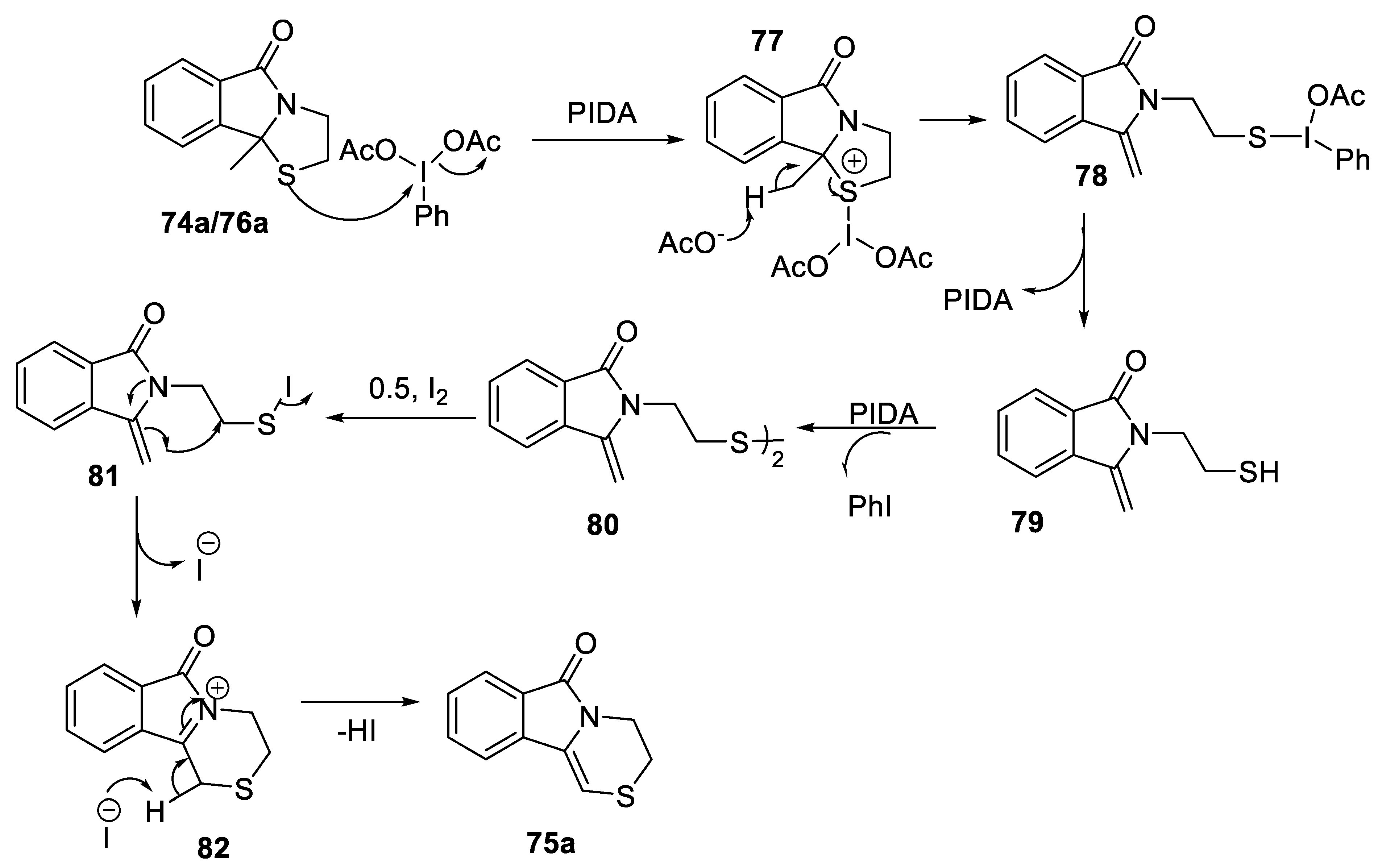
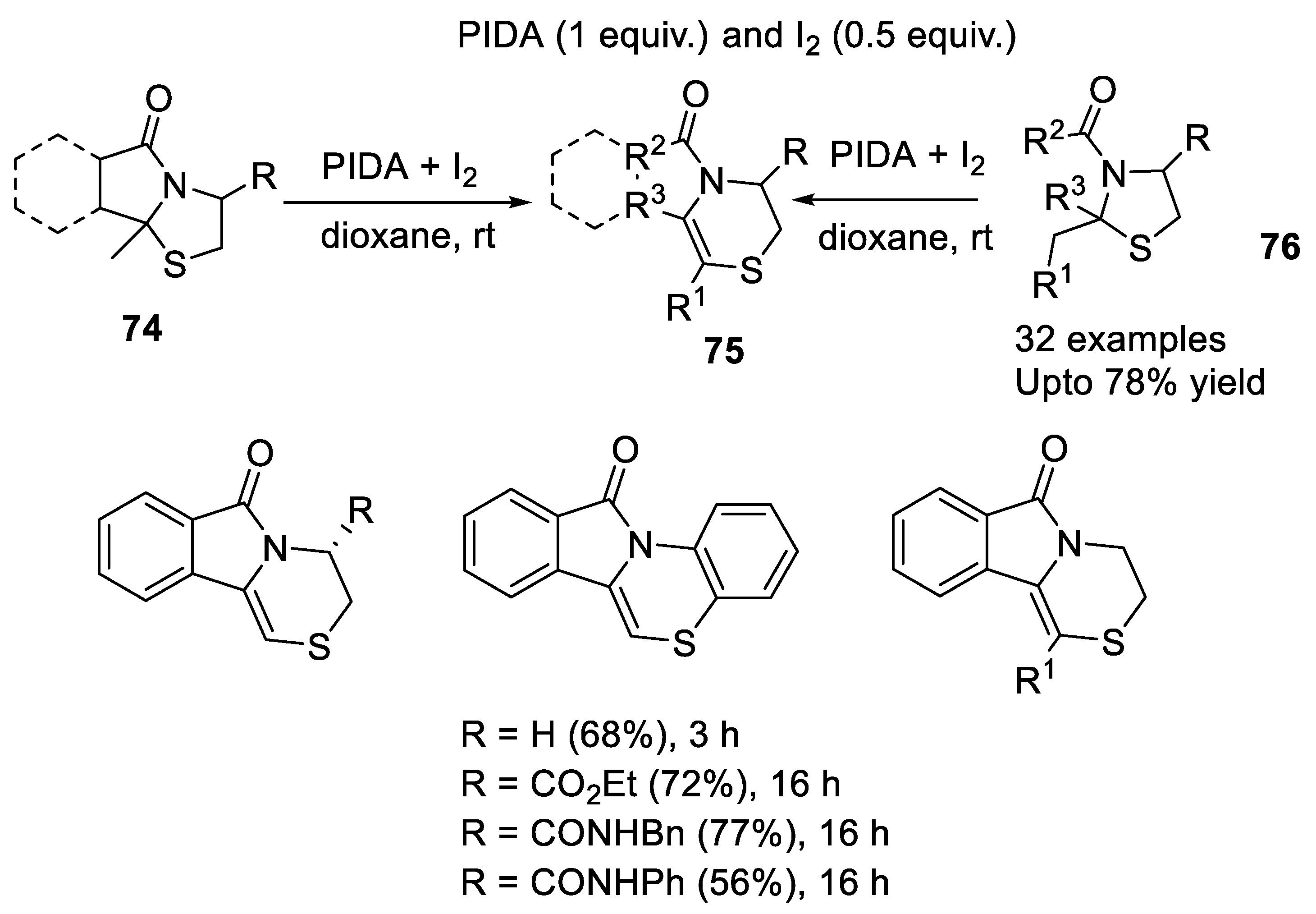
- Danton, F.; Othman, M.; Lawson, A.M.; Moncol, J.; Ghinet, A.; Rigo, B.; Daich, A. Phenyliodine(III) diacetate/i2-mediated domino approach for pyrrolo[1,4]thiazines and 1,4-thiazines by a one-pot morin rearrangement of N,S-acetals. Chem. Eur. J. 2019, 25, 6113–6118. [Google Scholar] [CrossRef]
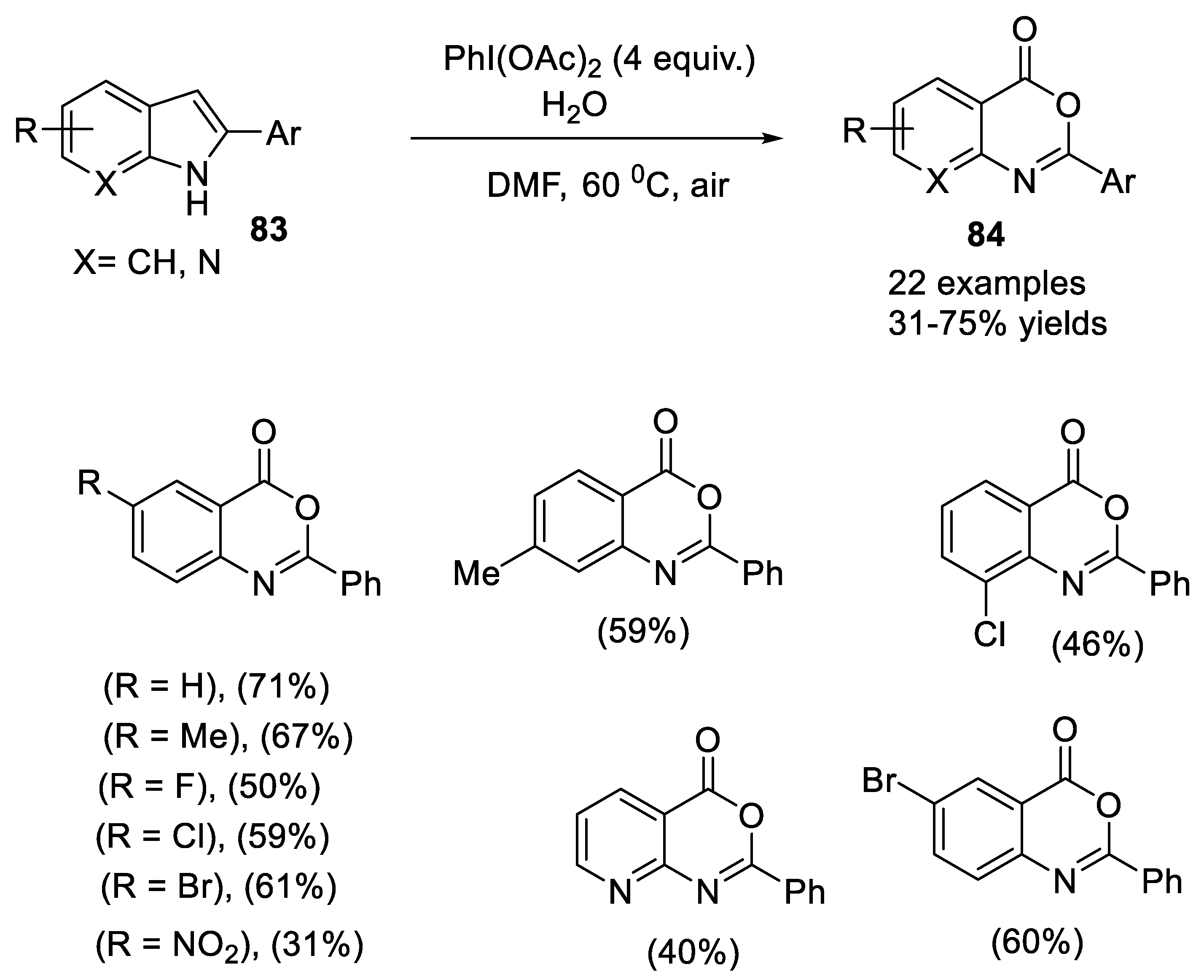
- Shang, X.-X.; Vu, H.-M.; Li, X.-Q. One-pot synthesis of 2-arylbenzoxazinones from 2-arylindoles with (diacetoxyiodo)benzene as the sole oxidant. Synthesis 2018, 50, 377–383. [Google Scholar] [CrossRef]
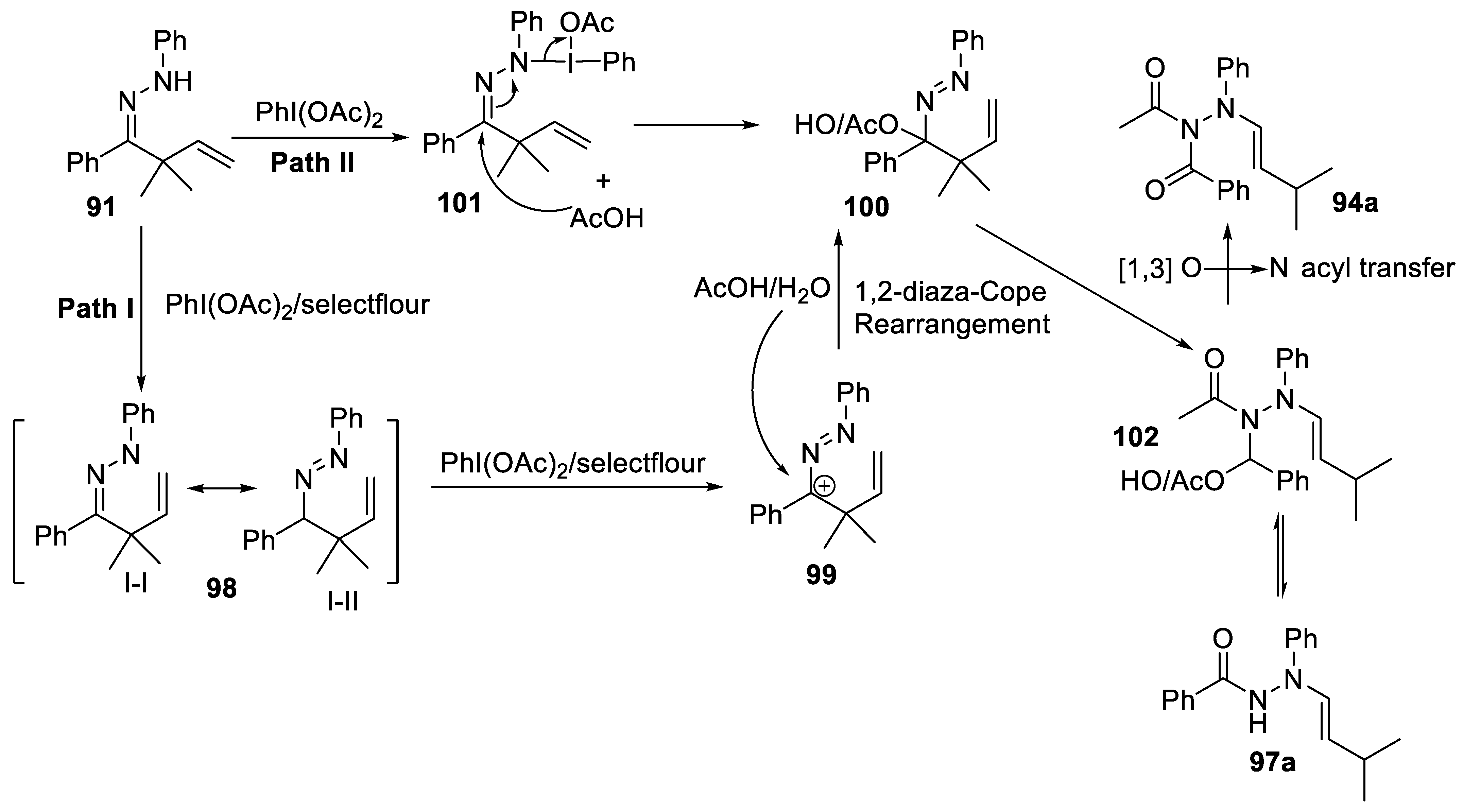
- Fang, Z.-Y.; Qi, L.; Song, J.-Y.; Ren, P.-X.; Hou, C.-Y.; Ji, S.-C.; Wang, L.-J.; Li, W. PhI(OAc)2-promoted 1,2-diaza-cope rearrangement of β,γ-unsaturated Hydrazones with Acetate/H2O: Access to diacyl/acyl N-allylhydrazines. Eur. J. Org. Chem. 2020, 2020, 5464–5468. [Google Scholar] [CrossRef]
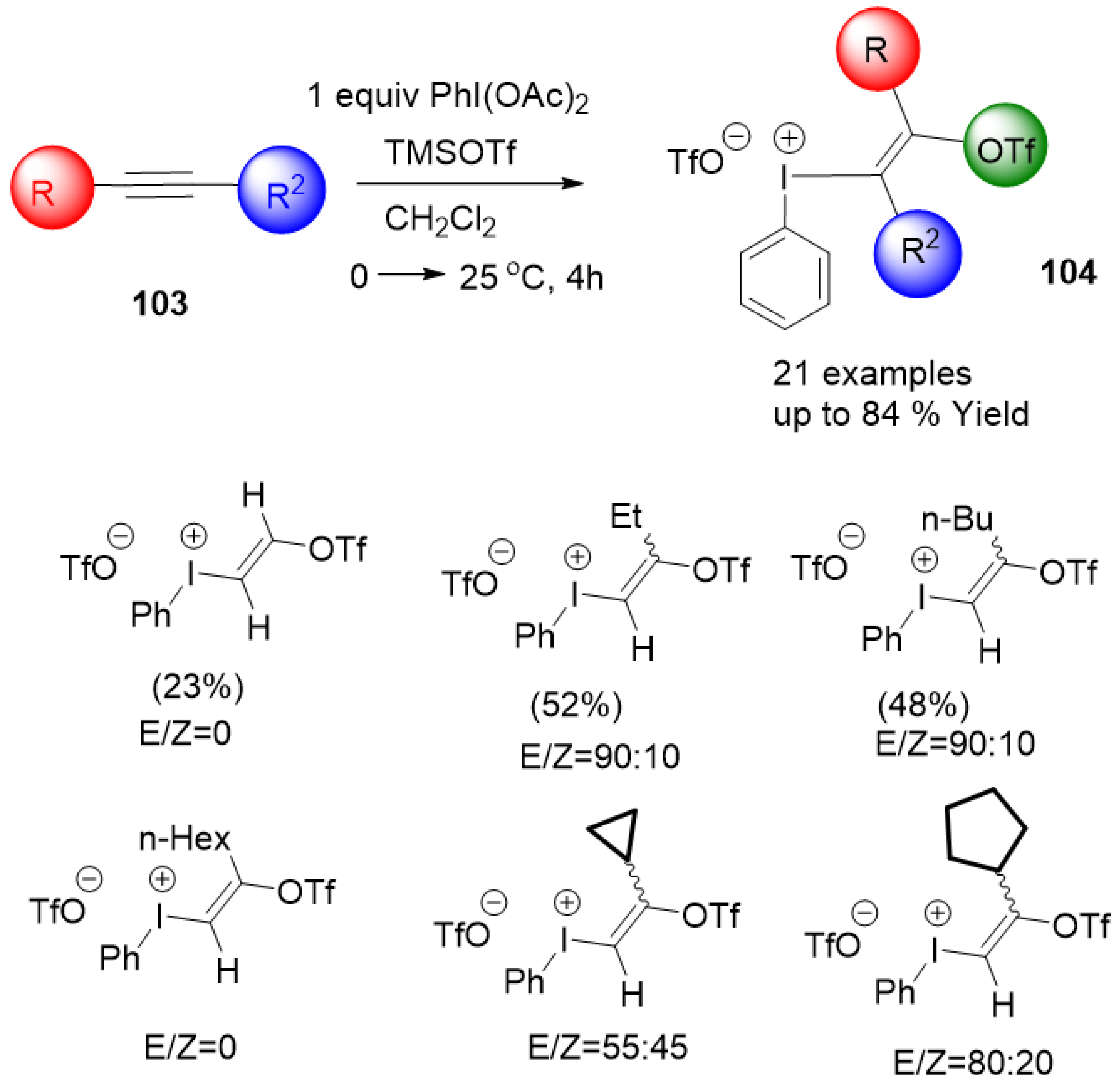
- Toth, B.L.; Beke, F.; Egyed, O.; Benyei, A.; Stirling, A.; Novak, Z. Synthesis of multifunctional aryl(trifloxyalkenyl)iodonium triflate salts. ACS Omega 2019, 4, 9188–9197. [Google Scholar] [CrossRef]
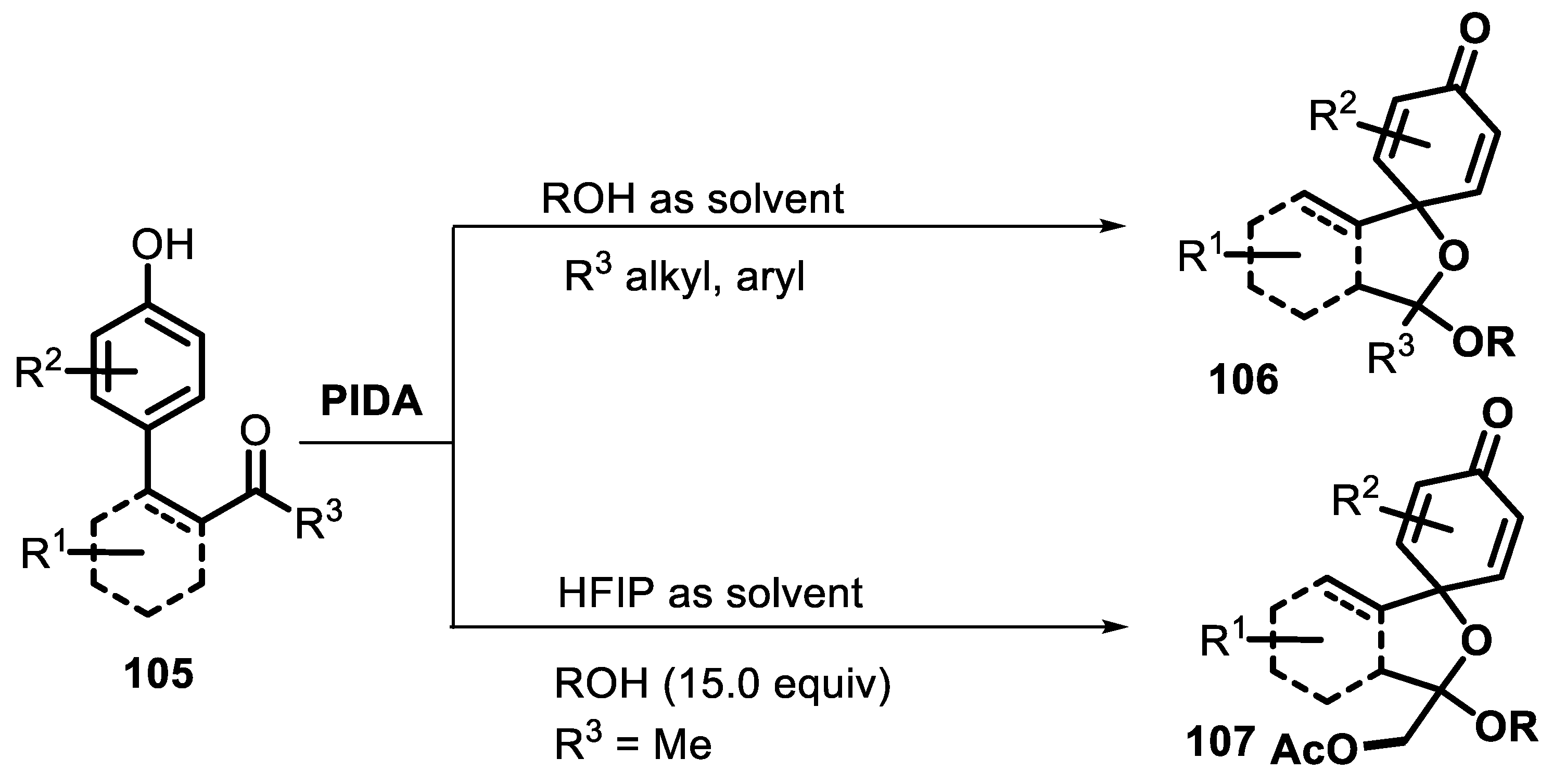
- Peng, S.; He, J.; Yang, L.; Zhang, H.; Li, H.; Lang, M.; Chen, C.; Wang, J. PIDA-promoted/hfip-controlled dearomative spirocyclization of phenolic ketones via a spirocyclohexadienone-oxocarbenium cation species. J. Org. Chem. 2022, 87, 6247–6262. [Google Scholar] [CrossRef] [PubMed]
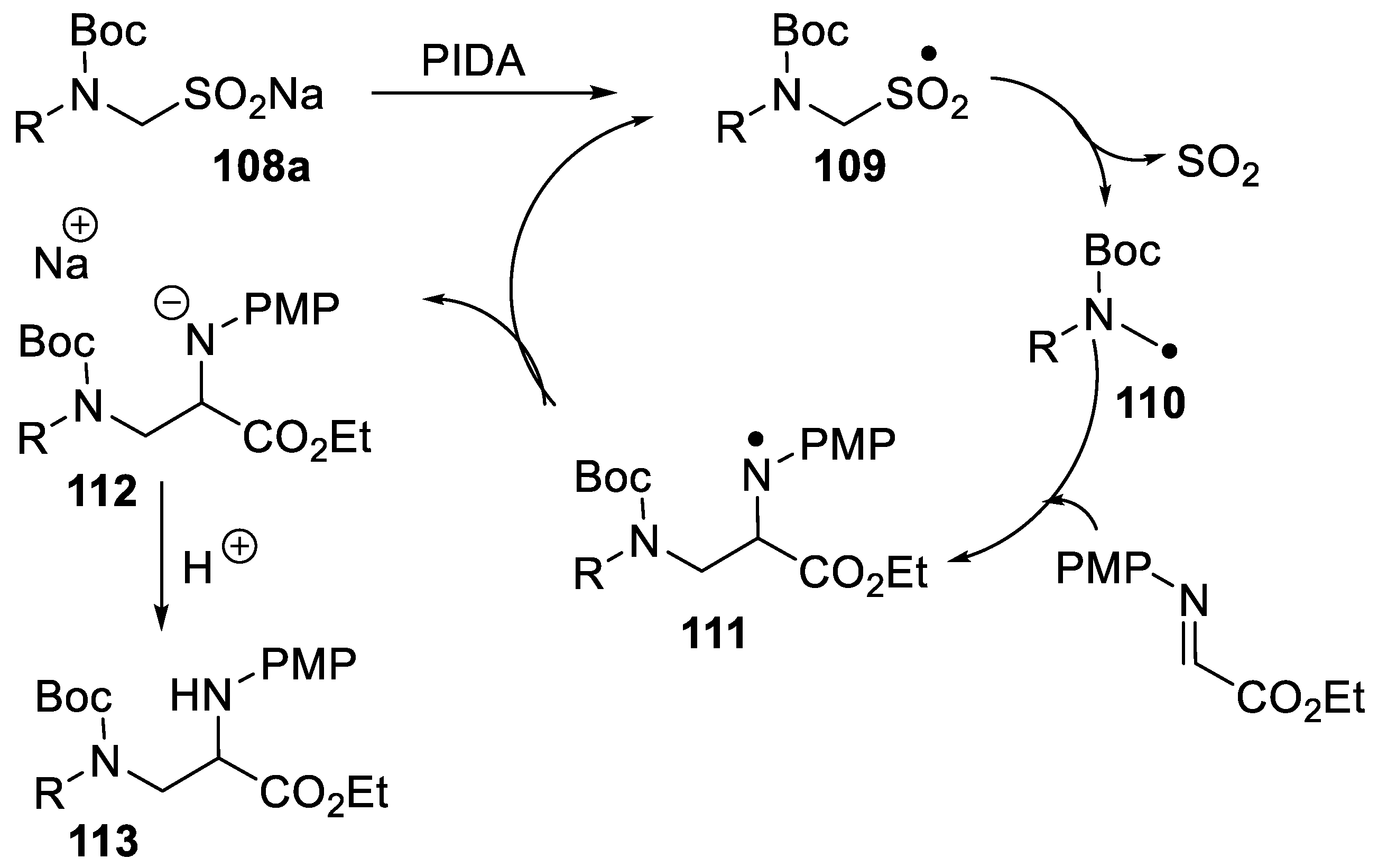
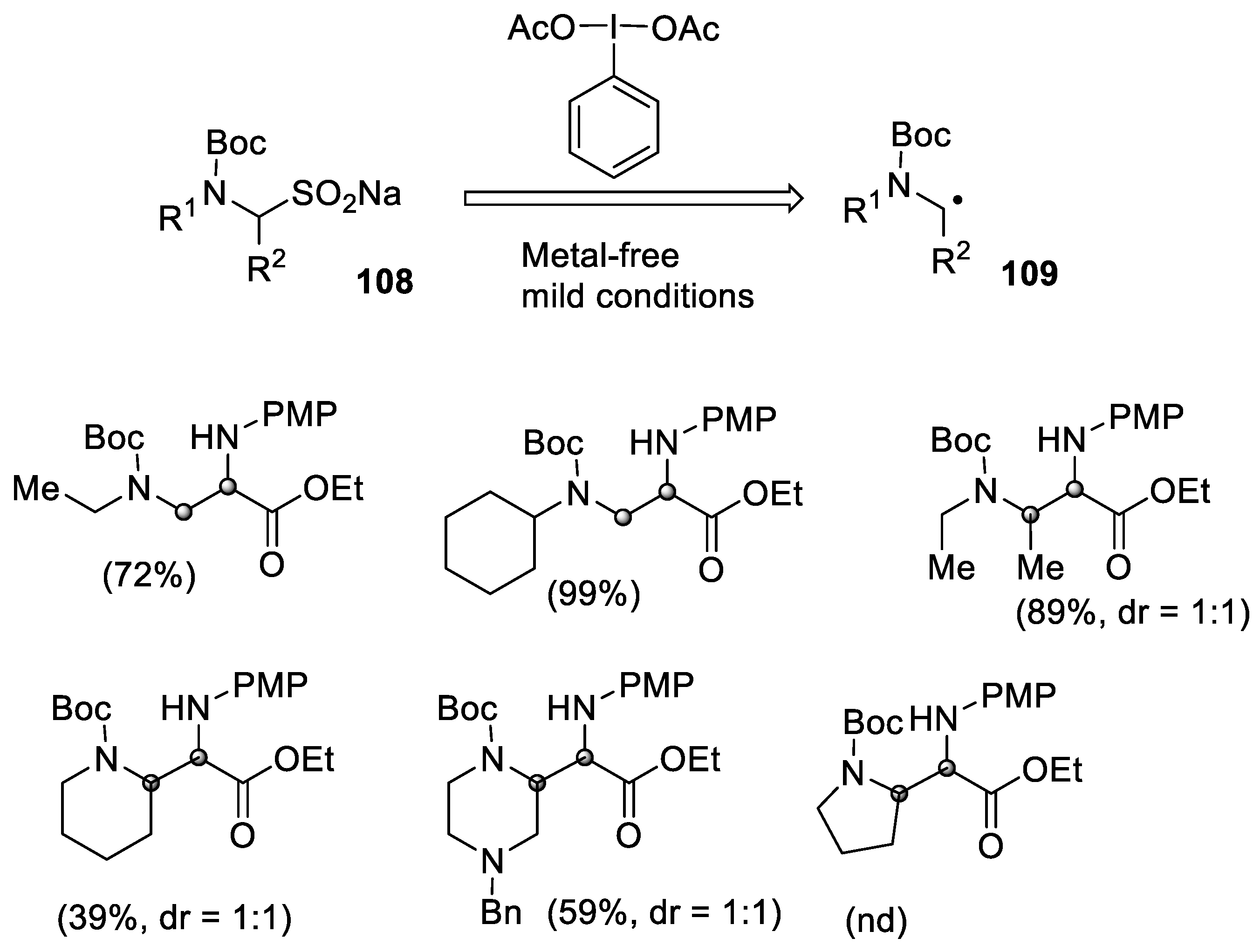
- Sakamoto, R.; Tomomi, Y.; Takada, H.; Maruoka, K. A Synthetic route to sodium α-aminoalkanesulfinates and their application in the generation of α-aminoalkyl radicals for radical addition reactions. Org. Lett. 2018, 20, 2080–2083. [Google Scholar] [CrossRef] [PubMed]
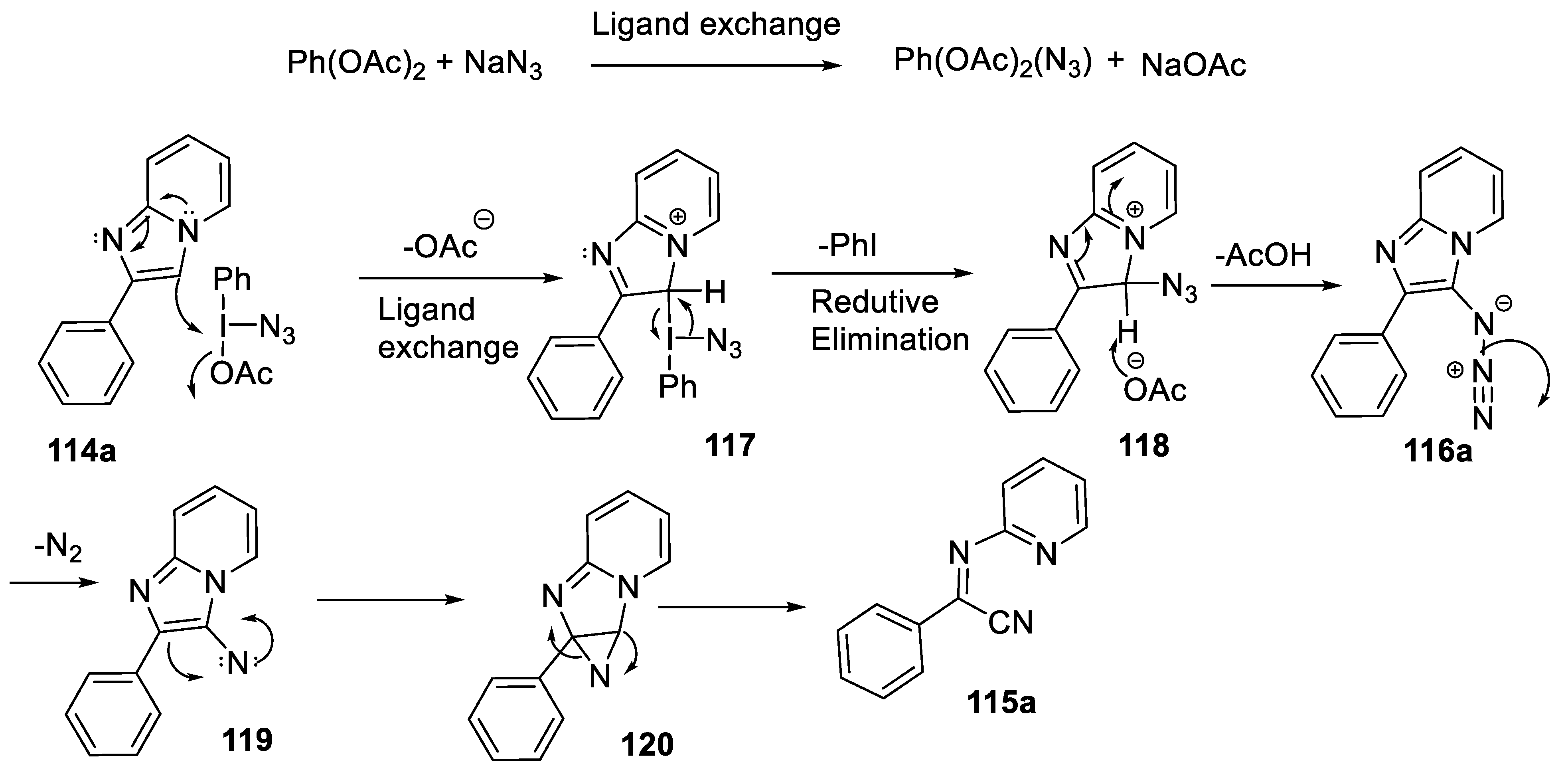
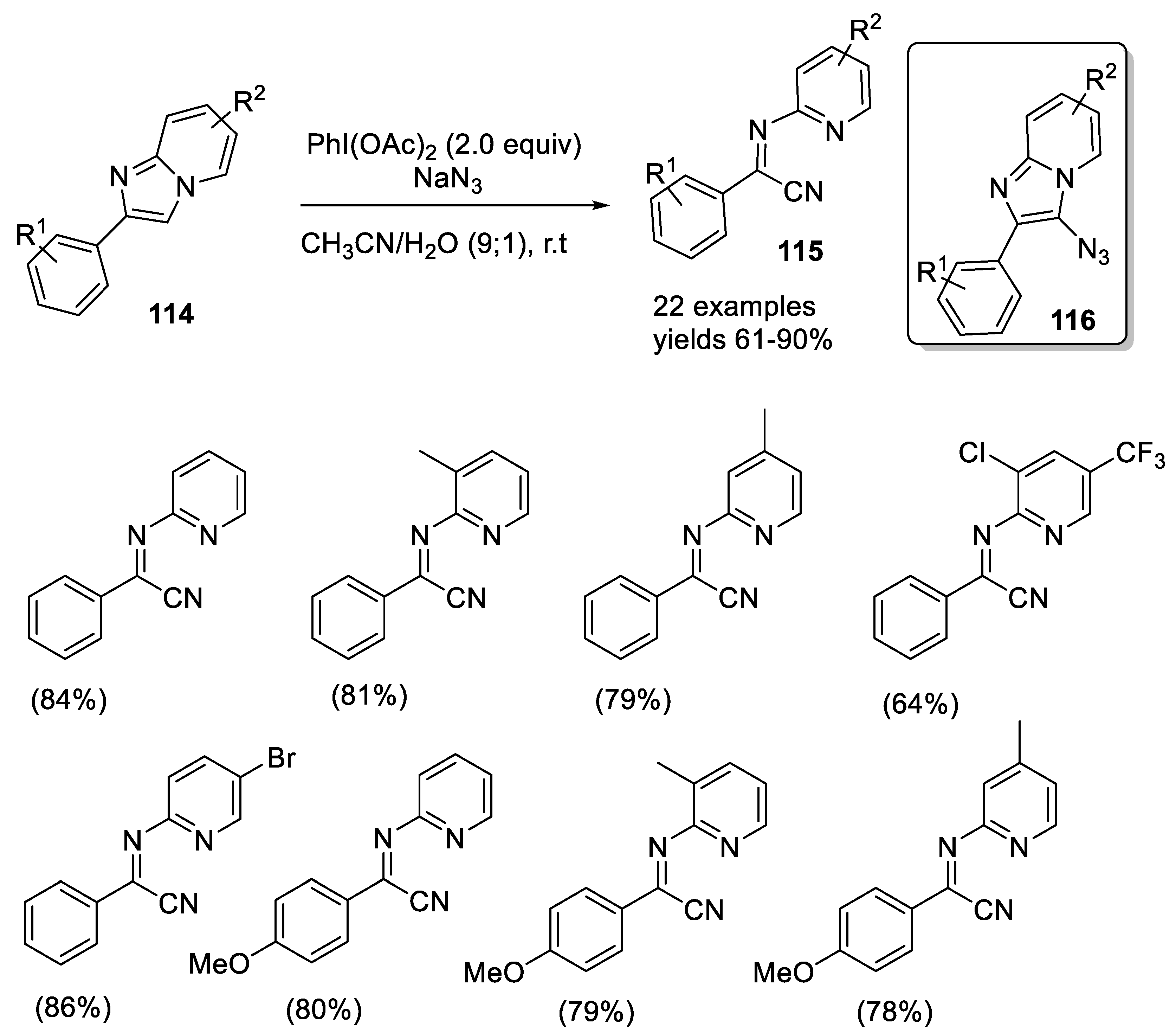
- Kalbandhe, A.H.; Kavale, A.C.; Karade, N.N. Ring-opening reaction of imidazo[1,2-a]pyridines using (diacetoxyiodo)benzene and NaN3: The synthesis of α-iminonitriles. Eur. J. Org. Chem. 2017, 10, 1318–1322. [Google Scholar] [CrossRef]
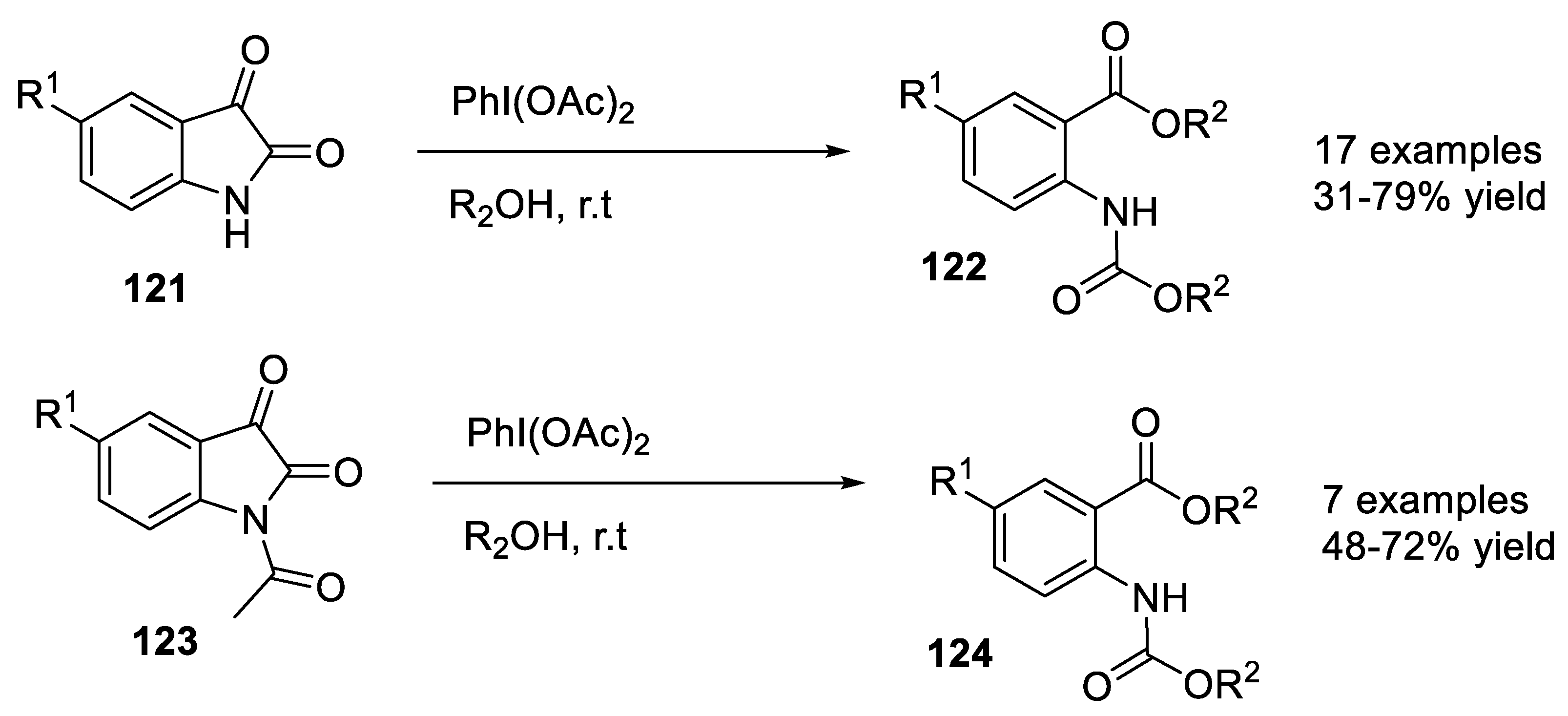
- Kalbandhe, A.H.; Kavale, A.C.; Thorat, P.B.; Karade, N.N. Oxidative cleavage of c2-c3 bond in isatin using (diacetoxyiodo)benzene: A facile synthesis of carbamates of alkyl anthranilates. Synlett 2016, 27, 763–768. [Google Scholar] [CrossRef]
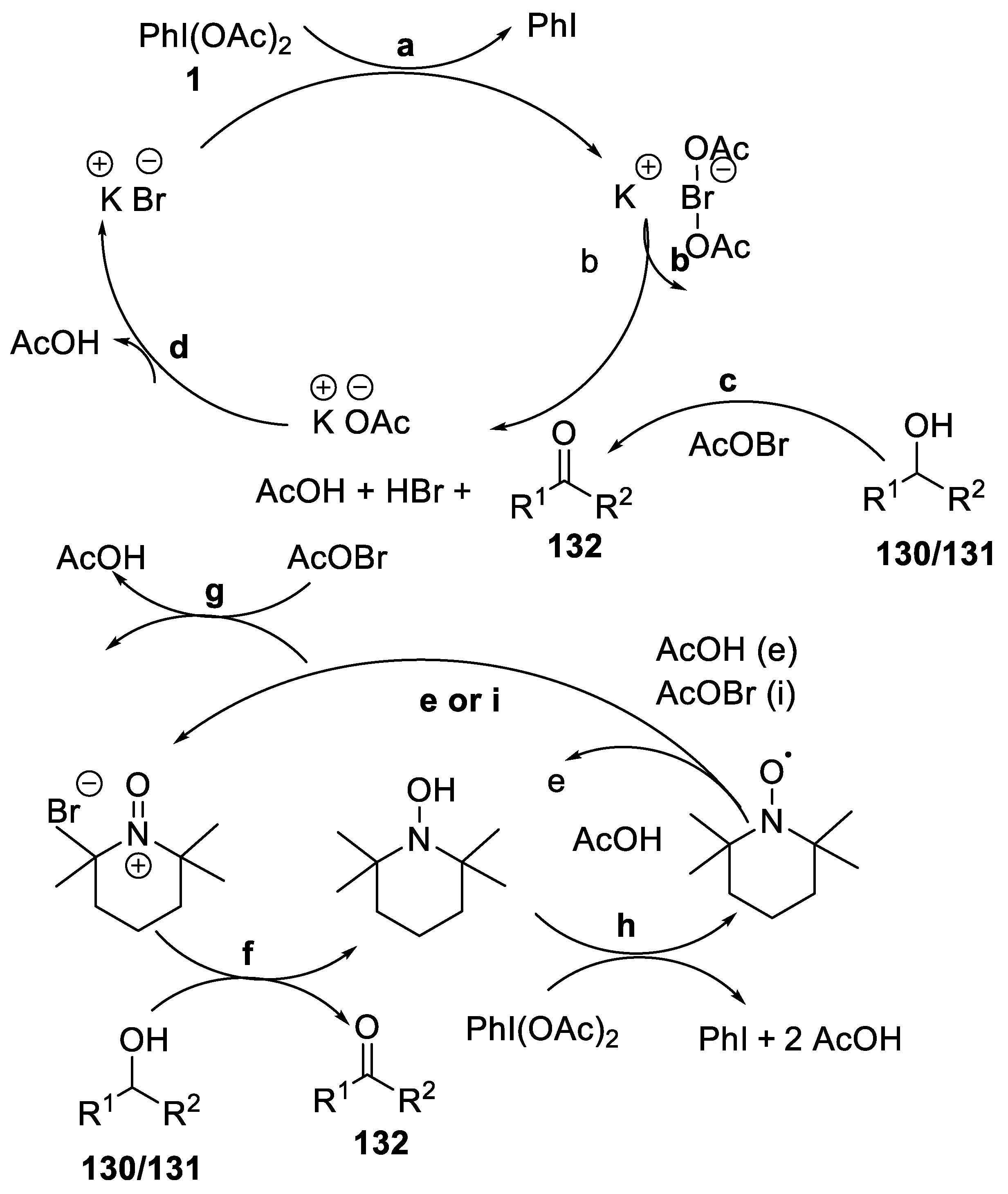
- Salvo, A.M.P.; Campisciano, V.; Beejapur, H.A.; Giacalone, F.; Gruttadauria, M. A simple procedure for the oxidation of alcohols using [bis(acetoxy) iodo]benzene and a catalytic amount of bromide ions in ethyl acetate. Synlett 2015, 26, 1179–1184. [Google Scholar] [CrossRef]
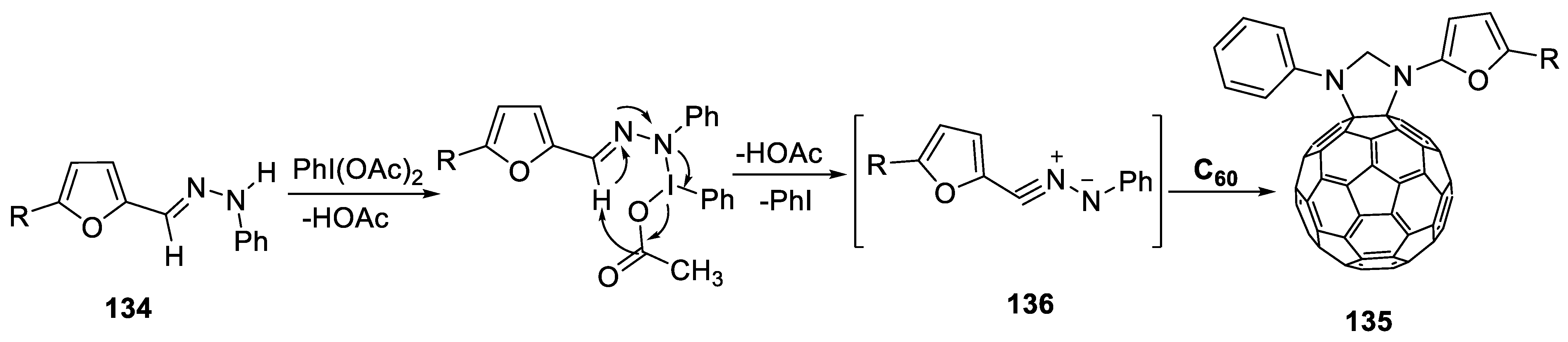
- Al-Matar, H.M.; Bin, S.; Mohammad, H.; Shalaby, M.A. Synthesis and electrochemistry of new furylpyrazolino[60]fullerene derivatives by efficient microwave radiation. Molecules 2019, 24, 4435. [Google Scholar] [CrossRef] [Green Version]
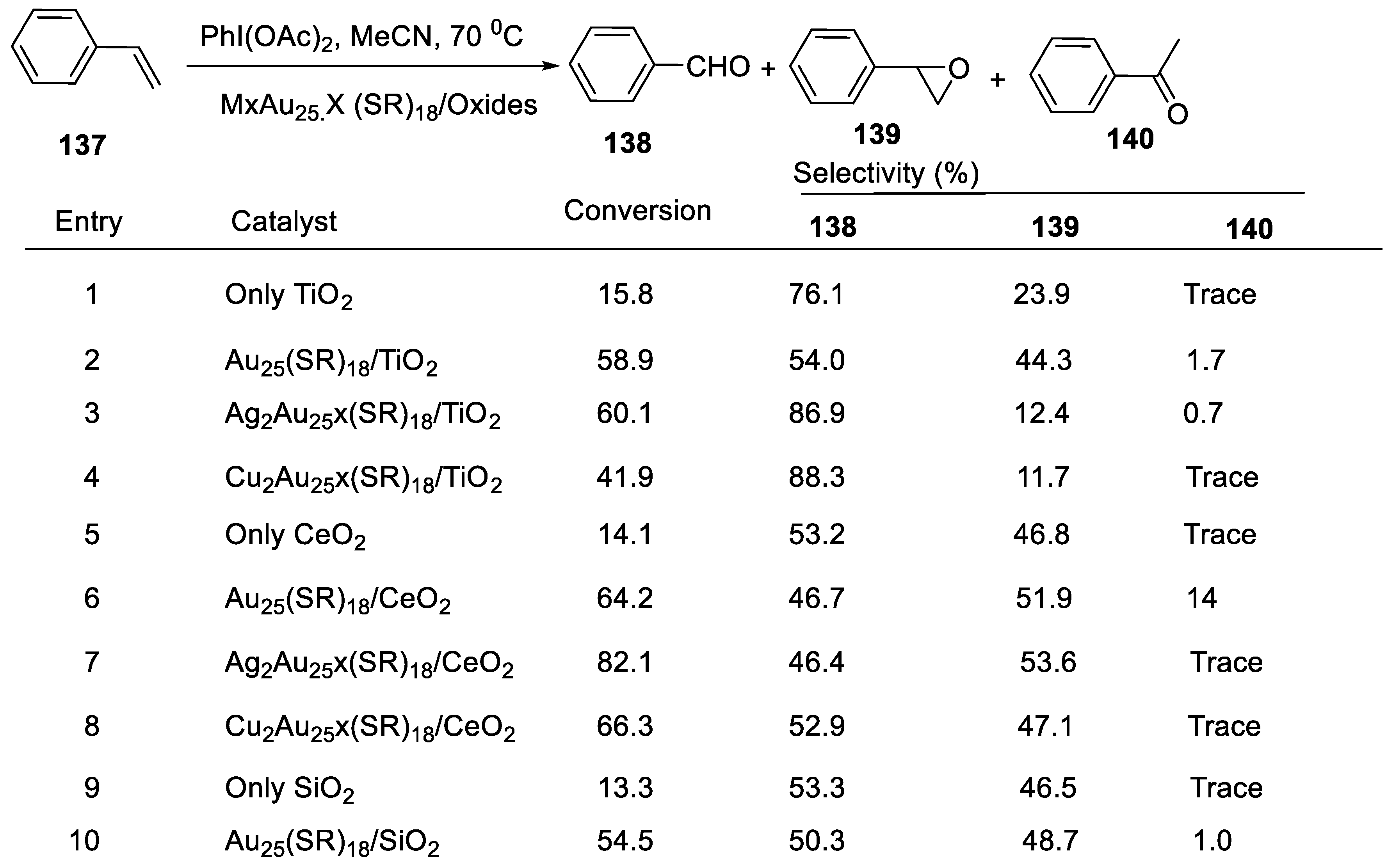
- Li, G.; Jin, R. Atomic level tuning of the catalytic properties: Doping effects of 25-atom bimetallic nanoclusters on styrene oxidation. Catal. 2016, 278, 187–191. [Google Scholar] [CrossRef] [Green Version]
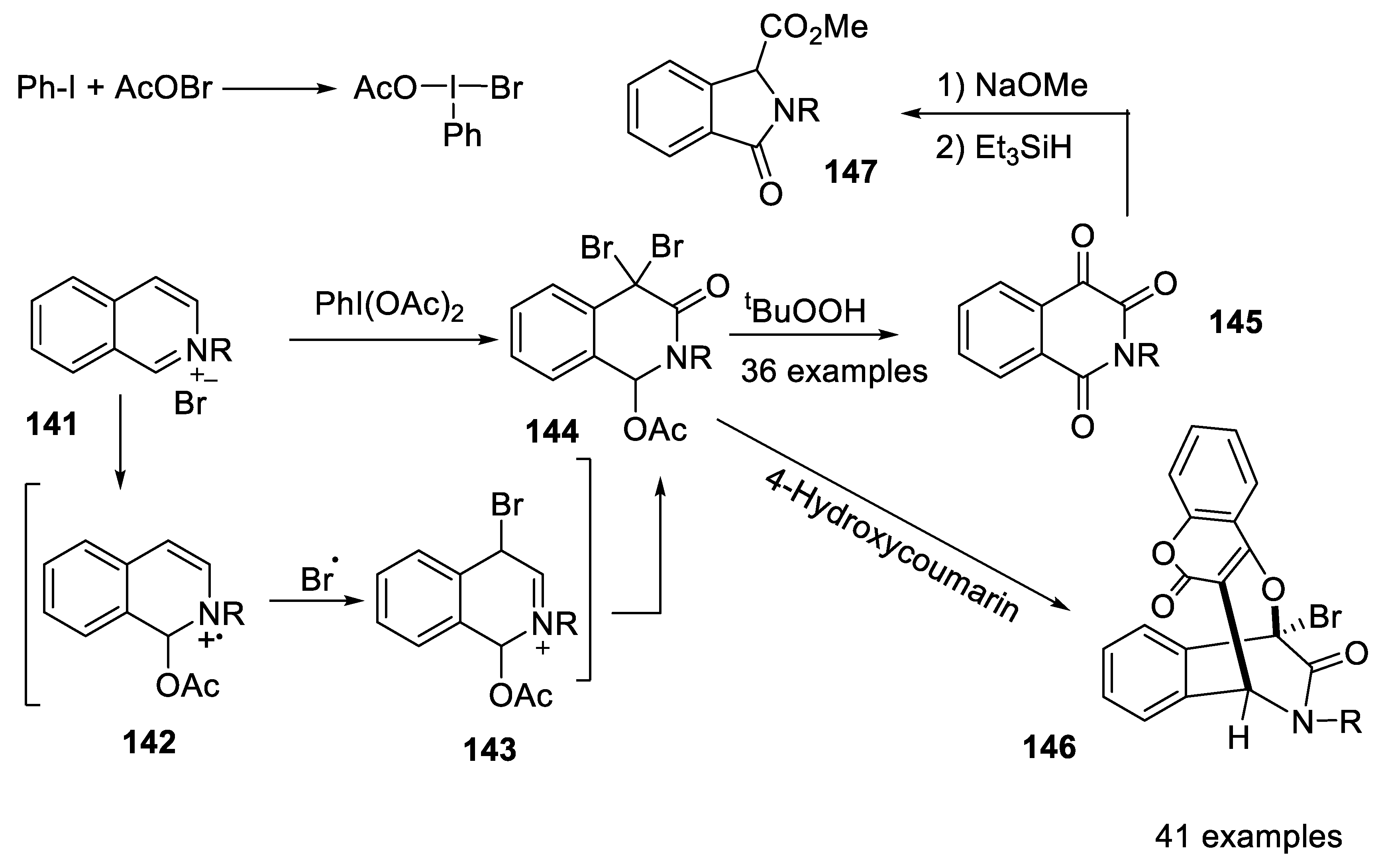
- Lin, X.-L.; Yu, Y.; Zhang, L.; Leng, L.; Xiao, D.-R.; Cai, T.; Luo, Q.-L. Switchable synthesis of 1,4-bridged dihydroisoquinoline-3-ones and isoquinoline-1,3,4-triones through radical oxidation of isoquinolinium salts with phenyliodine(III) diacetate. Org. Chem. Front. 2022, 9, 4676–4684. [Google Scholar] [CrossRef]
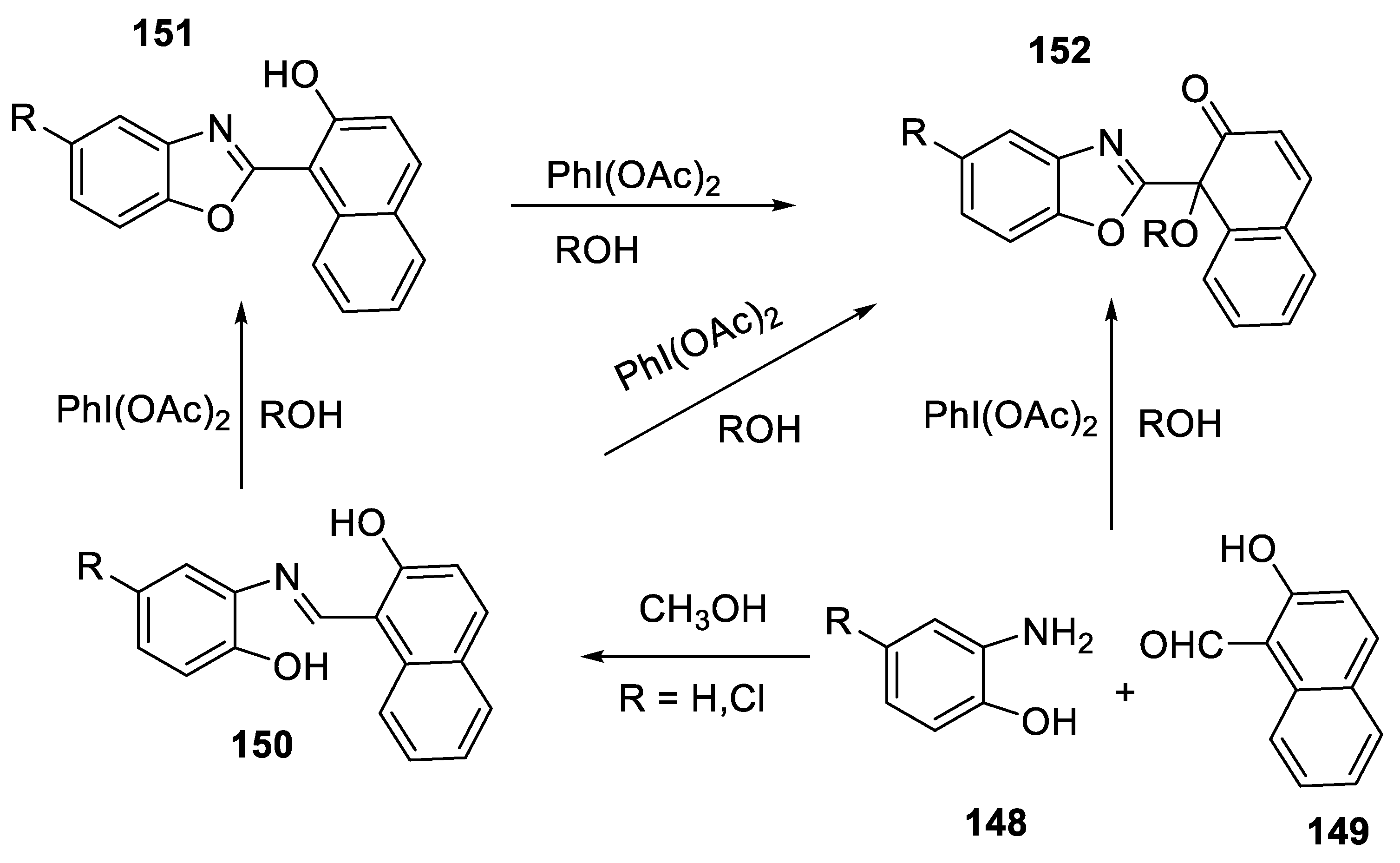
- Yadav, G.; Kumar, S.; Kataria, R.; Kumar, D. Phenyliodine(III) diacetate-induced regioselective synthesis of 1-(benzoxazol-2-yl)-1-alkoxynaphthalen-2(1H)- ones from 2-(2-hydroxynaphthyl)benzoxazoles. Synth. Commun. 2022, 52, 1412–1420. [Google Scholar] [CrossRef]
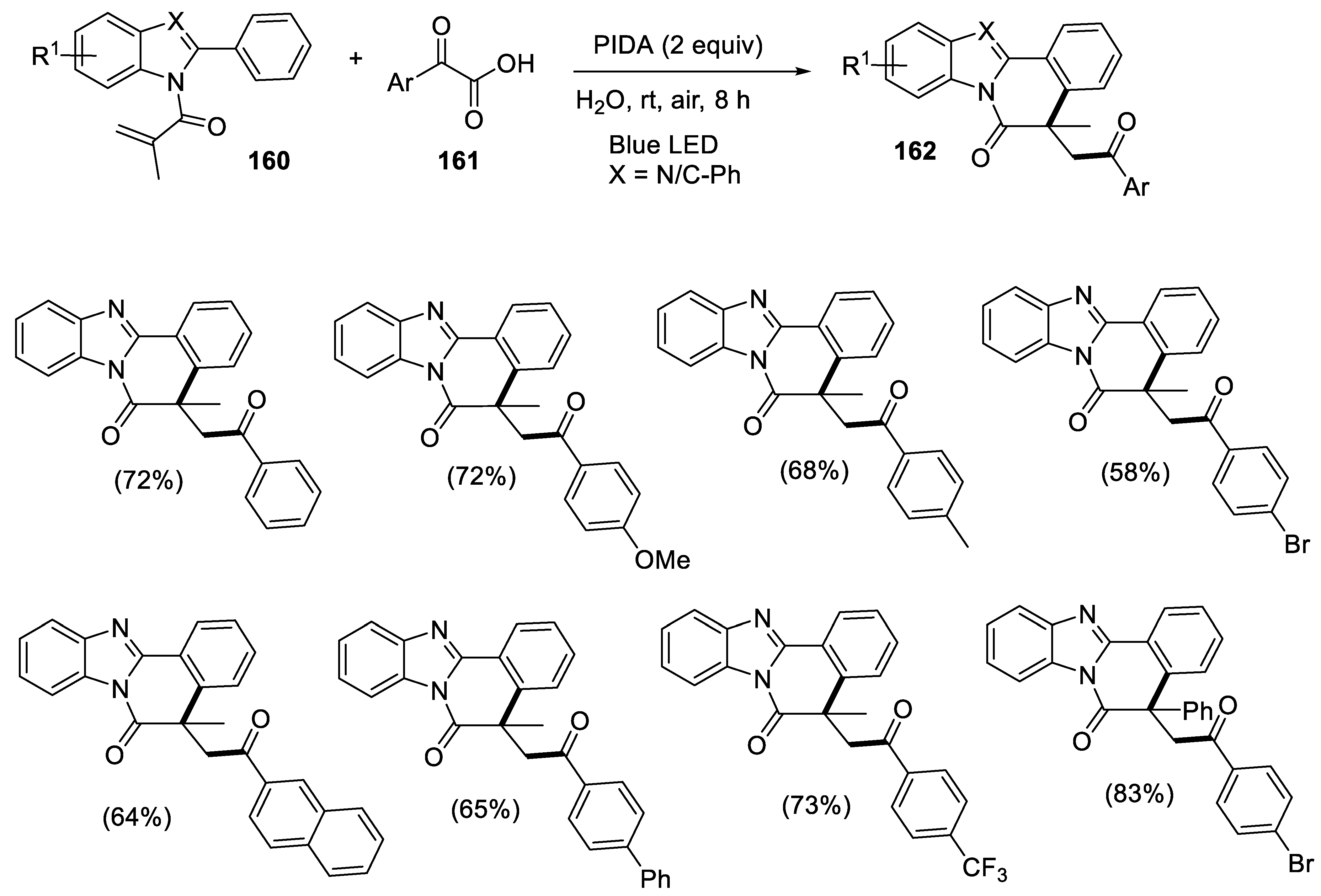
- Tang, L.; Ouyang, Y.; Sun, K.; Yu, B. Visible-light-promoted decarboxylative radical cascade cyclization to acylated benzimidazo/indolo[2,1-a]isoquinolin-6(5H)-ones in water. RSC Adv. 2022, 12, 19736–19740. [Google Scholar] [CrossRef]


















































Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Varala, R.; Seema, V.; Dubasi, N. Phenyliodine(III)diacetate (PIDA): Applications in Organic Synthesis. Organics 2023, 4, 1-40. https://doi.org/10.3390/org4010001
Varala R, Seema V, Dubasi N. Phenyliodine(III)diacetate (PIDA): Applications in Organic Synthesis. Organics. 2023; 4(1):1-40. https://doi.org/10.3390/org4010001
Chicago/Turabian StyleVarala, Ravi, Vittal Seema, and Narsimhaswamy Dubasi. 2023. "Phenyliodine(III)diacetate (PIDA): Applications in Organic Synthesis" Organics 4, no. 1: 1-40. https://doi.org/10.3390/org4010001