

Histological Assessment of Endochondral Ossification and Bone Mineralization
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
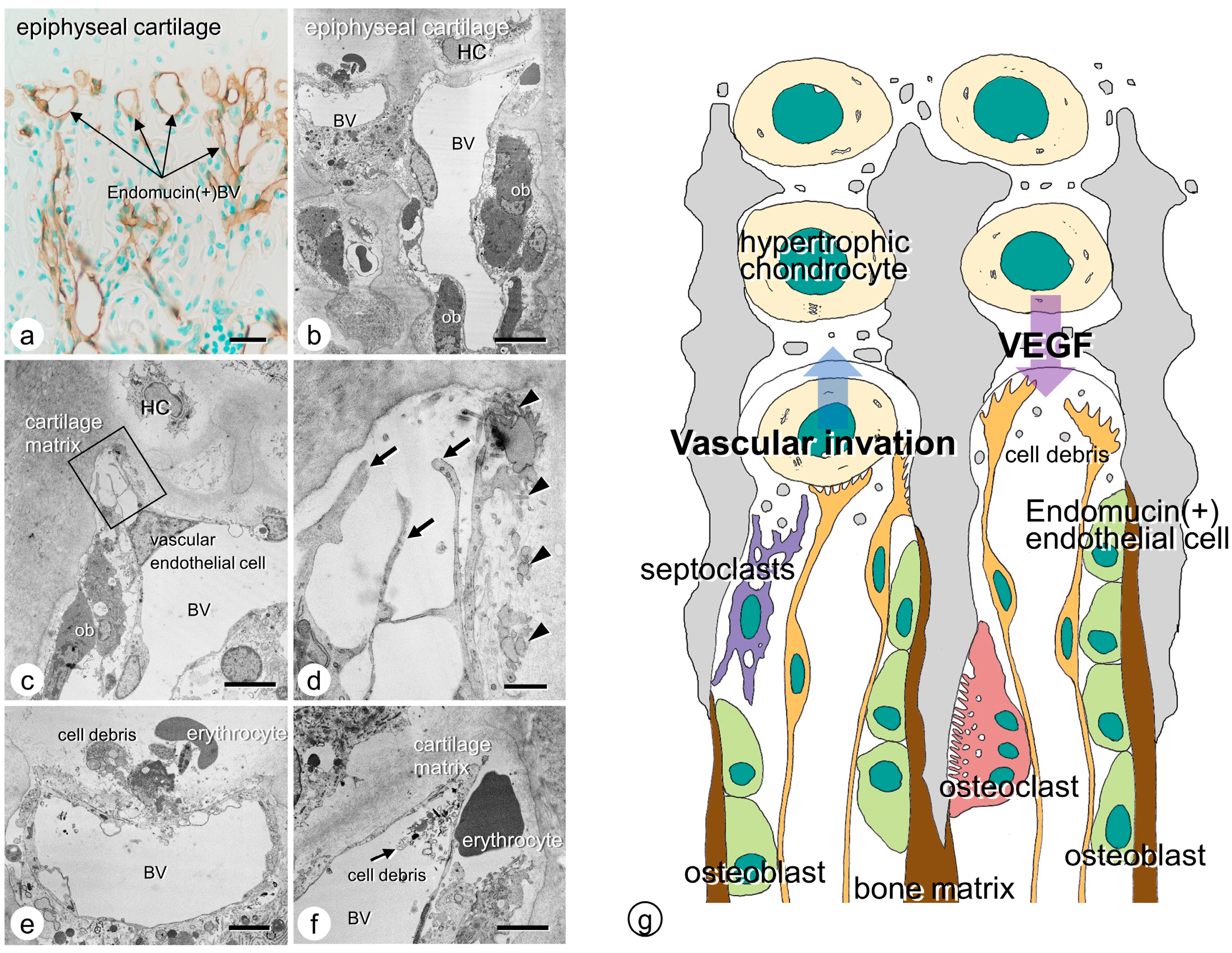
2. Histological Aspects on Endochondral Ossification
2.1. Cartilage Mineralization by Hypertrophic Chondrocytes
2.2. Vascular Invasion at the Chondro-Osseous Junction
2.3. Osteoclasts’ Function at the Chondro-Osseous Junction
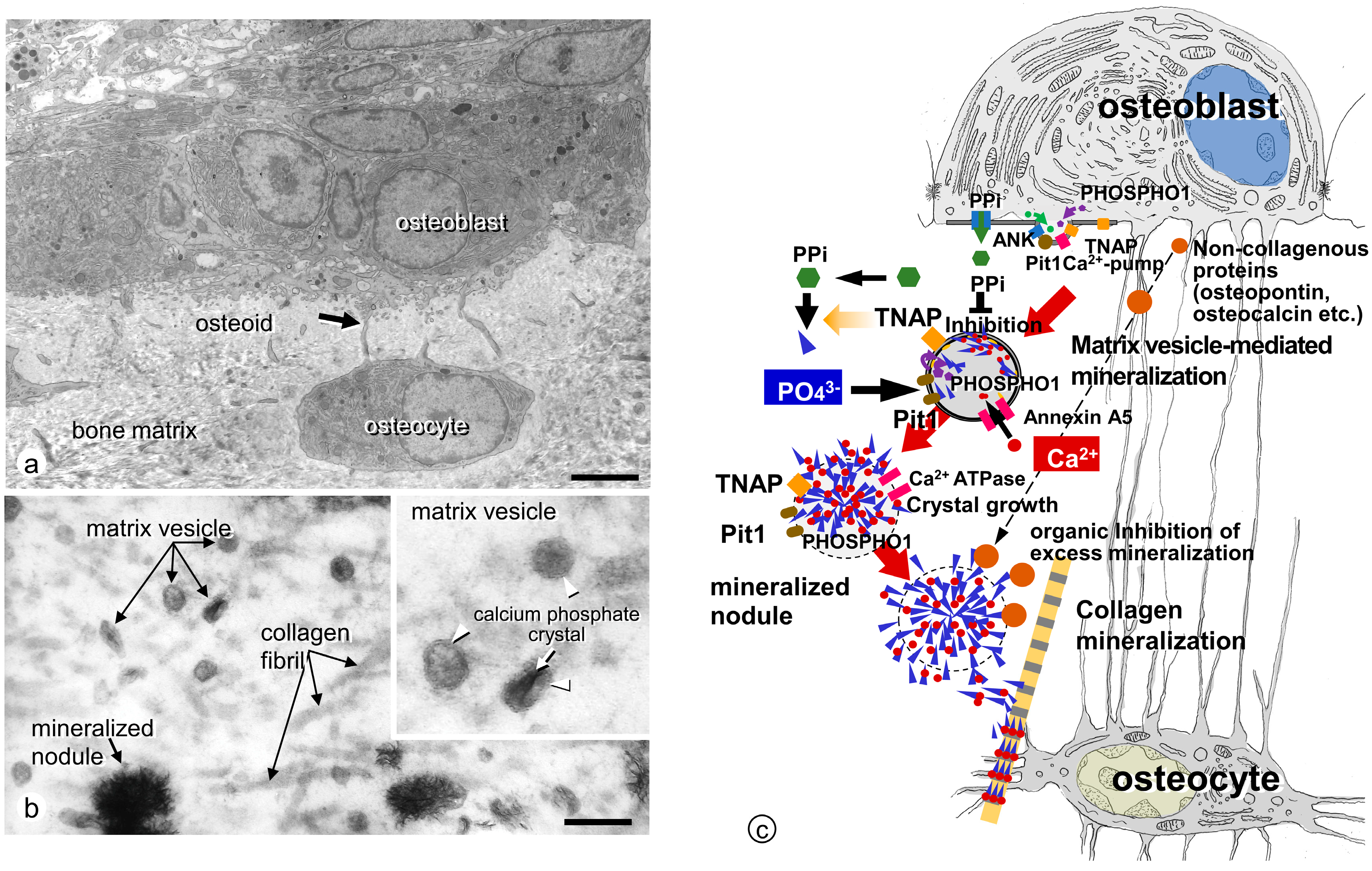
3. Ultrastructural Aspects of Matrix Vesicle-Mediated Mineralization in Bone
3.1. Formation of Crystalline Calcium Phosphates in Matrix Vesicles
3.2. Mineralized Nodules Develop from Matrix Vesicles
3.3. Enzymes and Membrane Transporter Necessary for Matrix Vesicle-Mediated Mineralization in Bone
3.3.1. TNAP
3.3.2. ENPP1
3.3.3. ANK
3.3.4. PHOSPHO1
3.3.5. Annexins
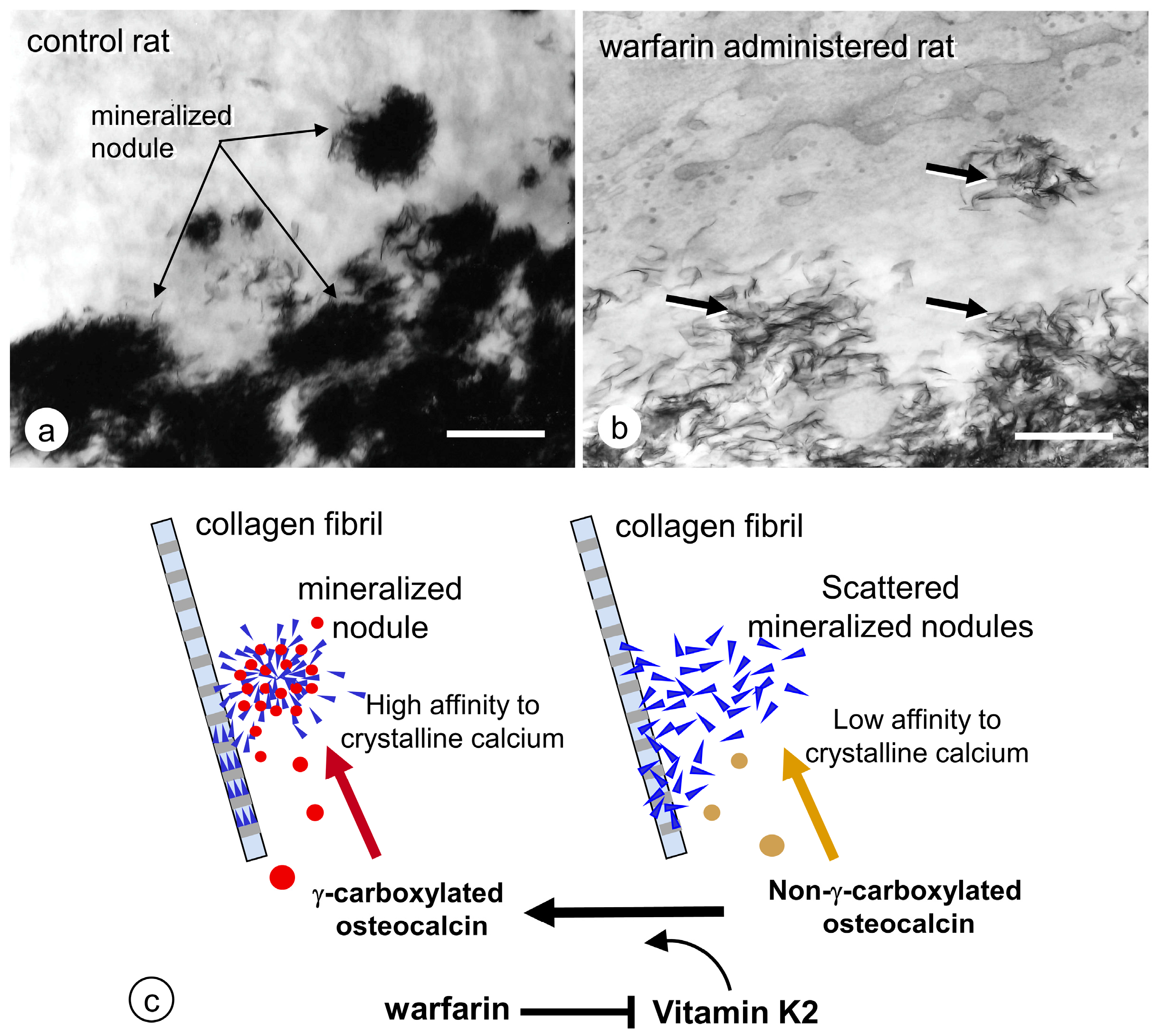
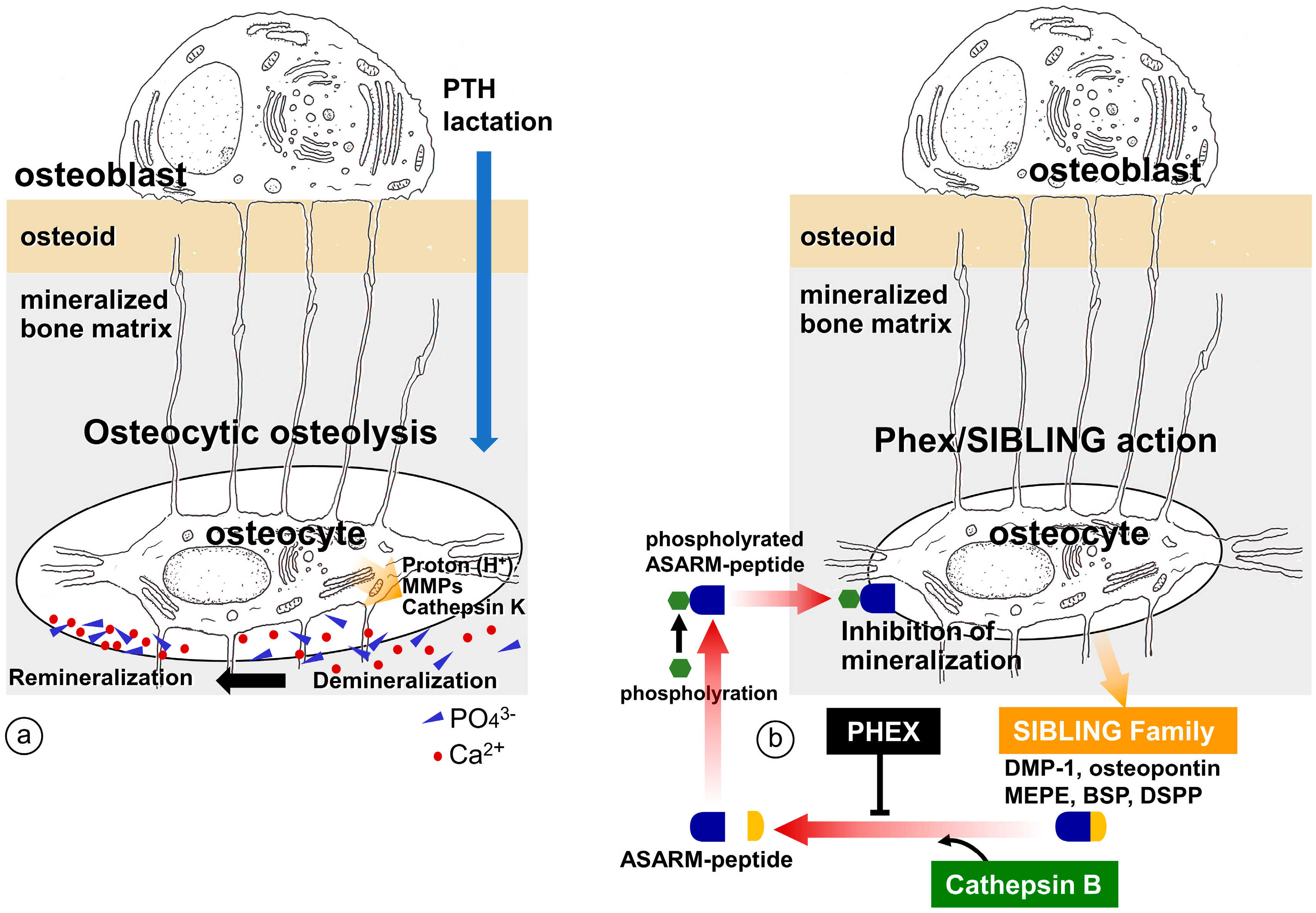
4. Regulation of Bone Mineralization by Osteocyte
4.1. Erosion of Bone Minerals in the Vicinity of Osteocytes
4.2. Regulation of Mineralization by Mediating SIBLING Family
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Amizuka, N.; Hasegawa, T.; Oda, K.; Freitas, P.H.L.; Hoshi, K.; Li, M.; Ozawa, H. Histology of epiphyseal cartilage calcification and endochondral ossification. Front. Biosci. 2012, 4, 2085–2100. [Google Scholar] [CrossRef]
- Ali, S.Y.; Sajdera, S.W.; Anderson, H.C. Isolation and characterization of calcifying matrix vesicles from epiphyseal cartilage. Proc. Natl. Acad. Sci. USA 1970, 67, 1513–1520. [Google Scholar] [CrossRef]
- Anderson, H.C. Vesicles associated with calcification in the matrix of epiphyseal cartilage. J. Cell Biol. 1969, 41, 59–72. [Google Scholar] [CrossRef]
- Bonucci, E. Fine structure of early cartilage calcification. J. Ultrastruct. Res. 1967, 20, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Bonucci, E. Fine structure and histochemistry of “calcifying globules” in epiphyseal cartilage. Z. Zellforsch. Mikrosk. Anat. 1970, 103, 192–217. [Google Scholar] [CrossRef]
- Ozawa, H.; Yamada, M.; Yajima, T. The ultrastructural and cytochemical aspects of matrix vesicles and calcification processes. In Formation and Calcification of Hard Tissues; Talmage, R.V., Ozawa, H., Eds.; Shakai Hoken Pub: Tokyo, Japan, 1978; pp. 9–57. [Google Scholar]
- Ozawa, H.; Yamada, M.; Yamamoto, T. Ultrastructural observations on the location of lead and calcium in the mineralizing dentine of rat incisor. In Matrix Vesicles; Ascenzi, A., Bonucci, E., de Bernard, B., Eds.; Wiching Editore srl: Milano, Italy, 1981; pp. 179–187. [Google Scholar]
- Wuthier, R.E. Lipid composition of isolated epiphyseal cartilage cells, membranes and matrix vesicles. Biochim. Biophys. Acta 1975, 409, 128–143. [Google Scholar] [CrossRef] [PubMed]
- Ohba, S. Hedgehog Signaling in Skeletal Development: Roles of Indian Hedgehog and the Mode of Its Action. Int. J. Mol. Sci. 2020, 21, 6665. [Google Scholar] [CrossRef] [PubMed]
- Tavormina, P.L.; Shiang, R.; Thompson, L.M.; Zhu, Y.Z.; Wilkin, D.J.; Lachman, R.S.; Wilcox, W.R.; Rimoin, D.L.; Cohn, D.H.; Wasmuth, J.J. Thanatophoric dysplasia (types I and II) caused by distinct mutations in fibroblast growth factor receptor 3. Nat. Genet. 1995, 9, 321–328. [Google Scholar] [CrossRef] [PubMed]
- Su, W.C.; Kitagawa, M.; Xue, N.; Xie, B.; Garofalo, S.; Cho, J.; Deng, C.; Horton, W.A.; Fu, X.Y. Activation of Stat1 by mutant fibroblast growth-factor receptor in thanatophoric dysplasia type II dwarfism. Nature 1997, 386, 288–292. [Google Scholar] [CrossRef]
- Peters, K.; Ornitz, D.; Werner, S.; Williams, L. Unique expression pattern of the FGF receptor 3 gene during mouse organogenesis. Dev. Biol. 1993, 155, 423–430. [Google Scholar] [CrossRef]
- Deng, C.; Wynshaw-Boris, A.; Zhou, F.; Kuo, A.; Leder, P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell 1996, 84, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Amizuka, N.; Davidson, D.; Liu, H.; Valverde-Franco, G.; Chai, S.; Maeda, T.; Ozawa, H.; Hammond, V.; Ornitz, D.M.; Goltzman, D.; et al. Signalling by fibroblast growth factor receptor 3 and parathyroid hormone-related peptide coordinate cartilage and bone development. Bone 2004, 34, 13–25. [Google Scholar] [CrossRef]
- Greenspan, J.S.; Blackwood, H.J. Histochemical studies of chondrocyte function in the cartilage of the mandibular codyle of the rat. J. Anat. 1966, 100, 615–626. [Google Scholar] [PubMed]
- Ikeda, T.; Nomura, S.; Yamaguchi, A.; Suda, T.; Yoshiki, S. In situ hybridization of bone matrix proteins in undecalcified adult rat bone sections. J. Histochem. Cytochem. 1992, 40, 1079–1088. [Google Scholar] [CrossRef]
- Oshima, O.; Leboy, P.S.; McDonald, S.A.; Tuan, R.S.; Shapiro, I.M. Developmental expression of genes in chick growth cartilage detected by in situ hybridization. Calcif. Tissue Int. 1989, 45, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Poole, A.R.; Pidoux, I.; Rosenberg, L. Role of proteoglycans in endochondral ossification: Immunofluorescent localization of link protein and proteoglycan monomer in bovine fetal epiphyseal growth plate. J. Cell Biol. 1982, 92, 249–260. [Google Scholar] [CrossRef]
- Schmid, T.M.; Linsenmayer, T.F. Immunohistochemical localization of short chain cartilage collagen (type X) in avian tissues. J. Cell Biol. 1985, 100, 598–605. [Google Scholar] [CrossRef]
- Gerber, H.P.; Vu, T.H.; Ryan, A.M.; Kowalski, J.; Werb, Z.; Ferrara, N. VEGF couples hypertrophic cartilage remodeling, ossification and angiogenesis during endochondral bone formation. Nat. Med. 1999, 5, 623–628. [Google Scholar] [CrossRef]
- Tsuchiya, E.; Hasegawa, T.; Hongo, H.; Yamamoto, T.; Abe, M.; Yoshida, T.; Zhao, S.; Tsuboi, K.; Udagawa, N.; Freitas, P.H.L.; et al. Histochemical assessment on the cellular interplay of vascular endothelial cells and septoclasts during endochondral ossification in mice. Microscopy 2021, 70, 201–214. [Google Scholar] [CrossRef]
- Kojima, T.; Hasegawa, T.; Freitas, P.H.L.; Yamamoto, T.; Sasaki, M.; Horiuchi, K.; Hongo, H.; Yamada, T.; Sakagami, N.; Saito, N.; et al. Histochemical aspects of the vascular invasion at the erosion zone of the epiphyseal cartilage in MMP-9-deficient mice. Biomed. Res. 2013, 34, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Vu, T.H.; Shipley, J.M.; Bergers, G.; Berger, J.E.; Helms, J.A.; Hanahan, D.; Shapiro, S.D.; Senior, R.M.; Werb, Z. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell 1998, 93, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Engsig, M.T.; Chen, Q.J.; Vu, T.H.; Pedersen, A.C.; Therkidsen, B.; Lund, L.R.; Henriksen, K.; Lenhard, T.; Foged, N.T.; Werb, Z.; et al. Matrix metalloproteinase 9 and vascular endothelial growth factor are essential for osteoclast recruitment into developing long bones. J. Cell Biol. 2000, 151, 879–889. [Google Scholar] [CrossRef]
- Marks, S.C., Jr.; Odgren, P.R. The structure and development of the skeleton. In Principles of Bone Biology; Bilezikian, J.P., Raisz, L.G., Rodan, G.A., Eds.; Academic Press: New York, NY, USA, 2002; pp. 3–15. [Google Scholar]
- Nakamura, H.; Ozawa, H. Ultrastructural, enzyme-, lectin, and immunohistochemical studies of the erosion zone in rat tibiae. J. Bone Miner. Res. 1996, 11, 1158–1164. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.R.; Lamplugh, L.; Shepard, N.L.; Mort, J.S. The septoclast, a cathepsin B-rich cell involved in the resorption of growth plate cartilage. J. Histochem. Cytochem. 1995, 43, 525–536. [Google Scholar] [CrossRef]
- Gartland, A.; Mason-Savas, A.; Yang, M.; MacKay, C.A.; Birnbaum, M.J.; Odgren, P.R. Septoclast deficiency accompanies postnatal growth plate chondrodysplasia in the toothless (tl) osteopetrotic, colony-stimulating factor-1 (CSF-1)-deficient rat and is partially responsive to CSF-1 injections. Am. J. Pathol. 2009, 175, 2668–2675. [Google Scholar] [CrossRef]
- Bando, Y.; Yamamoto, M.; Sakiyama, K.; Inoue, K.; Takizawa, S.; Owada, Y.; Iseki, S.; Kondo, H.; Amano, O. Expression of epidermal fatty acid binding protein (E-FABP) in septoclasts in the growth plate cartilage of mice. J. Mol. Histol. 2014, 45, 507–518. [Google Scholar] [CrossRef]
- Bando, Y.; Yamamoto, M.; Sakiyama, K.; Sakashita, H.; Taira, F.; Miyake, G.; Iseki, S.; Owada, Y.; Amano, O. Retinoic acid regulates cell-shape and -death of E-FABP (FABP5)-immunoreactive septoclasts in the growth plate cartilage of mice. Histochem. Cell Biol. 2017, 148, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Weiner, S. Organization of extracellularly mineralized tissues: A comparative study of biological crystal growth. CRC Crit. Rev. Biochem. 1986, 20, 365–408. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, H. Ultrastructural Concepts on Biological Calcification; Focused on Matrix Vesicles. J. Oral Biosci. 1985, 27, 751–774. [Google Scholar]
- Bosky, A.L.; Maresca, M.; Ullrich, W.; Doty, S.B.; Butler, W.T.; Prince, C.W. Osteopontin-hydroxyapatite interactions in vitro: Inhibition of hydroxyapatite formation and growth in a gelatin-gel. Bone Miner. 1993, 22, 147–159. [Google Scholar] [CrossRef]
- Hunter, G.K.; Hauschka, P.V.; Poole, A.R.; Rosenberg, L.C.; Goldberg, H.A. Nucleation and inhibition of hydroxyapatite formation by mineralized tissue proteins. Biochem. J. 1996, 317, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Mark, M.P.; Butler, W.T.; Prince, C.W.; Finkleman, R.D.; Ruch, J.V. Developmental expression of 44-kDa phosphoprotein (osteopontin) and bone-carboxyglutamic acid (Gla)-containing protein (osteocalcin) in calcifying tissues of rat. Differentiation 1988, 37, 123–136. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.G.; Pauli, R.M.; Wilson, K.M. Maternal and fetal sequelae of anti-coagulation during pregnancy. Am. J. Med. 1980, 68, 122–140. [Google Scholar] [CrossRef]
- Hauschka, P.V.; Lian, J.B.; Gallop, P.M. Direct identification of the calcium-binding amino acid, gamma-carboxyglutamate, in mineralized tissue. Proc. Natl. Acad. Sci. USA 1975, 72, 3925–3929. [Google Scholar] [CrossRef]
- Price, P.A.; Otsuka, A.A.; Poser, J.W.; Kristaponis, J.; Raman, N. Characterization of a gamma-carboxyglutamic acid-containing protein from bone. Proc. Natl. Acad. Sci. USA 1976, 73, 1447–1451. [Google Scholar] [CrossRef]
- Amizuka, N.; Li, M.; Hara, K.; Kobayashi, M.; Freitas, P.H.L.; Ubaidus, S.; Oda, K.; Akiyama, Y. Warfarin administration disrupts the assembly of mineralized nodules in the osteoid. J. Electron. Microsc. 2009, 58, 55–65. [Google Scholar] [CrossRef]
- Azuma, K.; Shiba, S.; Hasegawa, T.; Ikeda, K.; Urano, T.; Horie-Inoue, K.; Ouchi, Y.; Amizuka, N.; Inoue, S. Osteoblast-specific γ-glutamyl carboxylase-deficient mice display enhanced bone formation with aberrant mineralization. J. Bone Miner. Res. 2015, 30, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, T.; Hongo, H.; Yamamoto, T.; Abe, M.; Yoshino, H.; Haraguchi-Kitakamae, M.; Ishizu, H.; Shimizu, T.; Iwasaki, N.; Amizuka, N. Matrix vesicle-mediated mineralization and osteocytic regulation of bone mineralization. Int. J. Mol. Sci. 2022, 23, 9941. [Google Scholar] [CrossRef]
- Hasegawa, T. Ultrastructure and biological function of matrix vesicles in bone mineralization. Histochem. Cell Biol. 2018, 149, 289–304. [Google Scholar] [CrossRef] [PubMed]
- de Bernard, B.; Bianco, P.; Bonucci, E.; Costantini, M.; Lunazzi, G.C.; Martinuzzi, P.; Modricky, C.; Moro, L.; Panfili, E.; Pollesello, P. Biochemical and immunohistochemical evidence that in cartilage an alkaline phosphatase is a Ca2+-binding glycoprotein. J. Cell Biol. 1986, 103, 1615–1623. [Google Scholar] [CrossRef]
- Matsuzawa, T.; Anderson, H.C. Phosphatases of epiphyseal cartilage studied by electron microscopic cytochemical methods. J. Histochem. Cytochem. 1971, 19, 801–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, M. Ultrastractural and cytochemical studies on the calcification of the tendon-bone joint. Arch. Histol. Jap. 1976, 39, 347–378. [Google Scholar] [CrossRef]
- Hoshi, K.; Ejiri, S.; Ozawa, H. Localizational alterations of calcium, phosphorus, and calcification-related organics such as proteoglycans and alkaline phosphatase during bone calcification. J. Bone Miner. Res. 2001, 16, 289–298. [Google Scholar] [CrossRef]
- Schmitz, J.P.; Schwartz, Z.; Sylvia, V.L.; Dean, D.D.; Calderon, F.; Boyan, B.D. Vitamin D3 regulation of stromelysin-1 (MMP-3) in chondrocyte cultures is mediated by protein kinase C. J. Cell Physiol. 1996, 168, 570–579. [Google Scholar] [CrossRef]
- Fleish, H.; Neuman, W.F. Mechanisms of calcification: Role of collagen, polyphosphates, and phosphatase. Am. J. Physiol. 1961, 200, 1296–1300. [Google Scholar] [CrossRef] [PubMed]
- Fleish, H.; Neuman, W. The role of phosphatase and polyphosphates in calcification of collagen. Helv. Physiol. Pharmacol. Acta 1961, 19, C17–C18. [Google Scholar] [CrossRef] [PubMed]
- Takano, Y.; Ozawa, H.; Crenshaw, M.A. Ca-ATPase and ALPase activities at the initial calcification sites of dentine and enamel in the rat incisor. Cell Tissue Res. 1986, 243, 91–99. [Google Scholar] [CrossRef]
- Terkeltaub, R.; Rosenbach, M.; Fong, F.; Goding, J. Causal link between nucleotide pyrophosphohydrolase overactivity and increased intracellular inorganic pyrophosphate generation demonstrated by transfection of cultured fibroblasts and osteoblasts with plasma cell membrane glycoprotein-1. Arthritis Rheum. 1994, 37, 934–941. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.; Vaingankar, S.; Chen, Y.; Moffa, A.; Goldring, M.B.; Sano, K.; Jin-Hua, P.; Sali, A.; Goding, J.; Terkeltaub, R. Differential mechanisms of inorganic pyrophosphate production by plasma cell membrane glycoprotein-1 and B10 in chondrocytes. Arthritis Rheum. 1999, 42, 1986–1997. [Google Scholar] [CrossRef]
- Johnson, K.; Moffa, A.; Chen, Y.; Pritzker, K.; Goding, J.; Terkeltaub, R. Matrix vesicle plasma membrane glycoprotein-1 regulates mineralization by murine osteoblastic MC3T3 cells. J. Bone Miner. Res. 1999, 14, 883–892. [Google Scholar] [CrossRef]
- Ho, A.M.; Johnson, M.D.; Kingsley, D.M. Role of the mouse ank gene in control of tissue calcification and arthritis. Science 2000, 289, 265–270. [Google Scholar] [CrossRef]
- Szeri, F.; Niaziorimi, F.; Donnelly, S.; Fariha, N.; Tertyshnaia, M.; Patel, D.; Lundkvist, S.; van de Wetering, K. The Mineralization Regulator ANKH Mediates Cellular Efflux of ATP, Not Pyrophosphate. J. Bone Miner. Res. 2022, 37, 1024–1031. [Google Scholar] [CrossRef]
- Houston, B.; Stewart, A.J.; Farquharson, C. PHOSPHO1-A novel phosphatase specifically expressed at sites of mineralisation in bone and cartilage. Bone 2004, 34, 629–637. [Google Scholar] [CrossRef]
- Roberts, S.; Narisawa, S.; Harmey, D.; Millán, J.L.; Farquharson, C. Functional involvement of PHOSPHO1 in matrix vesicle–mediated skeletal mineralization. J. Bone. Miner. Res. 2007, 22, 617–627. [Google Scholar] [CrossRef]
- Ciancaglini, P.; Yadav, M.C.; Simão, A.M.; Narisawa, S.; Pizauro, J.M.; Farquharson, C.; Hoylaerts, M.F.; Millán, J.L. Kinetic analysis of substrate utilization by native and TNAP-, NPP1-, or PHOSPHO1-deficient matrix vesicles. J. Bone. Miner. Res. 2010, 25, 716–723. [Google Scholar] [CrossRef]
- Huesa, C.; Yadav, M.C.; Finnilä, M.A.; Goodyear, S.R.; Robins, S.P.; Tanner, K.E.; Aspden, R.M.; Millán, J.L.; Farquharson, C. PHOSPHO1 is essential for mechanically competent mineralization and the avoidance of spontaneous fractures. Bone 2011, 48, 1066–1074. [Google Scholar] [CrossRef] [PubMed]
- Boyde, A.; Staines, K.A.; Javaheri, B.; Millan, J.L.; Pitsillides, A.A.; Farquharson, C. A distinctive patchy osteomalacia characterizes PHOSPHO1-deficient mice. J. Anat. 2017, 231, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Yadav, M.C.; Simão, A.M.S.; Narisawa, S.; Huesa, C.; McKee, M.D.; Farquharson, C.; Millán, J.L. Loss of skeletal mineralization by the simultaneous ablation of PHOSPHO1 and alkaline phosphatase function—A unified model of the mechanisms of initiation of skeletal calcification. J. Bone Miner. Res. 2011, 26, 286–297. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, T.; Nah, H.D.; Shapiro, I.M.; Pacifici, M. Regulated production of mineralization-competent matrix vesicles in hypertrophic chondrocytes. J. Cell Biol. 1997, 137, 1149–1160. [Google Scholar] [CrossRef]
- Nakano, Y.; Beertsen, W.; van den Bos, T.; Kawamoto, T.; Oda, K.; Takano, Y. Site-specific localization of two distinct phosphatases along the osteoblast plasma membrane: Tissue non-specific alkaline phosphatase and plasma membrane calcium ATPase. Bone 2004, 35, 1077–1085. [Google Scholar] [CrossRef]
- Narisawa, S.; Fröhlander, N.; Millian, J.L. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev. Dyn. 1997, 208, 432–446. [Google Scholar] [CrossRef]
- Waymire, K.G.; Mahuren, J.D.; Jaje, J.M.; Guilarte, T.R.; Coburn, S.P.; MacGregor, G.R. Mice lacking tissue non-specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat. Genet. 1995, 11, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Tesch, W.; Vandenbos, T.; Roschgr, P.; Fratzl-Zelman, N.; Klaushofer, K.; Beertsen, W.; Fratzl, P. Orientation of mineral crystallites and mineral density during skeletal development in mice deficient in tissue nonspecific alkaline phosphatase. J. Bone Miner. Res. 2003, 18, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, C.; Jakob, F.; Seefried, L.; Mentrup, B.; Graser, S.; Plotkin, H.; Girschick, H.J.; Liese, J. Recombinant enzyme replacement therapy in hypophosphatasia. Subcell. Biochem. 2015, 76, 323–341. [Google Scholar] [PubMed]
- Whyte, M.P.; Rockman-Greenberg, C.; Ozono, K.; Riese, R.; Moseley, S.; Melian, A.; Thompson, D.D.; Bishop, N.; Hofmann, C. Asfotase alfa treatment improves survival for perinatal and infantile hypophosphatasia. J. Clin. Endocrinol. Metab. 2016, 101, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Kato, K.; Nishimasu, H.; Okudaira, S.; Mihara, E.; Ishitani, R.; Takagi, J.; Aoki, J.; Nureki, O. Crystal structure of Enpp1, an extracellular glycoprotein involved in bone mineralization and insulin signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 16876–16881. [Google Scholar] [CrossRef]
- Andrilli, L.H.S.; Sebinelli, H.G.; Favarin, B.Z.; Cruz, M.A.E.; Ramos, A.P.; Bolean, M.; Millán, J.L.; Bottini, M.; Ciancaglini, P. NPP1 and TNAP hydrolyze ATP synergistically during biomineralization. Purinergic. Signal. 2022; in press. [Google Scholar] [CrossRef]
- Rutsch, F.; Vaingankar, S.; Johnson, K.; Goldfine, I.; Maddux, B.; Schauerte, P.; Kalhoff, H.; Sano, K.; Boisvert, W.A.; Superti-Furga, A.; et al. PC-1 Nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am. J. Pathol. 2001, 158, 543–554. [Google Scholar] [CrossRef]
- Rutsch, F.; Ruf, N.; Vaingankar, S.; Toliat, M.R.; Suk, A.; Höhne, W.; Schauer, G.; Lehmann, M.; Roscioli, T.; Schnabel, D.; et al. Mutations in ENPP1 are associated with ‘idiopathic’ infantile arterial calcification. Nat. Genet. 2003, 34, 379–381. [Google Scholar] [CrossRef]
- Yamamoto, T.; Hasegawa, T.; Mae, T.; Hongo, H.; Yamamoto, T.; Abe, M.; Nasoori, A.; Morimoto, Y.; Maruoka, H.; Kubota, K.; et al. Comparative immunolocalization of tissue nonspecific alkaline phosphatase and ectonucleotide pyrophosphatase/phosphodiesterase 1 in murine bone. J. Oral Biosci. 2021, 63, 259–264. [Google Scholar] [CrossRef]
- Okawa, A.; Nakamura, I.; Goto, S.; Moriya, H.; Nakamura, Y.; Ikegawa, S. Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat. Genet. 1998, 19, 271–273. [Google Scholar] [CrossRef]
- Johnson, K.; Pritzker, K.; Goding, J.; Terkeltaub, R. The nucleoside triphosphate pyrophosphohydrolase isozyme PC-1 directly promotes cartilage calcification through chondrocyte apoptosis and increased calcium precipitation by mineralizing vesicles. J. Rheumatol. 2001, 28, 2681–2691. [Google Scholar] [PubMed]
- Johnson, K.; Hashimoto, S.; Lotz, M.; Pritzker, K.; Goding, J.; Terkeltaub, R. Up-regulated expression of the phosphodiesterase nucleotide pyrophosphatase family member PC-1 is a marker and pathogenic factor for knee meniscal cartilage matrix calcification. Arthritis Rheum. 2001, 44, 1071–1081. [Google Scholar] [CrossRef]
- Johnson, K.; Goding, J.; Van Etten, D.; Sali, A.; Hu, S.I.; Farley, D.; Krug, H.; Hessle, L.; Millán, J.L.; Terkeltaub, R. Linked deficiencies in extracellular PP(i) and osteopontin mediate pathologic calcification associated with defective PC-1 and ANK expression. J. Bone Miner. Res. 2003, 18, 994–1004. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, N.C.; Zhu, D.; Milne, E.M.; van ‘t Hof, R.; Martin, A.; Darryl Quarles, L.; Millán, J.L.; Farquharson, C.; MacRae, V.E. Altered bone development and an increase in FGF-23 expression in Enpp1(-/-) mice. PLoS ONE 2012, 7, e32177. [Google Scholar] [CrossRef]
- Nam, H.K.; Emmanouil, E.; Hatch, N.E. Deletion of the pyrophosphate generating enzyme ENPP1 rescues craniofacial abnormalities in the TNAP-/- mouse model of Hypophosphatasia and reveals FGF23 as a marker of phenotype severity. Front. Dent. Med. 2022, 3, 846962. [Google Scholar] [CrossRef]
- Bergwitz, C.; Juppner, H. FGF23 and syndromes of abnormal renal phosphate handling. Adv. Exp. Med. Biol. 2012, 728, 41–64. [Google Scholar]
- Ho, B.B.; Bergwitz, C. FGF23 signalling and physiology. J. Mol. Endocrinol. 2021, 66, R23–R32. [Google Scholar] [CrossRef]
- Lederer, E. Regulation of serum phosphate. J. Physiol. 2014, 592, 3985–3995. [Google Scholar] [CrossRef]
- Abhishek, A.; Doherty, M. Pathophysiology of articular chondrocalcinosis--role of ANKH. Nat. Rev. Rheumatol. 2011, 7, 96–104. [Google Scholar] [PubMed]
- Stewart, A.J.; Leong, D.T.K.; Farquharson, C. PLA 2 and ENPP6 may act in concert to generate phosphocholine from the matrix vesicle membrane during skeletal mineralization. FASEB J. 2018, 32, 20–25. [Google Scholar] [CrossRef]
- Cao, X.; Genge, B.R.; Wu, L.N.; Buzzi, W.R.; Showman, R.M.; Wuthier, R.E. Characterization, cloning and expression of the 67-kDA annexin from chicken growth plate cartilage matrix vesicles. Biochem. Biophys. Res. Commun. 1993, 197, 556–561. [Google Scholar] [CrossRef]
- Kirsch, T.; Nah, H.D.; Demuth, D.R.; Harrison, G.; Golub, E.E.; Adams, S.L.; Pacifici, M. Annexin V-mediated calcium flux across membranes is dependent on the lipid composition: Implications for cartilage mineralization. Biochemistry 1997, 36, 3359–3367. [Google Scholar] [CrossRef] [PubMed]
- Kirsch, T.; Claassen, H. Matrix vesicles mediate mineralization of human thyroid cartilage. Calcif. Tissue Int. 2000, 66, 292–297. [Google Scholar] [CrossRef] [PubMed]
- Majeska, R.J.; Holwerda, D.L.; Wuthier, R.E. Localization of phosphatidylserine in isolated chick epiphyseal cartilage matrix vesicles with trinitrobenzenesulfonate. Calcif. Tissue Int. 1979, 27, 41–46. [Google Scholar] [CrossRef]
- Taylor, M.G.; Simkiss, K.; Simmons, J.; Wu, L.N.; Wuthier, R.E. Structural studies of a phosphatidyl serine-amorphous calcium phosphate complex. Cell. Mol. Life Sci. 1998, 54, 196–202. [Google Scholar] [CrossRef]
- Hasegawa, T.; Yamamoto, T.; Hongo, H.; Qiu, Z.; Abe, M.; Kanesaki, T.; Tanaka, K.; Endo, T.; Freitas, P.H.L.; Li, M. Three-dimensional ultrastructure of osteocytes assessed by focused ion beam-scanning electron microscopy (FIB-SEM). Histochem. Cell Biol. 2018, 149, 423–432. [Google Scholar] [CrossRef] [PubMed]
- Bélanger, L.F. Osteocytic osteolysis. Calcif. Tissue. Res. 1969, 4, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bai, L.; Collins, J.F.; Xu, H.; Xu, L.; Ghishan, F.K. Molecular cloning of a murine type III sodium-dependent phosphate cotransporter (Pit-2) gene promoter. Biochim. Biophys. Acta 2001, 1522, 42–45. [Google Scholar] [CrossRef]
- Qing, H.; Bonewald, L.F. Osteocyte remodeling of perilacunar and pericanalicular matrix. Int. J. Oral Sci. 2009, 1, 59–65. [Google Scholar] [CrossRef]
- Teit, A.; Zallone, A. Do osteocytes contribute to bone mikneral homeostasis? Osteocytic osteolysis revisited. Bone 2009, 44, 11–16. [Google Scholar] [CrossRef]
- Bonewald, L.F. The amazing osteocyte. J. Bone Miner. Res. 2011, 26, 229–238. [Google Scholar] [CrossRef]
- Qing, H.; Ardeshirpour, L.; Pajevic, P.D.; Dusevich, V.; Jähn, K.; Kato, S.; Wysolmerski, J.; Bonewald, L.F. Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. J. Bone Miner. Res. 2012, 27, 1018–1029. [Google Scholar] [CrossRef]
- Whysolmerski, J.J. Osteocytic osteolysis: Time for a second look? BoneKEy Rep. 2012, 1, 229. [Google Scholar] [CrossRef]
- Whysolmerski, J.J. Osteocytes remove and replace perilacunar minewral during reproductive cycles. Bone 2013, 54, 230–236. [Google Scholar] [CrossRef]
- Sano, H.; Kikuta, J.; Furuya, M.; Kondo, N.; Endo, N.; Ishii, M. Intravital bone imaging by two-photon excitation microscopy to identify osteocytic osteolysis in vivo. Bone 2015, 74, 134–139. [Google Scholar] [CrossRef] [PubMed]
- Bonucci, E.; Gherardi, G.; Faraggiana, T. Bone changes in hemodialyzed uremic subjects. Comparative light and electron microscope investigations. Virchows Arch. A Pathol. Anat. Histol. 1976, 371, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Mosekilde, L.; Melsen, F. A tetracycline-based histomorphometric evaluation of bone resorption and bone turnover in hyperthyroidism and hyperparathyroidism. Acta Med. Scand. 1978, 204, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Jähn, K.; Kelkar, S.; Zhao, H.; Xie, Y.; Tiede-Lewis, L.M.; Dusevich, V.; Dallas, S.L.; Bonewald, L.F. Osteocytes acidify their microenvironment in response to PTHrP in vitro and in lactating mice in vivo. J. Bone Miner. Res. 2017, 32, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Rolvien, T.; Krause, M.; Jeschke, A.; Yorgan, T.; Püschel, K.; Schinke, T.; Busse, B.; Demay, M.B.; Amling, M. Vitamin D regulates osteocyte survival and perilacunar remodeling in human and murine bone. Bone 2017, 103, 78–87. [Google Scholar] [CrossRef]
- Kogawa, M.; Wijenayaka, A.R.; Ormsby, R.T.; Thomas, G.P.; Anderson, P.H.; Bonewald, L.F.; Findlay, D.M.; Atkins, G.J. Sclerostin regulates release of bone mineral by osteocytes by induction of carbonic anhydrase 2. J. Bone Miner. Res. 2013, 28, 2436–2448. [Google Scholar] [CrossRef]
- Nango, N.; Kubota, S.; Hasegawa, T.; Yashiro, W.; Momose, A.; Matsuo, K. Osteocyte-directed bone demineralization along canaliculi. Bone 2016, 84, 279–288. [Google Scholar] [CrossRef] [Green Version]
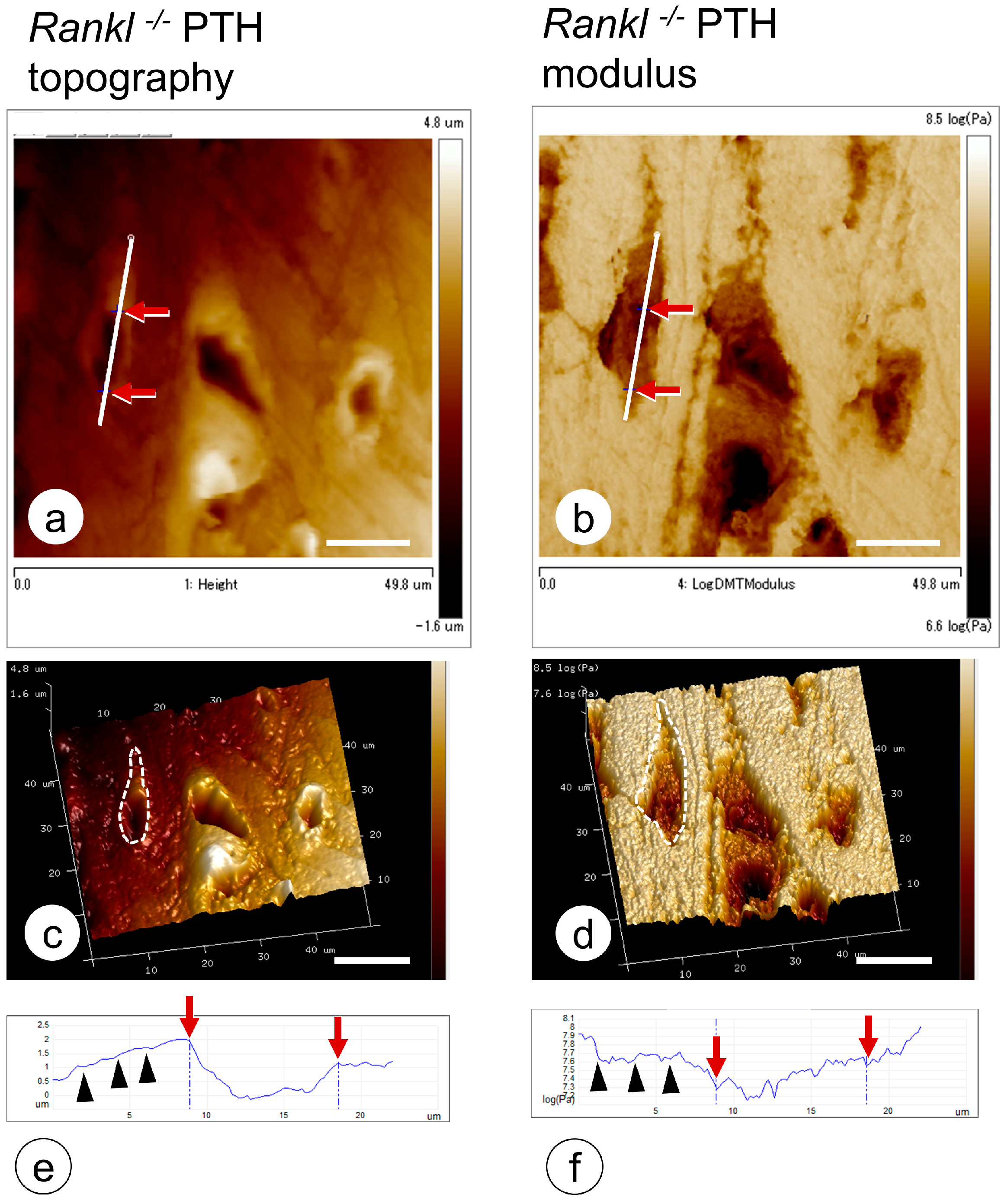
- Hongo, H.; Hasegawa, T.; Saito, M.; Tsuboi, K.; Yamamoto, T.; Sasaki, M.; Abe, M.; de Freitas, P.H.L.; Yurimoto, H.; Udagawa, N.; et al. Osteocytic osteolysis in PTH-treated wild-type and Rankl-/- mice examined by transmission electron microscopy, atomic force microscopy, and isotope microscopy. J. Histochem. Cytochem. 2020, 68, 651–668. [Google Scholar] [CrossRef] [PubMed]
- Jandl, N.M.; von Kroge, S.; Stürznickel, J.; Baranowsky, A.; Stockhausen, K.E.; Mushumba, H.; Beil, F.T.; Püschel, K.; Amling, M.; Rolvien, T. Large osteocyte lacunae in iliac crest infantile bone are not associated with impaired mineral distribution or signs of osteocytic osteolysis. Bone 2020, 135, 115324. [Google Scholar] [CrossRef] [PubMed]
- Misof, B.M.; Blouin, S.; Hofstaetter, J.G.; Roschger, P.; Zwerina, J.; Erben, R.G. No role of osteocytic osteolysis in the development and recovery of the bone phenotype induced by severe secondary hyperparathyroidism in vitamin D receptor deficient mice. Int. J. Mol. Sci. 2020, 21, 7989. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.A.; Kovacs, C.S. The puzzle of lactational bone physiology: Osteocytes masquerade as osteoclasts and osteoblasts. J. Clin. Investig. 2019, 129, 3041–3044. [Google Scholar] [CrossRef] [PubMed]
- Kaya, S.; Basta-Pljakic, J.; Seref-Ferlengez, Z.; Majeska, R.J.; Cardoso, L.; Bromage, T.G.; Zhang, Q.; Flach, C.R.; Mendelsohn, R.; Yakar, S.; et al. Lactation-induced changes in the volume of osteocyte lacunar-canalicular space alter mechanical properties in cortical bone tissue. J. Bone Miner. Res. 2017, 32, 688–697. [Google Scholar] [CrossRef]
- Emami, A.J.; Sebastian, A.; Lin, Y.Y.; Yee, C.S.; Osipov, B.; Loots, G.G.; Alliston, T.; Christiansen, B.A. Altered canalicular remodeling associated with femur fracture in mice. J. Orthop. Res. 2022, 40, 891–900. [Google Scholar] [CrossRef]
- Vahidi, G.; Rux, C.; Sherk, V.D.; Heveran, C.M. Lacunar-canalicular bone remodeling: Impacts on bone quality and tools for assessment. Bone 2021, 143, 115663. [Google Scholar]
- Sasaki, M.; Hasegawa, T.; Yamada, T.; Hongo, H.; de Freitas, P.H.; Suzuki, R.; Yamamoto, T.; Tabata, C.; Toyosawa, S.; Yamamoto, T.; et al. Altered distribution of bone matrix proteins and defective bone mineralization in klotho-deficient mice. Bone 2013, 57, 206–219. [Google Scholar] [CrossRef]
- Liu, S.; Rowe, P.S.; Vierthaler, L.; Zhou, J.; Quarles, L.D. Phosphorylated acidic serine-aspartate-rich MEPE-associated motif peptide from matrix extracellular phosphoglycoprotein inhibits phosphate regulating gene with homologies to endopeptidases on the X-chromosome enzyme activity. J. Endocrinol. 2007, 192, 261–267. [Google Scholar] [CrossRef]
- Staines, K.A.; MacRae, V.E.; Farquharson, C. The importance of the SIBLING family of proteins. J. Endocrinol. 2012, 214, 241–255. [Google Scholar] [CrossRef]
- Rowe, P.S.; de Zoysa, P.A.; Dong, R.; Wang, H.R.; White, K.E.; Econs, M.J.; Oudet, C.L. MEPE, a new gene expressed in bone marrow and tumors causing osteomalacia. Genomics 2000, 67, 54–68. [Google Scholar] [CrossRef]
- Rowe, P.S.; Kumagai, Y.; Gutierrez, G.; Garrett, I.R.; Blacher, R.; Rosen, D.; Cundy, J.; Navvab, S.; Chen, D.; Drezner, M.K.; et al. MEPE has the properties of an osteoblastic phosphatonin and minhibin. Bone 2004, 34, 303–319. [Google Scholar] [CrossRef] [PubMed]
- Rowe, P.S.; Garrett, I.R.; Schwarz, P.M.; Carnes, D.L.; Lafer, E.M.; Mundy, G.R.; Gutierrez, G.E. Surface plasmon resonance (SPR) confirms that MEPE binds to PHEX via the MEPE-ASARM motif: A model for impaired mineralization in X-linked rickets (HYP). Bone 2005, 36, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Addison, W.N.; Masica, D.L.; Gray, J.J.; McKee, M.D. Phosphorylation-dependent inhibition of mineralization by osteopontin ASARM peptides is regulated by PHEX cleavage. J. Bone Miner. Res. 2010, 25, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.Q.; Ward, L.M.; Liu, S.; Lu, Y.; Xie, Y.; Yuan, B.; Yu, X.; Rauch, F.; Davis, S.I.; Zhang, S.; et al. Loss of DMP1 causes rickets and osteomalacia and identifies a role for osteocytes in mineral metabolism. Nat. Genet. 2006, 38, 1310–1315. [Google Scholar] [CrossRef]
- Mäkitie, O.; Pereira, R.C.; Kaitila, I.; Turan, S.; Bastepe, M.; Laine, T.; Kröger, H.; Cole, W.G.; Jüppner, H. Long-term clinical outcome and carrier phenotype in autosomal recessive hypophosphatemia caused by a novel DMP1 mutation. J. Bone Miner. Res. 2010, 25, 2165–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasegawa, T.; Hongo, H.; Yamamoto, T.; Muneyama, T.; Miyamoto, Y.; Amizuka, N. Histological Assessment of Endochondral Ossification and Bone Mineralization. Endocrines 2023, 4, 66-81. https://doi.org/10.3390/endocrines4010006
Hasegawa T, Hongo H, Yamamoto T, Muneyama T, Miyamoto Y, Amizuka N. Histological Assessment of Endochondral Ossification and Bone Mineralization. Endocrines. 2023; 4(1):66-81. https://doi.org/10.3390/endocrines4010006
Chicago/Turabian StyleHasegawa, Tomoka, Hiromi Hongo, Tomomaya Yamamoto, Takafumi Muneyama, Yukina Miyamoto, and Norio Amizuka. 2023. "Histological Assessment of Endochondral Ossification and Bone Mineralization" Endocrines 4, no. 1: 66-81. https://doi.org/10.3390/endocrines4010006