1. Introduction
Severe Combined Immunodeficiency (SCID) is a clinical and immunological syndrome that arises from a variety of genetic defects that lead to an absence of lymphocyte development and function, the diagnosis of which constitutes a pediatric emergency. Affected children have extreme susceptibility to infections, which are almost always fatal in the first year of life without treatment [
1].
One of the common infections these patients suffer is caused by Pneumocystis jirovecii (PJ), which typically causes pneumonia. Pneumocystis jirovecii pneumonia (PJP) is a type of pneumonia originating from the fungus PJ, which is specific to humans [
2]. PJP antibodies can be serologically present in the absence of symptoms in healthy individuals suggesting that PJ is an opportunistic pathogen of ubiquitous distribution and low pathogenicity. Immunocompetent individuals with asymptomatic colonization have the potential to transmit the fungus to others including immunocompromised individuals [
3].
PJP typically appears as bilateral diffuse pulmonary infiltrates. Granulomatous PJP is an uncommon form of pneumocystis infection, occurring in only 3% to 5% of patients.
We report the case of a toddler affected by Severe Combined Immunodeficiency who presented a peculiar PJP characterized by 11 pulmonary calcified lesions that were identified at routine imaging performed prior to an allogeneic hematopoietic stem cell transplantation (HSCT).
2. Case Report
A 7-month-old boy was diagnosed with SCID following a PJP. His parents were not consanguineous and he had no relatives with a medical history compatible with congenital immunodeficiency. He had also failed to thrive in the past two months. SCID was diagnosed by the genetic analysis of the IL2RG that showed that the patient was heterozygous for the c.[74linsG] mutation that has not been reported in literature yet.
The child was admitted to the Burlo Garofolo children’s hospital for food refusal, restlessness, and worsening tachypnea without a cough. On physical examination, the infant exhibited a respiratory rate of 52 breaths/min with oxygen saturation of 86% on room air. The saturation improved to 94% upon receiving 4 L of oxygen via a face mask. His heart rate was 154 beats/min with poor peripheral perfusion. Pulmonary auscultation revealed crepitations with diminished breath sounds over both lung fields. The chest radiograph exhibited patchy and solid infiltrates spread over both lungs. Since no thymus shadow was observable in the thorax radiography, congenital immunodeficiency was suspected. The child started the therapy with broad-spectrum antibiotics and acyclovir. He also underwent non-invasive ventilation, using high-flow oxygen therapy with an AIRVO device because of increasing oxygen requirement.
After a few hours of hospitalization, the patient’s worsening respiratory distress required intubation with mechanical ventilation. He was transferred to the pediatric intensive care unit (PICU) for further management. A computed tomography (CT) scan, performed immediately after the move, showed numerous perihilar and peripheral parenchymal lung lesions. Diagnosis of PJP was documented with polymerase chain reaction analysis that revealed Pneumocystis DNA from the broncho-alveolar lavage (BAL) fluid. Other bacterial, viral, and fungal causes have been excluded by culture and direct polymerase chain reaction (PCR) on both peripheral blood and BAL fluid. The boy was treated with sulphamethoxazole trimethoprim at a dose of 20 mg/kg/day of trimethoprim iv. The patient was extubated on the 21st day after PICU admission and continued intravenous sulphamethoxazole trimethoprim treatment for 31 days with a resolution of symptoms. He has since then been on antifungal, antiviral, and anti-pneumocystis prophylaxis, doing fairly well apart from occasional episodes of tachypnoea not ascribable to infectious or metabolic causes. Furthermore, he presented impaired renal function with serum creatinine >0.60 mg/dL (normal range for his age 0.18–0.35 mg/dL).
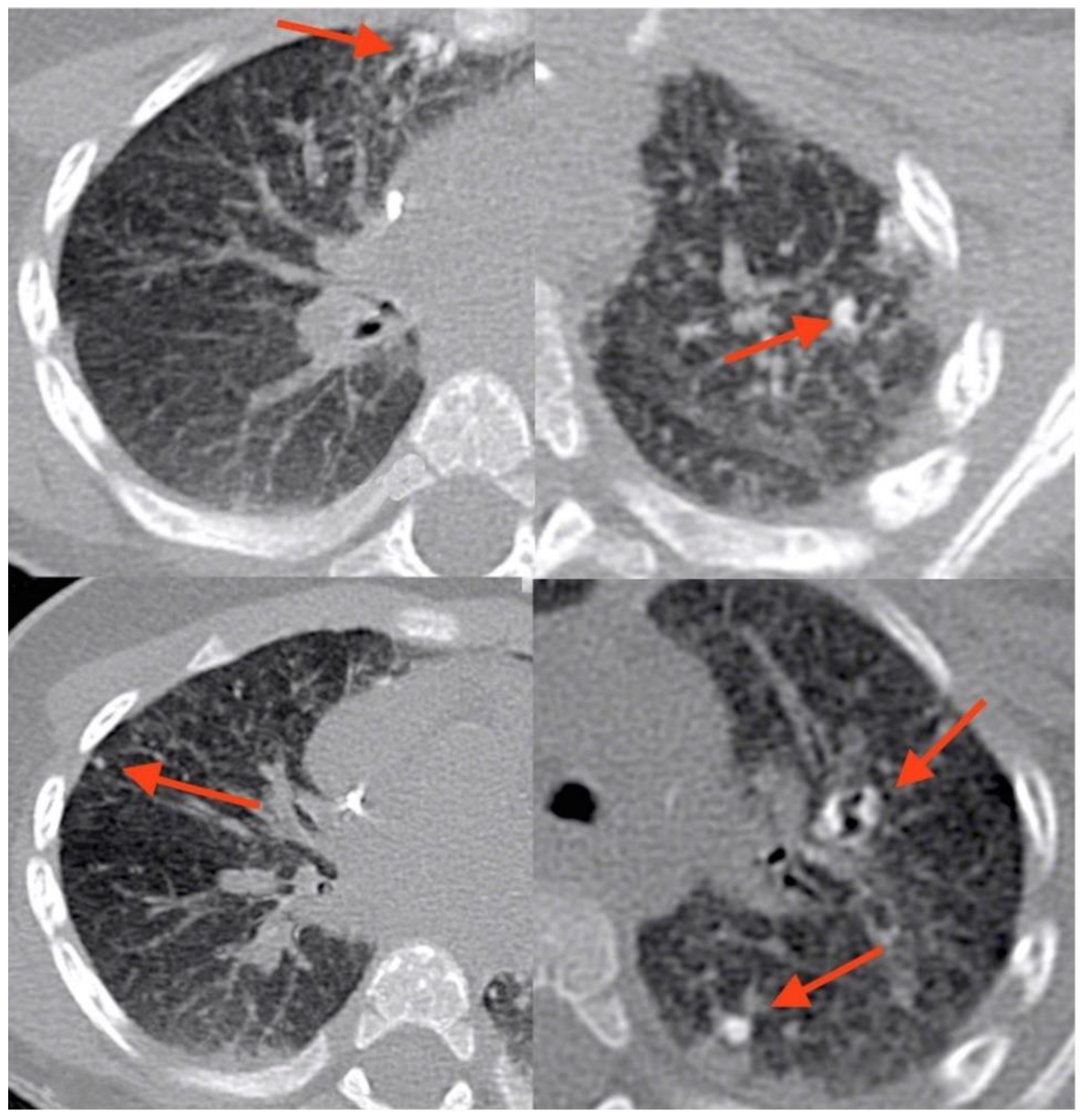
At age 9 months, prior to HSCT, he underwent a routine total-body magnetic resonance imaging (MRI). The examination was performed using an open bore 1,5T machine, Philips Ingenia. The sequences used for whole-body MRI are coronal T1 and T2 weighted and axial diffusion. A T2 HD axial sequence performed to study lungs showed significant alterations of the pulmonary parenchyma characterized by pulmonary consolidations and two cystic, thin-wall lesions. A CT (Philips Brilliance 40CT scan) of the lung was performed subsequently, showing the presence of 11 calcified granulomatous nodules. The calcifications were distributed perihilar and peripheral, both where previously consolidations were present, particularly the larger ones. (
Figure 1 and
Figure 2).
The boy underwent an allogeneic HSCT with a matched unrelated donor. The transplant was successful and the post-transplant period was complicated only by chronic proximal tubular acidosis. The possible genetic causes of proximal renal tubular acidosis (pRTA) were ruled out because no molecular genetic evidence of pRTA by directly sequencing the SLC4A4 gene was found. The most likely hypothesis is that the tubular acidosis was due to sulphamethoxazole trimethoprim treatment. The acidosis ameliorated significantly following the reduction of the drug schedule from daily therapeutic dosage to three times weekly prophylaxis and, after that, to one weekly prophylaxis.
3. Discussion
To the best of our knowledge, although PJP is a typical manifestation of SCID, granulomatous PJP with calcification has never been described in association with this condition. Cases have most commonly been described in human immunodeficiency virus (HIV)-infected patients with CD4+ counts <200 cells/mm
3, although the disease has also been described in adults with acquired immunodeficiency such as cancer. Calcification is exceptional [
4].
We hypothesized the reasons our patient developed such a peculiar presentation of the disease. After specifically asking the parents, we found out that the boy was receiving supplementation with Vitamin D (400 UI/die) throughout his first months of life and at the moment of infection presented Vitamin D levels of 74.3 mmol/L which is higher than average in healthy peers [
5]. On the other hand, it is very curious that our patient also presented chronic proximal tubular acidosis since several studies have underlined the possible connection between vitamin D levels and vascular calcification of chronic kidney disease [
6], and granulomatous PJP has been described in kidney transplant recipients with high Vitamin D levels [
7]. Vitamin D supplementation is considered a treatment-related factor contributing to the formation of overt vascular calcification in chronic kidney disease. It is therefore possible that the combination of underlying kidney disease and Vitamin D supplementation in the peculiar setting of PJP brought on the formation of rare calcified lesions.
X-linked Severe Combined Immunodeficiency (X-SCID) accounts for approximately half of all SCID cases and has an estimated incidence of 1:100,000 male births [
8]. It is caused by mutations in the interleukin (IL)-2 receptor gamma chain (IL2RG) gene and presents with absent or profoundly diminished peripheral T and NK cells and functionally defective B cells [
9,
10]. IL2RG is expressed by virtually all hematopoietic cells and is shared by the receptors for IL-2, 4, 7, 9, 15 and 21. The lack of normal IL-4, IL-7, IL-15 and IL-21 signaling explains the classical T
−B
+NK
− phenotype in X-SCID. IL-7 and IL-15 are required for T and NK cell development, respectively, and disturbed IL-4 and IL-21 signaling causes intrinsic B cell defect [
11]. Patients are highly susceptible to bacterial and viral infections showing various symptoms like chronic diarrhea, skin rashes, and the failure to thrive. Additionally, B-cell dysfunction and hypogammaglobulinemia are commonly found [
12]. The fact that our patient has an undescribed mutation of the IL2RG gene could relate to the unique clinical presentation of PJP.
4. Conclusions
BMT is often complicated by fungal infections, some of which may present as pulmonary granulomas or calcifications. It is, therefore, very important to be aware of any lesions caused by prior infections in these patients, to avoid dangerous misdiagnosis. This further enhances the importance of our observation, since no descriptions of calcified granulomas caused by PJ in patients with SCID have been reported in the literature.
We report a unique case of calcified granulomatous PJP in a toddler affected by SCID. Awareness of this rare yet possible presentation in patients with SCID is important given the potential clinical implications when managing a patient undergoing BMT and further enhances the importance of advanced radiologic imaging prior to BMT. MRI is a valid tool for the identification and follow-up of pulmonary lesions.
Author Contributions
Conceptualization, N.M. and F.Z.; writing—original draft preparation, N.G.; writing review and editing, N.M. and F.Z.; review, editing and supervision, N.G., N.M. and F.Z. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Written informed consent was obtained from the parents of the patient for publication of this case report and the accompanying images. A copy of the written consent is available for review by the editor of this journal.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Van der Burg, M.; Gennery, A.R. Educational paper. The expanding clinical and immunological spectrum of severe combined immunodeficiency. Eur. J. Pediatr. 2011, 170, 561–571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stringer, J.R. Pneumocystis. Int. J. Med. Microbiol. 2002, 292, 391–404. [Google Scholar] [CrossRef] [PubMed]
- Ponce, C.A.; Gallo, M.; Bustamante, R.; Vargas, S.L. Pneumocystis colonization is highly prevalent in the autopsied lungs of the general population. Clin. Infect. Dis. 2010, 50, 347–353. [Google Scholar] [CrossRef] [Green Version]
- Hartel, P.H.; Shilo, K.; Klassen-Fischer, M.; Neafie, R.C.; Ozbudak, I.H.; Galvin, J.R.; Franks, T.J. Granulomatous reaction to pneumocystis jirovecii: Clinicopathologic review of 20 cases. Am. J. Surg. Pathol. 2010, 34, 730–734. [Google Scholar] [CrossRef]
- Stagi, S.; Pelosi, P.; Strano, M.; Poggi, G.; Manoni, C.; de Martino, M.; Seminara, S. Determinants of Vitamin D Levels in Italian Children and Adolescents: A Longitudinal Evaluation of Cholecalciferol Supplementation versus the Improvement of Factors Influencing 25(OH)D Status. Int. J. Endocrinol. 2014, 2014, 583039. [Google Scholar] [CrossRef]
- Mizobuchi, M.; Ogata, H.; Koiwa, F.; Kinugasa, E.; Akizawa, T. Vitamin D and vascular calcification in chronic kidney disease. Bone 2009, 45, S26–S29. [Google Scholar] [CrossRef] [PubMed]
- Ramalho, J.; Bacelar Marques, I.D.; Aguirre, A.R.; Pierrotti, L.C.; de Paula, F.J.; Nahas, W.C.; David-Neto, E. Pneumocystis jirovecii pneumonia with an atypical granulomatous response after kidney transplantation. Transpl. Infect. Dis. 2014, 16, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Kwan, A.; Abraham, R.S.; Currier, R.; Brower, A.; Andruszewski, K.; Abbott, J.K.; Baker, M.; Ballow, M.; Bartoshesky, L.E.; Bonagura, V.R.; et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA 2014, 312, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Notarangelo, L.D. Primary immunodeficiencies. J. Allergy Clin. Immunol. 2010, 125, S182–S194. [Google Scholar] [CrossRef]
- Fischer, A. Severe combined immunodeficiencies (SCID). Clin. Exp. Immunol. 2000, 122, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Tuovinen, E.A.; Grönholm, J.; Öhman, T.; Pöysti, S.; Toivonen, R.; Kreutzman, A.; Heiskanen, K.; Trotta, L.; Toiviainen-Salo, S.; Routes, J.M.; et al. Novel Hemizygous IL2RG p.(Pro58Ser) Mutation Impairs IL-2 Receptor Complex Expression on Lymphocytes Causing X-Linked Combined Immunodeficiency. J. Clin. Immunol. 2020, 40, 503–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hou, Y.; Gratz, H.P.; Ureña-Bailén, G.; Gratz, P.G.; Schilbach-Stückle, K.; Renno, T.; Güngör, D.; Mader, D.A.; Malenke, E.; Antony, J.S.; et al. Somatic Reversion of a Novel IL2RG Mutation Resulting in Atypical X-Linked Combined Immunodeficiency. Genes 2021, 13, 35. [Google Scholar] [CrossRef] [PubMed]
| Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).


{kind=link}
{kind=link}

