

Bibliographical Synthesis on the Corrosion and Protection of Archaeological Iron by Green Inhibitors


Abstract
:1. Introduction
2. History of Iron and Metallurgy
3. Iron Corrosion
3.1. Corrosion of Iron in Aqueous Media
3.1.1. Behavior of Iron in Aqueous Medium
- -
- Solvated protons (H+(aq)).
- -
- Dissolved oxygen (O2).
- -
- In an aerated environment (aerobic):
- -
- In a deaerated (anaerobic) environment:
3.1.2. Thermodynamic Approach
- -
- A domain of immunity where the metal (Fe) is thermodynamically stable.
- -
- Corrosion areas where the ions resulting from the dissolution of iron either in acidic media (Fe2+ and Fe3+) or in very basic media (HFeO2−) are responsible for the corrosion.
- -
- Areas of passivation (possible) where the formation of a solid compound in a neutral and basic medium (Fe(OH)2 or Fe(OH)3) can be protective (or not).
3.1.3. Kinetic Approach
3.1.4. Iron Corrosion Products in Aqueous Media
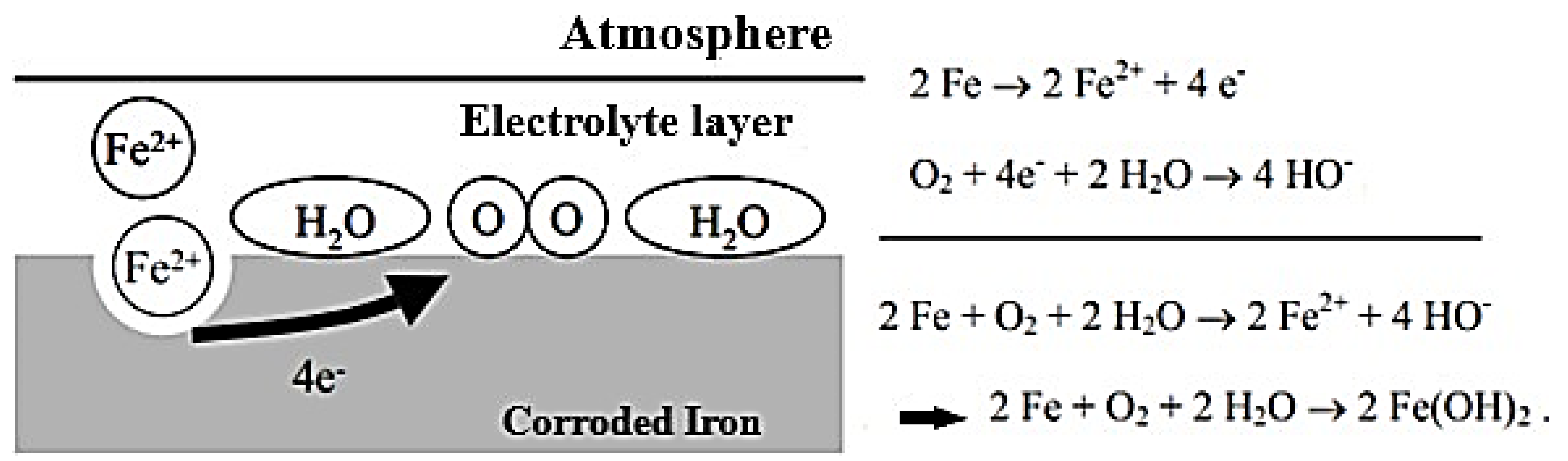
3.2. Atmospheric Corrosion of Iron
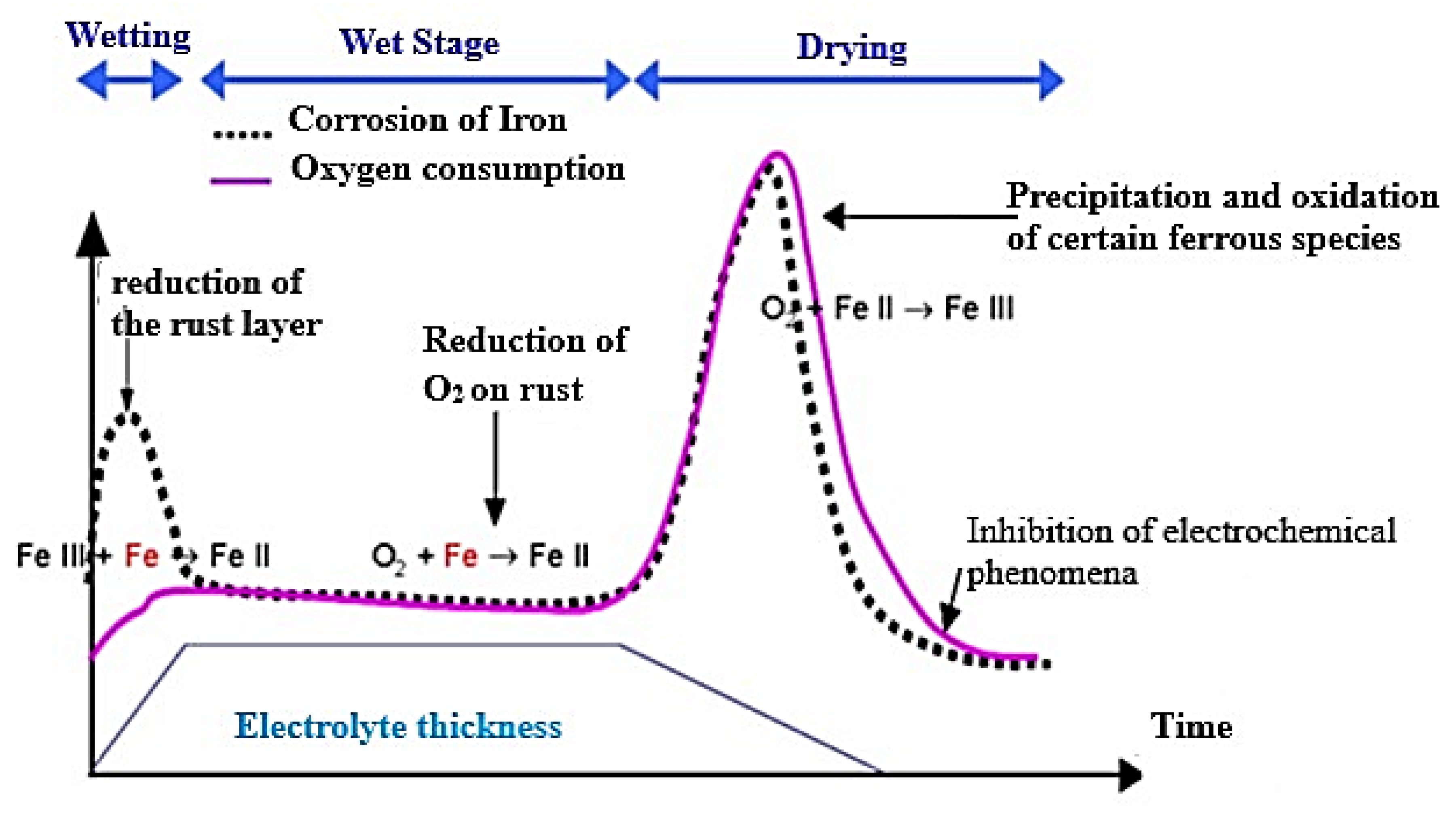
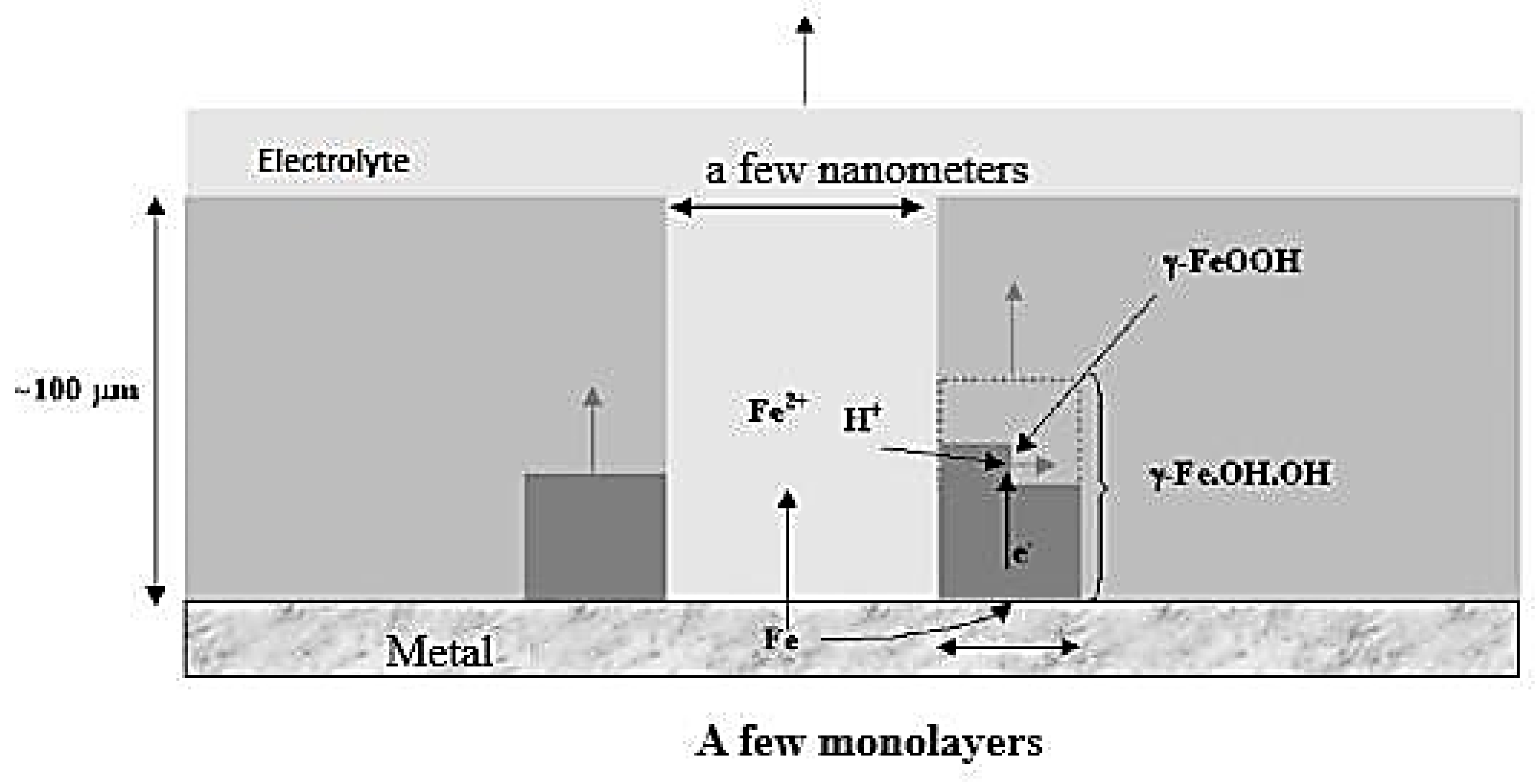
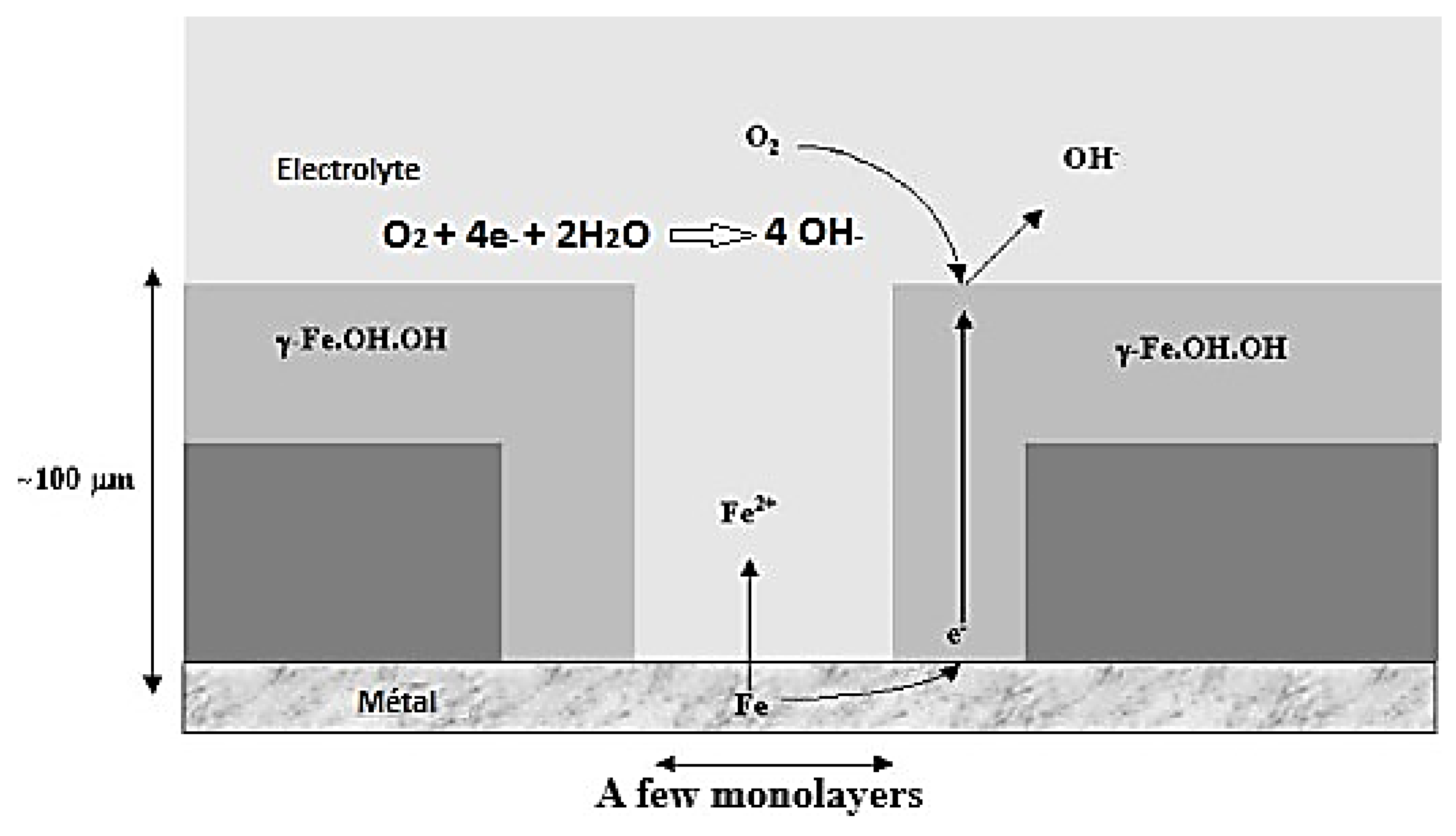
3.2.1. Mechanisms of Atmospheric Corrosion of Iron
Anchor Stage
Wet Stage
Drying Stage
3.2.2. Products of Atmospheric Corrosion of Iron
4. Protection of Iron by Corrosion Inhibitors
- Actions on the material, such as modifying its composition or microstructure or isolating it from its environment through a metallic or organic coating or anodization.
- Actions on the environment, such as incorporating corrosion inhibitors or avoiding moisture accumulation in the structure.
- Actions on the electrochemical corrosion process, such as cathodic protection.
4.1. Background
4.2. Definition
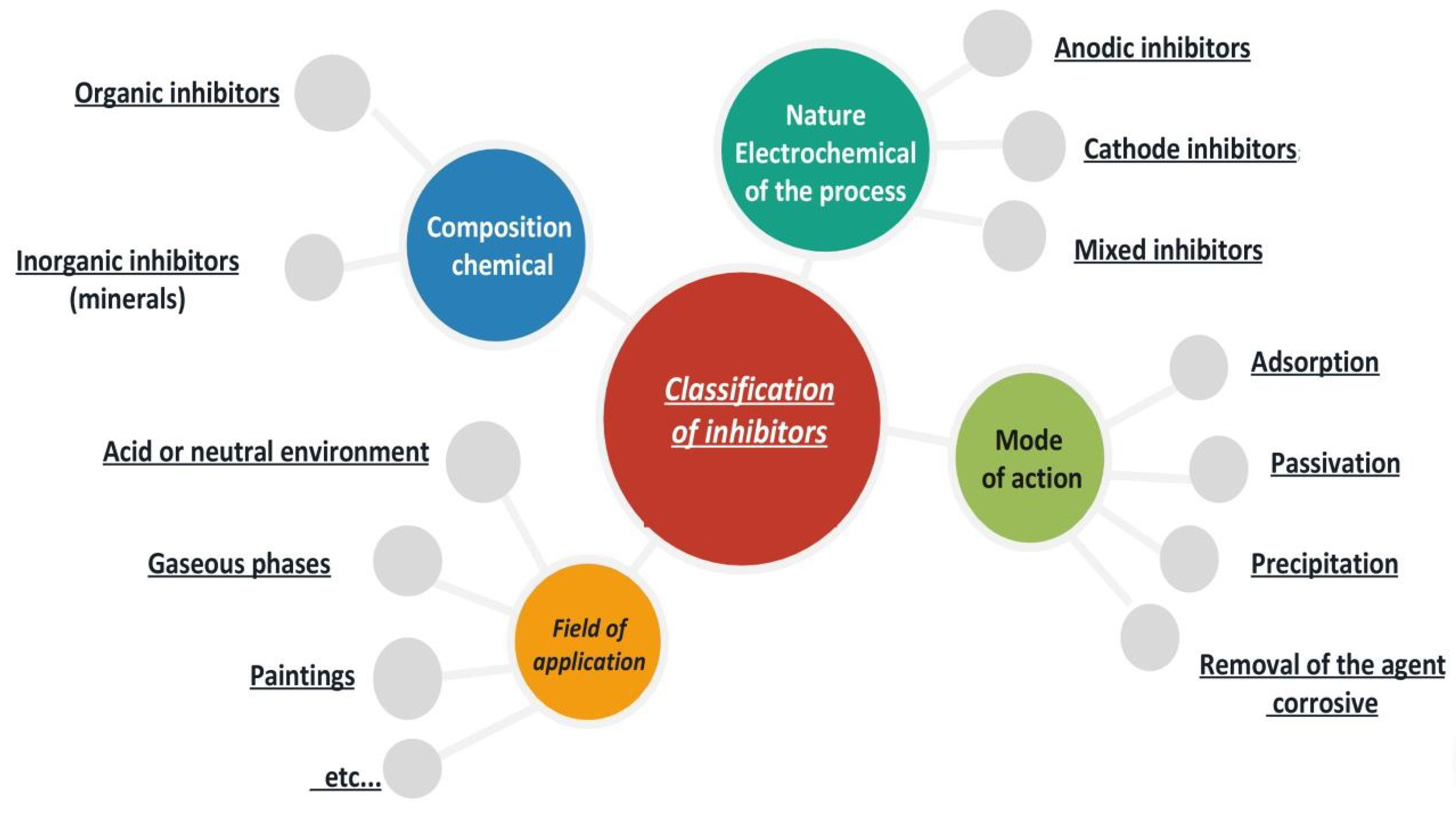
4.3. Classification
4.4. Inhibitors Specific to Ferrous Metals in an Acid Medium
4.4.1. Synthetic Inhibitors
4.4.2. Corrosion Inhibitors Based on Natural Substances
5. Conclusions
- Inhibitor effectiveness: the effectiveness of green corrosion inhibitors can be influenced by several factors, such as the type of metal, the corrosive environment, and the inhibitor concentration. Therefore, further research is needed to optimize the performance of these inhibitors under various conditions.
- Compatibility with other materials: green corrosion inhibitors may not be compatible with other materials used in the metal protection process, such as coatings or paints. Therefore, research efforts should focus on developing inhibitors that are compatible with other materials used in metal protection.
- Environmental impact: although green corrosion inhibitors are considered to be environmentally friendly, their impact on the environment should be carefully evaluated. For example, some natural inhibitors may cause eutrophication in water bodies or have other unintended consequences. Therefore, further research is needed to ensure that these inhibitors do not harm the environment.
- Cost: the cost of green corrosion inhibitors can be high, particularly for large-scale applications. Therefore, research efforts should focus on developing cost-effective inhibitors that can provide effective protection at a lower cost.
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Popov, B.N. Corrosion Engineering: Principles and Solved Problems, 1st ed.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 2–4. ISBN 978-0-444-62722-3. [Google Scholar]
- ISO 8044:2015; Corrosion of Metals and Alloys—Main Terms and Definitions. International Organization for Standardization: Geneva, Switzerland, 2015.
- Dwivedi, D.; Mata, J.P. Archaeometallurgical investigation of ancient artefacts’ degradation phenomenon. NPJ Mater. Degrad. 2019, 3, 35. [Google Scholar] [CrossRef] [Green Version]
- Hollner, S. Development of New Protective Treatments Based on Carboxylic Acid for the Conservation of Iron Objects of Cultural Heritage. Ph.D. Thesis, Université Henri-Poincaré Nancy I, Nancy, France, 2009. [Google Scholar]
- Hammouch, H. Development of a New Protective Treatment Based on Opuntia Ficus Indica for the Conservation of Archaeological Objects of Bronze and Iron. Ph.D. Thesis, Faculty of Science, Ibn Tofail-Kenitra University, Kenitra, Morocco, 2013. [Google Scholar]
- Tylecote, R.F. A History of Metallurgy, 2nd ed.; The Institute of Materials: London, UK, 1992; ISBN 0-9011462-88-8. [Google Scholar]
- France-Lanord, A. History of Iron: An Illustrated Guide to the Iron Museum; Centre de Recherches de L’histoire de la Sidérurgie: Nancy, France, 1977. [Google Scholar]
- Kordas, G. Corrosion Barrier Coatings: Progress and Perspectives of the Chemical Route. Corros. Mater. Degrad. 2022, 3, 376–413. [Google Scholar] [CrossRef]
- Selwyn, L. Overview of archaeological iron: The corrosion problem, key factors affecting treatment, and gaps in current knowledge. In Proceedings of the Metal 2004 National Museum of Australia Canberra ACT, Canberra, Australia, 4–8 October 2004. [Google Scholar]
- David, D. Matériaux: Analogues Archéologiques et Corrosion, Collection Science et Techniques; BIO Intelligence Service: Paris, France, 2002; p. 75. [Google Scholar]
- Pourbaix, M. Atlas of Electrochemical Equilibria; Gauthier—Villars et Cie Editeurs: Paris, France, 1963. [Google Scholar]
- Descostes, M. Evaluation of an Oxidizing Perturbation in Clay Media: Mechanism of Pyrite Oxidation. Ph.D. Thesis, University of Paris VII, Paris, France, 2001. [Google Scholar]
- Chivot, J. Sélection de données thermodynamiques concernant le système Fe-H2O. CEA/FAR SCECF, Technical Report SCECF n°481. 1998. Available online: www.andra.fr/sites/default/files/2017-12/236_0.pdf (accessed on 13 December 2022).
- Chivot, J. Les Diagrammes E-pH Révisés du Système Fer-H2O en Fonction de la Température; ANDRA: Paris, France, 1999. [Google Scholar]
- Monnier, J. Atmospheric Corrosion of Historical Ferrous Alloys under Shelter. Characterization of the System, Mechanisms and Contribution to Modelling. Ph.D. Thesis, Institut de Chimie et des Matériaux, University of Paris-Est, Champs-sur-Marne, France, 2008. [Google Scholar]
- Faiz, H. Etude du Mécanisme de Corrosion Atmosphérique à Long Terme des Aciers: Nouvelles Stratégies de Protection des Aciers du Patrimoine Culturel. Ph.D. Thesis, Faculté des Sciences et Technologies, Université Lorraine, Nancy, France, 2012. [Google Scholar]
- Accary, A.; Haijtink, B. La Paléo-Métallurgie-Outil de Prévision. In Journées de Paléométallurgie; Université de Technologie de Compiègne: Compiègne, France, 1983. [Google Scholar]
- Soerensen, B.; Gregory, D. In Situ Preservation of Artifacts in Nydam Mose, pg. 94 in Metal 98. In Proceedings of the International Conference on Metals Conservation, Draguignan-Figanieres, France, 27–29 May 1998. [Google Scholar]
- Johnson, A.B., Jr.; Francis, B. Durability of Metals from Archaeological Objects, Metal Meteorites and Native Metals; U.S. Department of Energy: Washington, DC, USA, 1980; p. 106.
- Azoulay, I. Long-Term Corrosion of Steels: Physicochemical Properties of Ferrous Hydroxysalts. Ph.D. Thesis, University of La Rochelle, La Rochelle, France, 2013. [Google Scholar]
- Misawa, T.; Hashimoto, K.; Shimodaira, S. The mechanism of formation of iron oxide and oxyhydroxides in aqueous solutions at room temperature. Corros. Sci. 1974, 14, 131–149. [Google Scholar] [CrossRef]
- Cornell, R.; Schwertmann, U. Iron Oxides in the Laboratory; Wiley-VCH: Weinheim, Germany, 2000; 137p. [Google Scholar]
- Vernon, W.H.J.; Hutton, R.S.; Patterson, W.S.; Evans, U.R.; Lee, A.R.; Martin, A.R.; Hudson, J.C.; Coste, J.H. A laboratory study of the atmospheric corrosion of metals. Discussion. Trans. Faraday Soc. 1931, 27, 582–594. [Google Scholar] [CrossRef]
- Schikorr, G. Über den Mechanismus des atmosphärischen Rostens des Eisens. Werkst. Und Korros. 1963, 14, 69–80. [Google Scholar] [CrossRef]
- Evans, U.; Taylor, C. Mechanism of atmospheric rusting. Corros. Sci. 1972, 12, 227–246. [Google Scholar] [CrossRef]
- Stratmann, M.; Bohnenkamp, K.; Engell, H. An electrochemical study of phasetransitions in rust layers. Corros. Sci. 1983, 23, 969–985. [Google Scholar] [CrossRef]
- Stratmann, M. The atmospheric corrosion of iron and steel. Metall. Odlew. 1990, 16, 46–52. [Google Scholar]
- Hoerlé, S.; Mazaudier, F.; Dillmann, P.; Santarini, G. Advances in understanding atmospheric corrosion of iron. II. Mechanistic modelling of wet–dry cycles. Corros. Sci. 2004, 46, 1431–1465. [Google Scholar] [CrossRef]
- Stratmann, M.; Streckel, H. On the atmospheric corrosion of metals which are covered with thin electrolyte layers—I, Verification of the experimental. Corros. Sci. 1990, 30, 681–696. [Google Scholar] [CrossRef]
- Antony, H. Etude Electrochimique des Composés du fer-apport à la Compréhension des Processus Environnementaux. Ph.D. Thesis, Université d’Evry, Evry, France, 2005; 213p. [Google Scholar]
- Dünnwald, J.; Otto, A. An investigation of phase transitions in rust layers using raman spectroscopy. Corros. Sci. 1989, 29, 1167–1176. [Google Scholar] [CrossRef]
- Yamashita, M.; Nagano, H.; Oriani, R. Dependence of corrosion potential and corrosion rate of a low-alloy steel upon depth of aqueous solution. Corros. Sci. 1998, 40, 1447–1453. [Google Scholar] [CrossRef]
- Cornell, R.; Schwertmann, U. The Iron Oxides—Structure, Properties, Occurrences and Uses, 2nd ed.; Wiley-VCH: Weinheim, Germany, 2003; 664p. [Google Scholar]
- Reguer, S. Phases Chlorées sur les Objets Archéologiques Ferreux Corrodés Dans les Sols: Caractérisations et Mécanismes de Formation. Ph.D. Thesis, Université Paris Sud-Paris XI, Bures-sur-Yvette, France, 2005. [Google Scholar]
- Nasrazadani, S. The application of infrared spectroscopy to a study of phosphoric and tannic acid interactions with magnetite, goethite and lepidocrocite. Corros. Sci. 1997, 39, 1845–1859. [Google Scholar] [CrossRef]
- Dillmann, P.; Mazaudier, F.; Hoerle, S. Advances in understanding atmospheric corrosion of iron. I. Rust characterisation of ancient ferrous artefacts exposed to indoor atmospheric corrosion. Corros. Sci. 2003, 46, 1401–1429. [Google Scholar] [CrossRef]
- Landolt, D. Treatise on Materials, 12 Corrosion and Chemistry of Metal Surfaces, Polytechnic and University Presses Romandes, 3rd ed.; Collection: Treatise on Materials; EPFL Press: Lausanne, Switzerland, 1993. [Google Scholar]
- Chan-Rosado, G.; Pech-Canul, M. Influence of native oxide film age on the passivation of carbon steel in neutral aqueous solutions with a dicarboxylic acid. Corros. Sci. 2019, 153, 19–31. [Google Scholar] [CrossRef]
- Sumoondur, A.; Shaw, S.; Ahmed, I.; Benning, L.G. Green rust as a precursor for magnetite: An in situ synchrotron based study. Miner. Mag. 2008, 72, 201–204. [Google Scholar] [CrossRef]
- Yamashita, M.; Konishi, H.; Kozakura, T.; Mizuki, J.; Uchida, H. In situ observation of initial rust for-mation process on carbon steel under Na2SO4 and NaCl solution films with wet/dry cycles using synchrotron radiation X-rays. Corros. Sci. 2005, 47, 2492–2498. [Google Scholar] [CrossRef]
- Lair, V.; Antony, H.; Legrand, L.; Chausse, A. Electrochemical reduction of ferric corrosion products and evaluation of galvanic coupling with iron. Corros. Sci. 2006, 48, 2050–2063. [Google Scholar] [CrossRef]
- Aliofkhazraei, M. Chapter 12. Corrosion Resistance Through the Application of Anti- Corrosion Coatings. In Developments in Corrosion Protection; BoD–Books on Demand: Norderstedt, Germany, 2014. [Google Scholar] [CrossRef] [Green Version]
- Serghini Idrissi, M. Study of the Electrochemical Behavior of C38 Steel and UR45N Stainless Steel in Different Media. Ph.D. Thesis, Faculty of Science, Mohammed V University, Rabat, Morocco, 10 December 2016. [Google Scholar]
- Bai, Q.; Bai, Y. Corrosion Prevention and Advanced CP Design. In Subsea Pipeline Design, Analysis, and Installation; Gulf Professional Publishing: Houston, TX, USA, 2014; pp. 451–464. [Google Scholar] [CrossRef]
- Marzorati, S.; Verotta, L.; Trasatti, S.P. Green Corrosion Inhibitors from Natural Sources and Biomass Wastes. Molecules 2018, 24, 48. [Google Scholar] [CrossRef] [Green Version]
- Zaki, A. Corrosion Control by Inhibition. In Principles of Corrosion Engineering and Corrosion Control; Elsevier: Amsterdam, The Netherlands, 2006; pp. 352–381. [Google Scholar] [CrossRef]
- Finšgar, M.; Jackson, J. Application of corrosion inhibitors for steels in acidic media for the oil and gas industry: A review. Corros. Sci. 2014, 86, 17–41. [Google Scholar] [CrossRef] [Green Version]
- Taghavikish, M.; Dutta, N.K.; Choudhury, N.R. Emerging Corrosion Inhibitors for Interfacial Coating. Coatings 2017, 7, 217. [Google Scholar] [CrossRef] [Green Version]
- Askari, M.; Aliofkhazraei, M.; Jafari, R.; Hamghalam, P.; Hajizadeh, A. Downhole corrosion inhibitors for oil and gas production—A review. Appl. Surf. Sci. Adv. 2021, 6, 100128. [Google Scholar] [CrossRef]
- Verma, C.; Ebenso, E.E.; Quraishi, M. Corrosion inhibitors for ferrous and non-ferrous metals and alloys in ionic sodium chloride solutions: A review. J. Mol. Liq. 2017, 248, 927–942. [Google Scholar] [CrossRef]
- Chen, L.; Lu, D.; Zhang, Y. Organic Compounds as Corrosion Inhibitors for Carbon Steel in HCl Solution: A Comprehensive Review. Materials 2022, 15, 2023. [Google Scholar] [CrossRef] [PubMed]
- Saji, V.S. A Review on Recent Patents in Corrosion Inhibitors. Recent Patents Corros. Sci. 2010, 2, 6–12. [Google Scholar] [CrossRef] [Green Version]
- Fekry, A.; Ameer, M. Corrosion inhibition of mild steel in acidic media using newly synthesized heterocyclic organic molecules. Int. J. Hydrogen Energy 2010, 35, 7641–7651. [Google Scholar] [CrossRef]
- ESutter, M.; Ammeloot, F.; Pouet, M.J.; Fiaud, C.; Couffignal, R. Heterocyclic compounds used as corrosion inhibitors: Correlation between 13C and 1H NMR spectroscopy and inhibition efficiency. Corros. Sci. 1999, 41, 105–115. [Google Scholar] [CrossRef]
- Abiola, O.; Oforka, N. Adsorption of (4-amino-2-methyl-5-pyrimidinyl methylthio) acetic acid on mild steel from hydrochloric acid solution (HCl)—Part 1. Mater. Chem. Phys. 2004, 83, 315–322. [Google Scholar] [CrossRef]
- Abiola, O.K. Adsorption of 3-(4-amino-2-methyl-5-pyrimidyl methyl)-4-methyl thiazolium chloride on mild steel. Corros. Sci. 2006, 48, 3078–3090. [Google Scholar] [CrossRef]
- Goyal, M.; Kumar, S.; Bahadur, I.; Verma, C.; Ebenso, E.E. Organic corrosion inhibitors for industrial cleaning of ferrous and non-ferrous metals in acidic solutions: A review. J. Mol. Liq. 2018, 256, 565–573. [Google Scholar] [CrossRef]
- Yara-Varón, E.; Li, Y.; Balcells, M.; Garayoa, R.C.; Fabiano-Tixier, A.-S.; Chemat, F. Vegetable Oils as Alternative Solvents for Green Oleo-Extraction, Purification and Formulation of Food and Natural Products. Molecules 2017, 22, 1474. [Google Scholar] [CrossRef]
- Zaferani, S.H.; Sharifi, M.; Zaarei, D.; Shishesaz, M.R. Application of eco-friendly products as corrosion inhibitors for metals in acid pickling processes—A review. J. Environ. Chem. Eng. 2013, 1, 652–657. [Google Scholar] [CrossRef]
- Zakeri, A.; Bahmani, E.; Aghdam, A.S.R. Plant extracts as sustainable and green corrosion inhibitors for protection of ferrous metals in corrosive media: A mini review. Corros. Commun. 2022, 5, 25–38. [Google Scholar] [CrossRef]
- AOstovari, A.; Hoseinieh, M.; Peikari, M.; Shadizadeh, S.; Hashemi, S. Corrosion inhibition of mild steel in 1M HCl solution by henna extract: A comparative study of the inhibition by henna and its constituents (Lawsone, Gallic acid, α-d-Glucose and Tannic acid). Corros. Sci. 2009, 51, 1935–1949. [Google Scholar] [CrossRef]
- Orubite, K.; Oforka, N. Inhibition of the corrosion of mild steel in hydrochloric acid solutions by the extracts of leaves of Nypa fruticans Wurmb. Mater. Lett. 2004, 58, 1768–1772. [Google Scholar] [CrossRef]
- Chauhan, L.; Gunasekaran, G. Corrosion inhibition of mild steel by plant extract in dilute HCl medium. Corros. Sci. 2007, 49, 1143–1161. [Google Scholar] [CrossRef]
- El-Etre, A. Khillah extract as inhibitor for acid corrosion of SX 316 steel. Appl. Surf. Sci. 2005, 252, 8521–8525. [Google Scholar] [CrossRef]
- Bouyanzer, A.; Hammouti, B.; Majidi, L. Pennyroyal oil from Mentha pulegium as corrosion inhibitor for steel in 1M HCl. Mater. Lett. 2006, 60, 2840–2843. [Google Scholar] [CrossRef]
- Rahim, A.A.; Rocca, E.; Steinmetz, J.; Kassim, M.J.; Adnan, R.; Ibrahim, M.S. Mangrove tannins and their flavanoid monomers as alternative steel corrosion inhibitors in acidic medium. Corros. Sci. 2007, 49, 402–417. [Google Scholar] [CrossRef]
- Amin, M.A.; Abd El-Rehim, S.S.; El-Sherbini, E.E.F.; Bayoumi, R.S. The inhibition of low carbon steel corrosion in hydrochloric acid solutions by succinic acid: Part I. Weight loss, polarization, EIS, PZC, EDX and SEM studies. Electrochim. Acta 2007, 52, 3588–3600. [Google Scholar] [CrossRef]
- El-Etre, A. Inhibition of acid corrosion of carbon steel using aqueous extract of olive leaves. J. Colloid Interface Sci. 2007, 314, 578–583. [Google Scholar] [CrossRef]
- Oguzie, E.E. Evaluation of the inhibitive effect of some plant extracts on the acid corrosion of mild steel. Corros. Sci. 2008, 50, 2993–2998. [Google Scholar] [CrossRef]
- Satapathy, A.; Gunasekaran, G.; Sahoo, S.; Amit, K.; Rodrigues, P. Corrosion inhibition by Justicia gendarussa plant extract in hydrochloric acid solution. Corros. Sci. 2009, 51, 2848–2856. [Google Scholar] [CrossRef]
- Okafor, P.C.; Ikpi, M.E.; Uwah, I.E.; Ebenso, E.E.; Ekpe, U.J. Inhibitory action of Phyllanthus amarus extracts on the corrosion of mild steel in acidic media. Corros. Sci. 2008, 50, 2310–2317. [Google Scholar] [CrossRef]
- AAbdel-Gaber, A.; Abd-El-Nabey, B.; Saadawy, M. The role of acid anion on the inhibition of the acidic corrosion of steel by lupine extract. Corros. Sci. 2009, 51, 1038–1042. [Google Scholar] [CrossRef]
- Da Rocha, J.C.; Gomes, J.A.D.C.P.; D’Elia, E. Corrosion inhibition of carbon steel in hydrochloric acid solution by fruit peel aqueous extracts. Corros. Sci. 2010, 52, 2341–2348. [Google Scholar] [CrossRef]
- Elkhotfi, Y.; Forsal, I.; Rakib, E.M.; Mernari, B. The Inhibition Action of Essential Oil of J. Juniperus Phoenicea on the Corrosion of Mild Steel in Acidic Media. Port. Electrochim. Acta 2018, 36, 77–87. [Google Scholar] [CrossRef]
- Kalaiselvi, P.; Chellammal, S.; Palanichamy, S.; Subramanian, G. Artemisia pallens as corrosion inhibitor for mild steel in HCl medium. Mater. Chem. Phys. 2010, 120, 643–648. [Google Scholar] [CrossRef]
- Abdallah, M. Guar Gum as Corrosion Inhibitor for Carbon Steel in Sulfuric Acid Solutions. Port. Electrochim. Acta 2004, 22, 161–175. [Google Scholar] [CrossRef]
- Abdel-Gaber, A.M.; Abd-El-Nabey, B.A.; Sidahmed, I.M.; El-Zayady, A.M.; Saadawy, M. Inhibitive action of some plant extracts on the corrosion of steel in acidic media. Corros. Sci. 2006, 48, 2765–2779. [Google Scholar] [CrossRef]
- Li, Y.; Zhao, P.; Liang, Q.; Hou, B. Berberine as a natural source inhibitor for mild steel in 1M H2SO4. Appl. Surf. Sci. 2005, 252, 1245–1253. [Google Scholar] [CrossRef]
- Noor, E.A. Temperature effects on the corrosion inhibition of mild steel in acidic solutions by aqueous extract of fenugreek leaves. Int. J. Electrochem. Sci. 2007, 2, 996–1017. [Google Scholar]
- Raja, P.B.; Sethuraman, M. Inhibitive effect of black pepper extract on the sulphuric acid corrosion of mild steel. Mater. Lett. 2008, 62, 2977–2979. [Google Scholar] [CrossRef]
- Ebenso, E.; Oguzie, E. Corrosion inhibition of mild steel in acidic media by some organic dyes. Mater. Lett. 2005, 59, 2163–2165. [Google Scholar] [CrossRef]
- Ebenso, E.E.; Alemu, H.; Umoren, S.A.; Obot, I.B. Inhibition of mild steel corrosion in sulphuric acid using alizarin yellow GG dye and synergistic iodide additive. Int. J. Electrochem. Sci. 2008, 3, 1325–1339. [Google Scholar]
- de Souza, F.; Spinelli, A. Caffeic acid as a green corrosion inhibitor for mild steel. Corros. Sci. 2009, 51, 642–649. [Google Scholar] [CrossRef]
- Shivakumar, M.; Dharmaprakash, M.S.; Manjappa, S.; Nagashree, K.L. Corrosion Inhibition Performance of Lignin Extracted from Black Liquor on Mild Steel in 0.5 M H2SO4 Acidic Media. Port. Electrochim. Acta 2017, 35, 351–359. [Google Scholar] [CrossRef]
- Abbout, S.; Zouarhi, M.; Chebabe, D.; Damej, M.; Berisha, A.; Hajjaji, N. Galactomannan as a new bio-sourced corrosion inhibitor for iron in acidic media. Heliyon 2020, 6, e03574. [Google Scholar] [CrossRef]
- Hammouch, H.; Dermaj, A.; Chebabe, D.; Decaro, P.; Hajjaji, N.; Bettach, N.; Takenouti, H.; Srhiri, A. Opuntia Ficus Indica seed oil: Characterization and application in corrosion inhibition of carbon steel in acid medium. Anal. Bioanal. Electrochem. 2013, 5, 236. [Google Scholar]
- Chellouli, M.; Chebabe, D.; Dermaj, A.; Erramli, H.; Bettach, N.; Hajjaji, N.; Casaletto, M.; Cirrincione, C.; Privitera, A.; Srhiri, A. Corrosion inhibition of iron in acidic solution by a green formulation derived from Nigella sativa L. Electrochim. Acta 2016, 204, 50–59. [Google Scholar] [CrossRef]
- Zouarhi, M.; Chellouli, M.; Abbout, S.; Hammouch, H.; Dermaj, A.; Hassane, S.O.S.; Decaro, P.; Bettach, N.; Hajjaji, N.; Srhiri, A. Inhibiting Effect of a Green Corrosion Inhibitor Containing Jatropha Curcas Seeds Oil for Iron in an Acidic Medium. Port. Electrochim. Acta 2018, 36, 179–195. [Google Scholar] [CrossRef]
- Abbout, S.; Chellouli, M.; Zouarhi, M.; Benzidia, B.; Hammouch, H.; Chebabe, D.; Dermaj, A.; Erramli, H.; Bettach, N.; Hajjaji, N. New formulation based on Ceratonia Siliqua L seed oil, as a green corrosion inhibitor of iron in acidic medium. Anal. Bioanal. Electrochem. 2018, 10, 789–804. [Google Scholar]
- Zouarhi, M.; Abbout, S.; Benzidia, B.; Chellouli, M.; Hammouch, H.; Erramli, H.; Hassane, S.O.S.; Bettach, N.; Hajjaji, N. Evaluation of a New Formulation Derived from Aleurites moluccana Seeds Oil as a Green Corrosion Inhibitor for Iron in Acidic Medium. Anal. Bioanal. Electrochem. 2019, 11, 1651–1668. [Google Scholar]
- Rehioui, M.; Abbout, S.; Benzidia, B.; Hammouch, H.; Erramli, H.; Daoud, N.A.; Badrane, N.; Hajjaji, N. Corrosion inhibiting effect of a green formulation based on Opuntia Dillenii seed oil for iron in acid rain solution. Heliyon 2021, 7, e06674. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Names | Formula | Space Group | Color | |
|---|---|---|---|---|
| Oxyhydroxides | Goethite | α-FeOOH |  | Crystals: black brown Powder: yellow brown |
| Akaganeite | β-FeOOH |  | Light brown | |
| Lepidocrocite | γ-FeOOH |  | Crystals: brown-red Powder: orange-yellow | |
| Feroxyhyte | δ-FeOOH |  | Brown | |
| Ferrihydrite | Fe2O3, nH2O |  | Yellow-brown to black-brown | |
| Oxides | Hematite | α-Fe2O3 |  | Crystals: Dark grey Powder: Dark red |
| Maghemite | γ-Fe2O3 |  | Brown | |
| Magnetite | Fe3O4 |  | Crystals: dark grey Powder: black |
| Compounds | References | |
|---|---|---|
| Nitrogenous | Hexamethylenetetramine or methenamine | [52] |
| Diazoles: imidazole and its derivatives | ||
| Triazoles and its derivatives such as benzotriazole | ||
| Bipyrazole derivatives | ||
| Oxadiazoles | ||
| Quinoline derivatives | ||
| Quinone derivatives such as Quinoxaline-2,3-dione | ||
| Pyridine derivatives | ||
| Mixed compounds such as Schiff’s Bases which result from the condensation of an amine with an aldehyde | ||
| Aliphatic amines (ethylamine, dimethylamine, butylamine, diethylamine, butyldiethylamine) Octylamine derivatives | [53,54] | |
| Sulphur | Thiourea and its derivatives | [52] |
| Thiadiazole and its derivatives | ||
| Oxygenated | Lactones | |
| Carboxylic compounds in combination with heterocycles (furans, imidazoles, thiophenes, isoxazole derivatives and several other moieties) Linear sodium carboxylates | [55,56,57] |
| Non-Toxic Inhibitors | Type of Inhibitor | IE (%) | Environment | Substrate | Adsorption Mechanisms | References | ||
|---|---|---|---|---|---|---|---|---|
| Henna, L. inermis | Cathodic | 92.1 | HCl (1 M) | Mild steel | Chemisorption | [61] | ||
| N. fruticans Wurmb | Mixed | 75.1 | HCl | Mild steel | Physisorption | [62] | ||
| Eugenol derivatives | Mixed | 91 | HCl (1 M) | Steel | Chemisorption | [63] | ||
| Khillah extract (seeds (A. visnaga)) | Mixed | 99.3 | HCl (2 M) | Steel SX 316 | Chemical adsorption | [64] | ||
| Natural oil extracted from pennyroyal mint (Mentha pulegium, PM) | Cathodic | 80 | HCl (1 M) | Steel | Simple blocking of the available surface, intermolecular synergistic Active molecules of this oil | [65] | ||
| Plant extract of Z. alatum | Cathodic | 95 | HCl (5%) | Mild steel | Chemisorption | [63] | ||
| Flavonoids (Monomers) | Cathodic | >70 | HCl (0.5 M) aerate d | Steel | Chemisorption | [66] | ||
| Succinic acid (SA) | Anodic | 97.5 | HCl (1.0 M) aerate d unstirred | Low carbon steel | Film of inhibitor adsorbed on electrode surface | [67] | ||
| Aqueous extract of olive leaves (O. europaea L.) | Mixed | 91 | HCl (2 M) | Carbon steel | Physical adsorption | [68] | ||
| T. occidentalis, (TO) | Cathodic | 91–97 | HCl (1 M) | Mild steel | Physisorption | [69] | ||
| A. indica, (AI) | ||||||||
| H. sabdariffa, (HS) | ||||||||
| G. kola (GK) seed extract | ||||||||
| Extract from J. gendarussa (JGPE) | Mixed | 93 | HCl (1 M) | Mild steel | Physisorption | [70] | ||
| Extracts of leaves and seeds of P. amarus | Mixed | 80.1–94.1 | HCl (2 M) | Mild steel | Chemisorption | [71] | ||
| L. albus L. | Mixed | 77.6 | HCl (2 M) | Steel | Chemisorption | [72] | ||
| 85 | H2SO4 (1 M) | Physisorption | ||||||
| Pennyroyal oil from M. pulegium | Cathodic | 80 | HCl (1 M) | Steel | Chemisorption | [65] | ||
| Zest of (Mango, Orange, Passion, Cashew) | Mixed | 80–95 | HCl (1 M) | Carbon steel | Adsorption of organic compounds present in the extracts on the active sites of the electrode surface | [73] | ||
| Juniperus phoenicea (Cupressaceae) essential oil | Mixed | 83 | HCl (1 M) | Mild steel | adsorption of aromatic compounds on the metal surface | [74] | ||
| Methanolic extract of A. Pallens | Mixed | 96.5 | HCl (4 N) | Mild steel | Formation of a very tightly adhering adsorbent film on the metal surface | [75] | ||
| Guar gum | Mixed | 93.6 | H2SO4 (1 M) | Carbon steel | Formation of passive, active and continuously propagating centers. | [76] | ||
| Chamomile (C. mixtum L.) | Mixed | 90.2 | H2SO4 (1 M) | Steel | Adsorption of the stable complex to the steel surface | [77] | ||
| Halfab ar (C. proximus) | ||||||||
| Black cumin (N. sativa L.) | ||||||||
| Kidney bean (P. vulgaris L.) | ||||||||
| Berberine | Mixed | 98 | H2SO4 (1 M) | Mild steel | Chemical adsorption | [78] | ||
| Fenugreek leaves (AEFL) | Mixed | 88.3 | H2SO4 | Mild steel | Chemical adsorption of inhibitor molecules on mild steel | [79] | ||
| Black pepper extract | 90 | [80] | ||||||
| Saffron-o (SO) | 65 | [81] | ||||||
| Alizarin Yellow (GG) | Mixed | 85 | H2SO4 (2 M) | Mild steel | Physisorption | [82] | ||
| Caffeic acid | 83.9 | H2 SO4 (0.1 M) | [83] | |||||
| Lignin extracted from black liquor of the pulp and paper industry | Cathodic | 95 | H2SO4 (0.5 M) | Mild steel | Adsorption of more lignin molecules on the metal surface, preventing the electrochemical corrosion process | [84] | ||
| Galactomannan extracted from Carob seeds (Ceratonia Siliqua) | Mixed | 86.6 | HCl (1 M) | Archaeological iron | Establishment of inhibitor film on iron substrate surface | [85] | ||
| Formulations based on oils | Opuntia ficus indica (OTH) | Mixed | 99.6 | Acid rain- simula ted enviro nment pH = 3.6 | Archaeological iron | Establishment of inhibitor film on iron substrate surface | [86] | |
| extracted from the seeds of | Nigella sativa (FBN) | 99.3 | [87] | |||||
| Jatropha Curcas (JAC) | 97 | [88] | ||||||
| Ceratonia Siliqua L., (FCSL) | 98.6 | [89] | ||||||
| Aleurites moluccana (ALM) | 97 | [90] | ||||||
| Opuntia Dillenii (FOD) | 99 | [91] | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zouarhi, M. Bibliographical Synthesis on the Corrosion and Protection of Archaeological Iron by Green Inhibitors. Electrochem 2023, 4, 103-122. https://doi.org/10.3390/electrochem4010010
Zouarhi M. Bibliographical Synthesis on the Corrosion and Protection of Archaeological Iron by Green Inhibitors. Electrochem. 2023; 4(1):103-122. https://doi.org/10.3390/electrochem4010010
Chicago/Turabian StyleZouarhi, Meryem. 2023. "Bibliographical Synthesis on the Corrosion and Protection of Archaeological Iron by Green Inhibitors" Electrochem 4, no. 1: 103-122. https://doi.org/10.3390/electrochem4010010