

A General Hybrid Modeling Framework for Systems Biology Applications: Combining Mechanistic Knowledge with Deep Neural Networks under the SBML Standard

Abstract
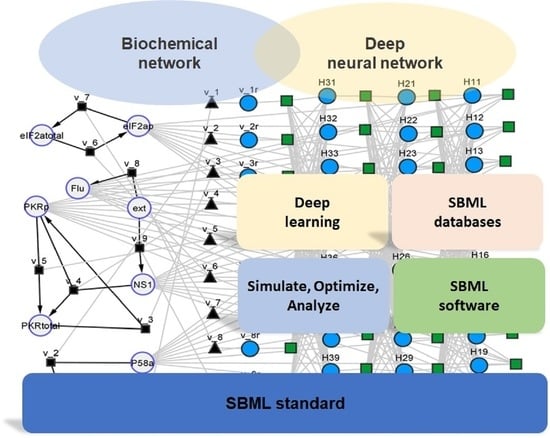
:
1. Introduction
2. Methods
2.1. General SBML Hybrid Model
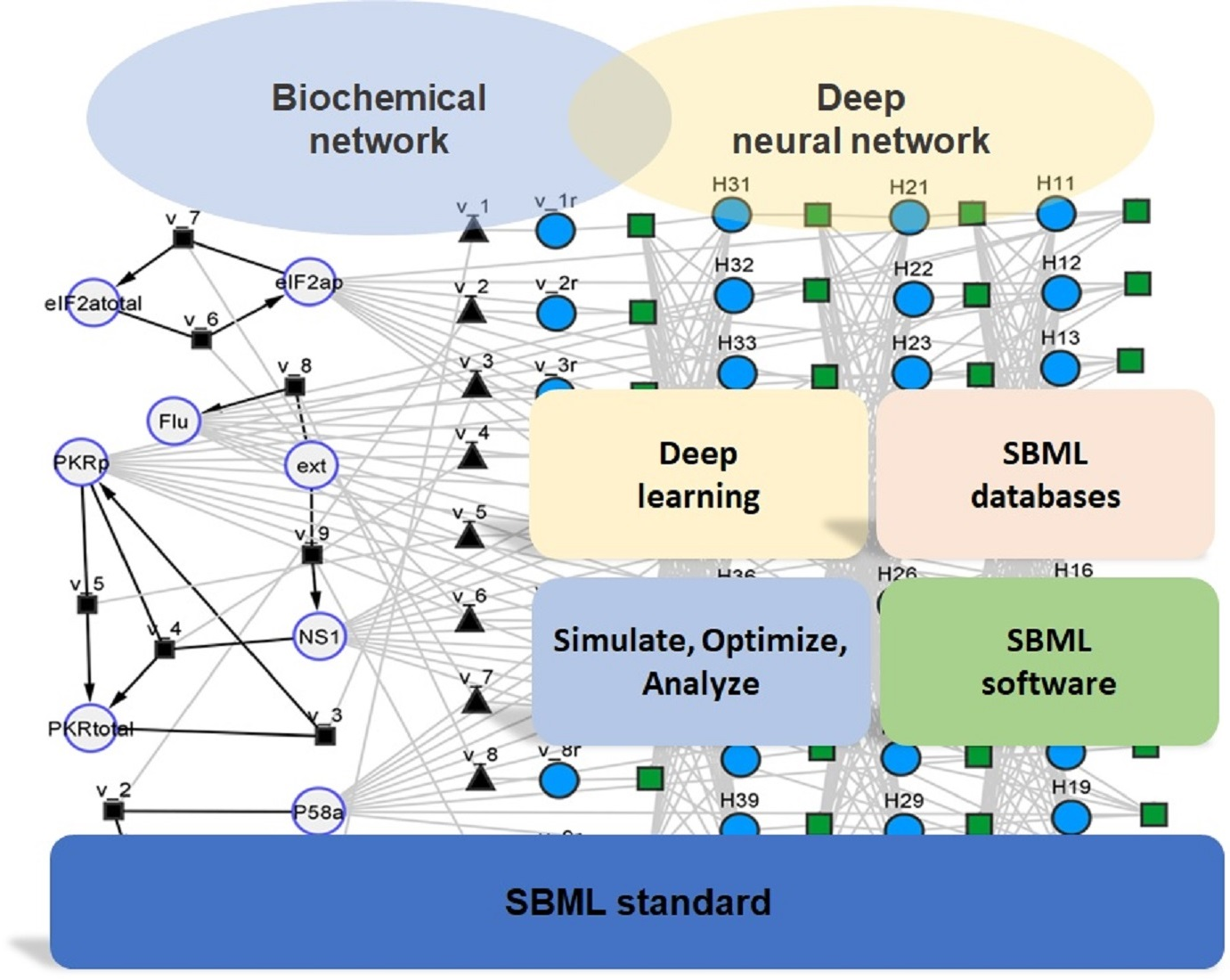
2.2. Interfacing with SBML Databases and SBML Modeling Tools
2.3. Case Studies
3. Results and Discussion
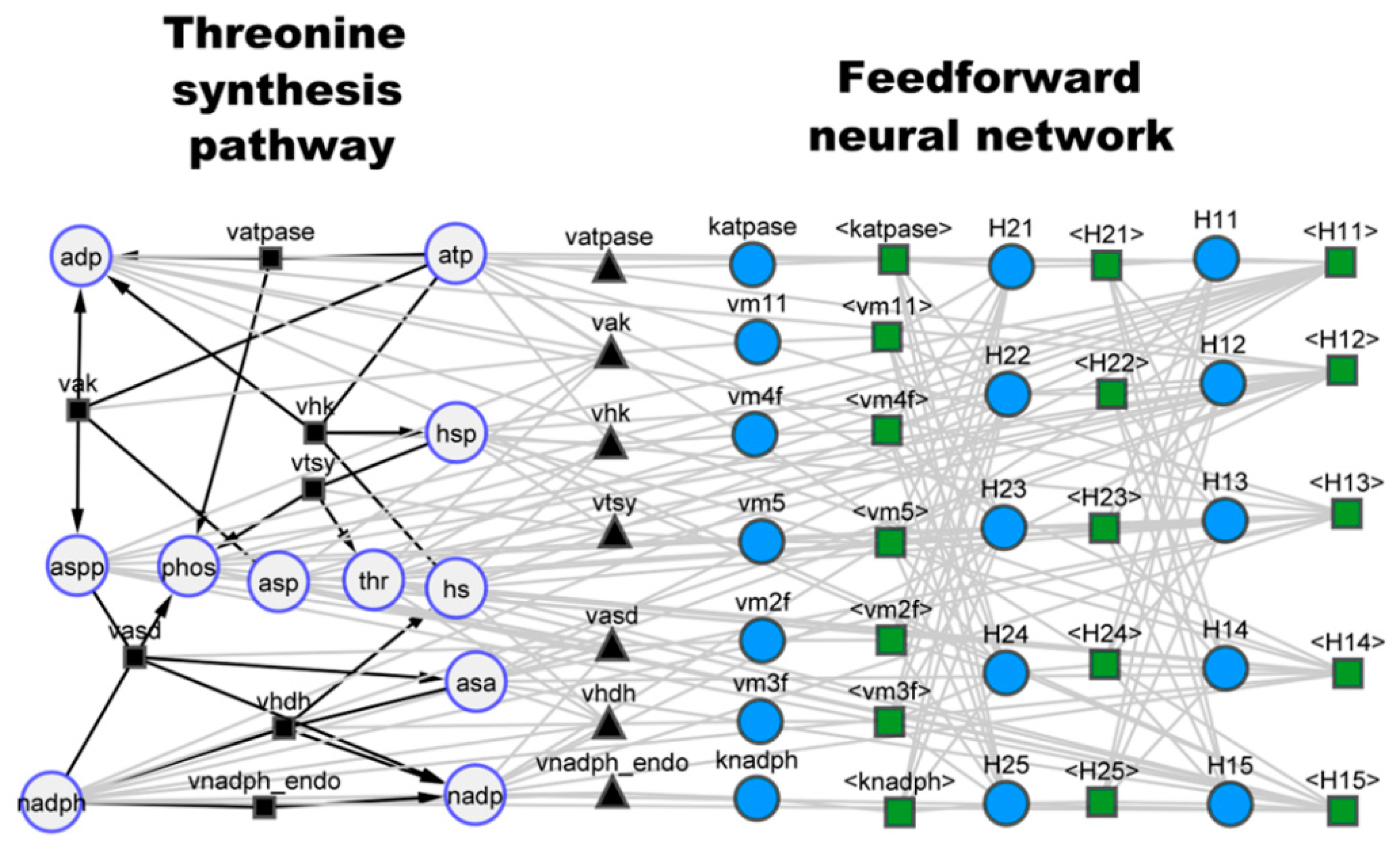
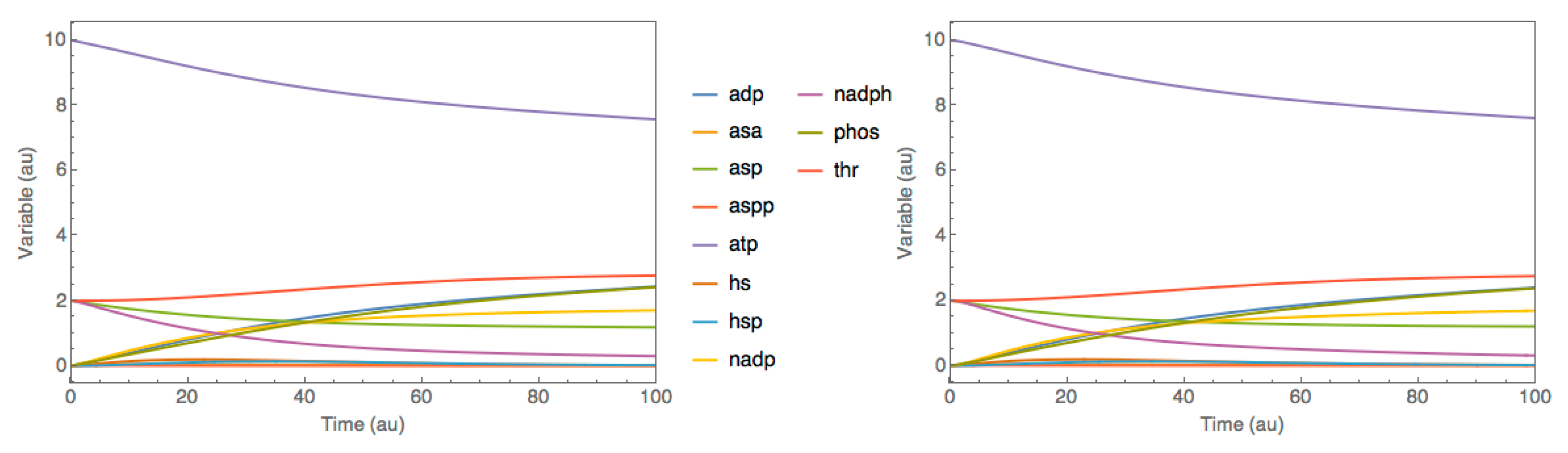
3.1. Case Study 1: Threonine Synthesis Pathway in E. coli
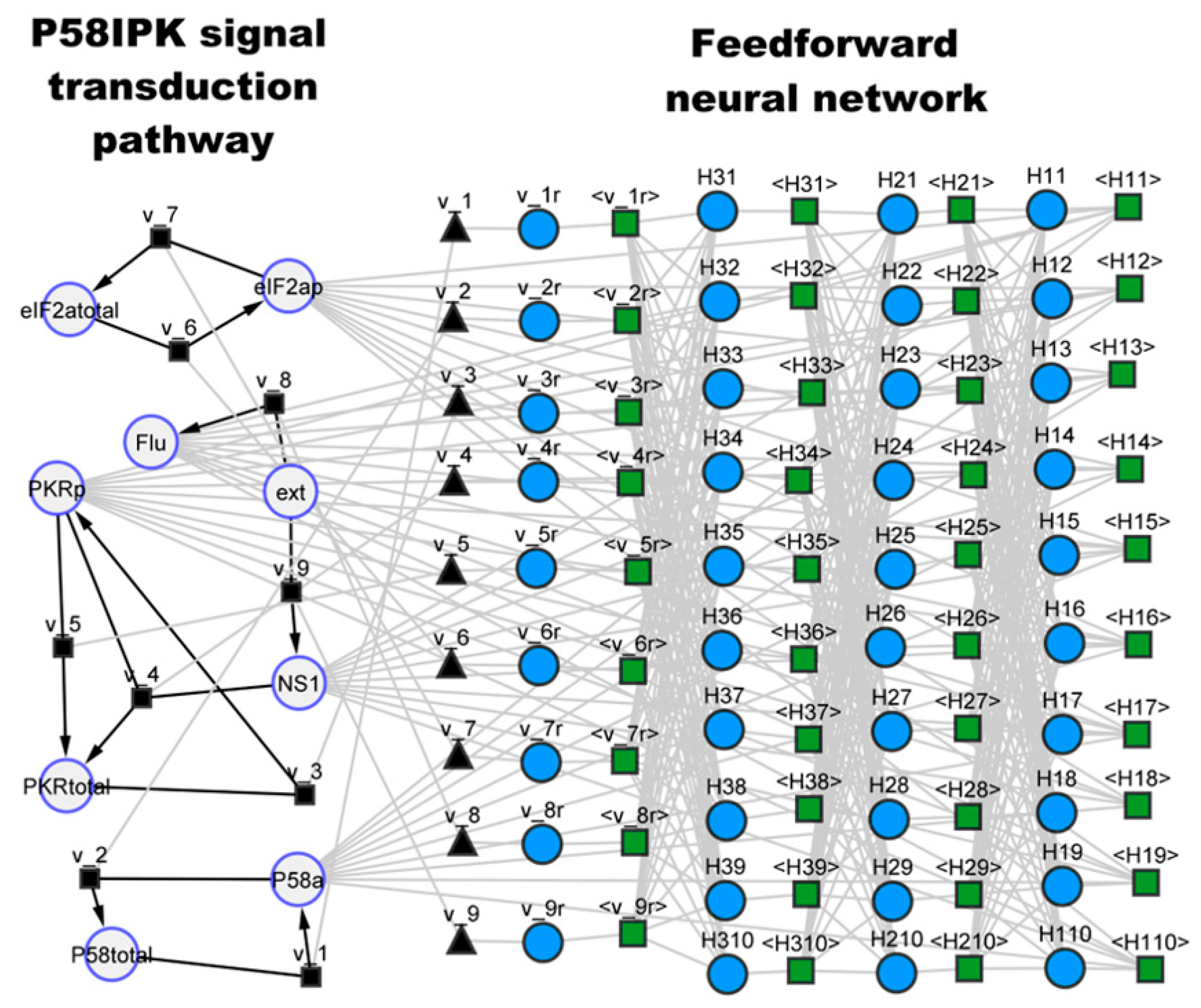
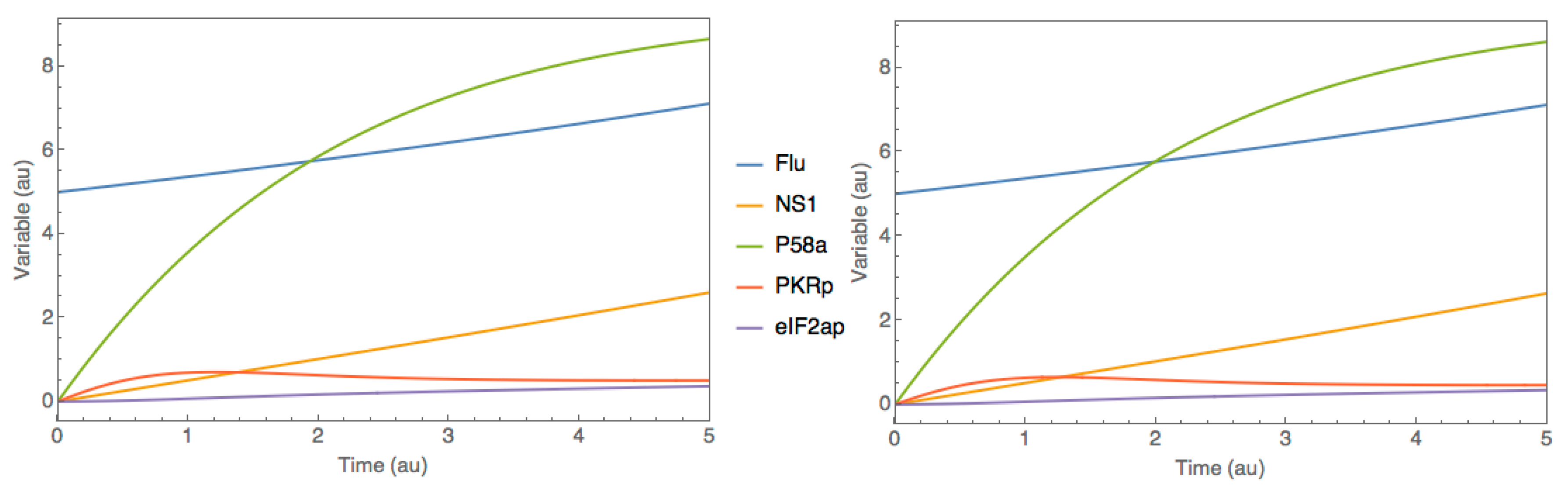
3.2. Case Study 2: P58IPK Signal Transduction Pathway
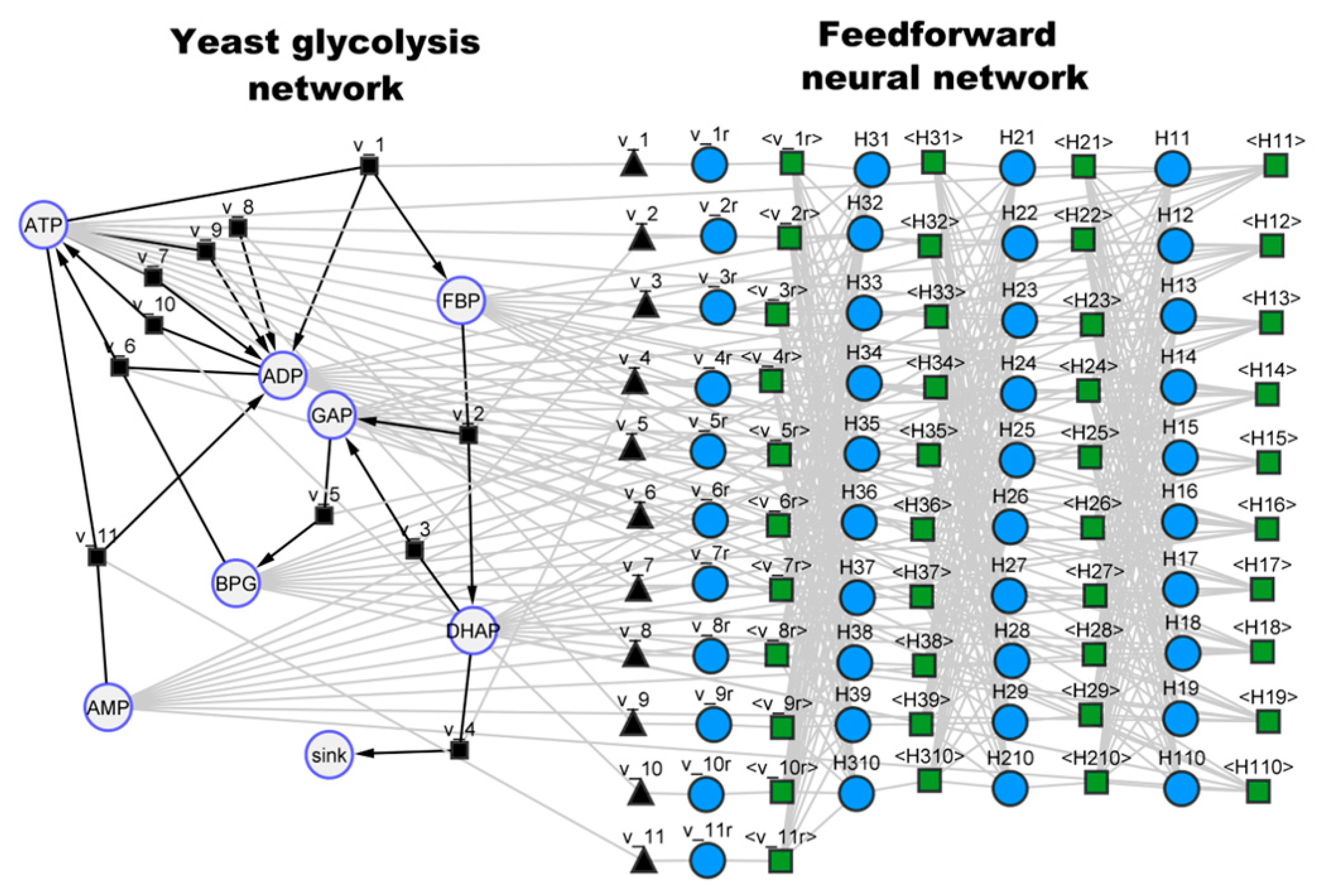
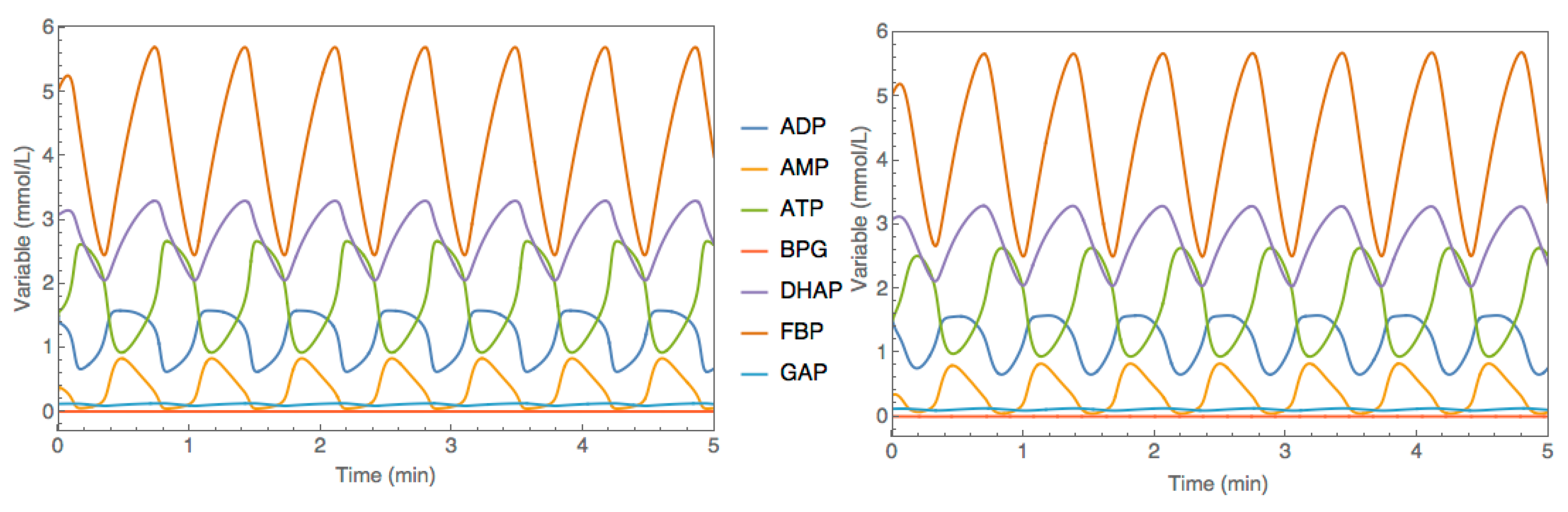
3.3. Case Study 3: Yeast Glycolytic Oscillations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- von Stosch, M.; Oliveira, R.; Peres, J.; de Azevedo, S.F. Hybrid semi-parametric modeling in process systems engineering: Past, present and future. Comput. Chem. Eng. 2014, 60, 86–101. [Google Scholar] [CrossRef] [Green Version]
- Psichogios, D.C.; Ungar, L.H. A Hybrid Neural Network-1st Principles Approach to Process Modeling. Aiche J. 1992, 38, 1499–1511. [Google Scholar] [CrossRef]
- Thompson, M.L.; Kramer, M.A. Modeling Chemical Processes Using Prior Knowledge and Neural Networks. Aiche J. 1994, 40, 1328–1340. [Google Scholar] [CrossRef]
- Schubert, J.; Simutis, R.; Dors, M.; Havlik, I.; Lubbert, A. Hybrid Modeling of Yeast Production Processes—Combination of a-Priori Knowledge on Different Levels of Sophistication. Chem. Eng. Technol. 1994, 17, 10–20. [Google Scholar] [CrossRef]
- Teixeira, A.P.; Clemente, J.J.; Cunha, A.E.; Carrondo, M.J.T.; Oliveira, R. Bioprocess iterative batch-to-batch optimization based on hybrid parametric/nonparametric models. Biotechnol. Prog. 2006, 22, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, A.P.; Alves, C.; Alves, P.M.; Carrondo, M.J.; Oliveira, R. Hybrid elementary flux analysis/nonparametric modeling: Application for bioprocess control. BMC Bioinform. 2007, 8, 30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Stosch, M.; Oliveira, R.; Peres, J.; de Azevedo, S.F. A novel identification method for hybrid (N)PLS dynamical systems with application to bioprocesses. Expert Syst. Appl. 2011, 38, 10862–10874. [Google Scholar] [CrossRef]
- Pinto, J.; de Azevedo, C.R.; Oliveira, R.; von Stosch, M. A bootstrap-aggregated hybrid semi-parametric modeling framework for bioprocess development. Bioprocess Biosyst. Eng. 2019, 42, 1853–1865. [Google Scholar] [CrossRef]
- Rajulapati, L.; Chinta, S.; Shyamala, B.; Rengaswamy, R. Integration of machine learning and first principles models. Aiche J. 2022, 68, e17715. [Google Scholar] [CrossRef]
- Glassey, J.; von Stosch, M. Hybrid Modeling in Process Industries, 1st ed.; Taylor&Francis, Ed.; CRC Press: Boca Raton, FL, USA, 2018. [Google Scholar]
- Agharafeie, R.; Oliveira, R.; Ramos, J.; Mendes, J. Application of Hybrid Neural Models to Bioprocesses: A Systematic Literature Review. Authorea 2023. [Google Scholar] [CrossRef]
- Le, N.Q.K.; Do, D.T.; Hung, T.N.K.; Lam, L.H.T.; Huynh, T.T.; Nguyen, N.T.K. A Computational Framework Based on Ensemble Deep Neural Networks for Essential Genes Identification. Int. J. Mol. Sci. 2020, 21, 9070. [Google Scholar] [CrossRef] [PubMed]
- Le, N.Q.K. Potential of deep representative learning features to interpret the sequence information in proteomics. Proteomics 2022, 22, e2100232. [Google Scholar] [CrossRef]
- Greener, J.G.; Kandathil, S.M.; Moffat, L.; Jones, D.T. A guide to machine learning for biologists. Nat. Rev. Mol. Cell Biol. 2022, 23, 40–55. [Google Scholar] [CrossRef]
- Cuperlovic-Culf, M.; Nguyen-Tran, T.; Bennett, S.A.L. Machine Learning and Hybrid Methods for Metabolic Pathway Modeling. Methods Mol. Biol. 2023, 2553, 417–439. [Google Scholar] [CrossRef] [PubMed]
- Antonakoudis, A.; Barbosa, R.; Kotidis, P.; Kontoravdi, C. The era of big data: Genome-scale modelling meets machine learning. Comput. Struct Biotec. 2020, 18, 3287–3300. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Kim, G.B.; Lee, S.Y. Machine learning applications in genome-scale metabolic modeling. Curr. Opin. Syst. Biol. 2021, 25, 42–49. [Google Scholar] [CrossRef]
- Carinhas, N.; Bernal, V.; Teixeira, A.P.; Carrondo, M.J.T.; Alves, P.M.; Oliveira, R. Hybrid metabolic flux analysis: Combining stoichiometric and statistical constraints to model the formation of complex recombinant products. BMC Syst. Biol. 2011, 5, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isidro, I.A.; Portela, R.M.; Clemente, J.J.; Cunha, A.E.; Oliveira, R. Hybrid metabolic flux analysis and recombinant protein prediction in Pichia pastoris X-33 cultures expressing a singlechain antibody fragment. Bioprocess Biosyst. Eng. 2016, 39, 1351–1363. [Google Scholar] [CrossRef]
- Ferreira, A.R.; Dias, J.M.L.; von Stosch, M.; Clemente, J.; Cunha, A.E.; Oliveira, R. Fast development of Pichia pastoris GS115 Mut(+) cultures employing batch-to-batch control and hybrid semi-parametric modeling. Bioprocess Biosyst. Eng. 2014, 37, 629–639. [Google Scholar] [CrossRef]
- Teixeira, A.P.; Dias, J.M.L.; Carinhas, N.; Sousa, M.; Clemente, J.J.; Cunha, A.E.; von Stosch, M.; Alves, P.M.; Carrondo, M.J.T.; Oliveira, R. Cell functional enviromics: Unravelling the function of environmental factors. BMC Syst. Biol. 2011, 5, 92. [Google Scholar] [CrossRef] [Green Version]
- von Stosch, M.; Peres, J.; de Azevedo, S.F.; Oliveira, R. Modelling biochemical networks with intrinsic time delays: A hybrid semi-parametric approach. BMC Syst. Biol. 2010, 4, 131. [Google Scholar] [CrossRef] [Green Version]
- Folch-Fortuny, A.; Marques, R.; Isidro, I.A.; Oliveira, R.; Ferrer, A. Principal elementary mode analysis (PEMA). Mol. Biosyst. 2016, 12, 737–746. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- von Stosch, M.; Hamelink, J.M.; Oliveira, R. Hybrid modeling as a QbD/PAT tool in process development: An industrial E-coli case study. Bioprocess Biosyst. Eng. 2016, 39, 773–784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, D.; Jayaraman, A.; Kwon, J.S. Development of a hybrid model for a partially known intracellular signaling pathway through correction term estimation and neural network modeling. PLoS Comput. Biol. 2020, 16, e1008472. [Google Scholar] [CrossRef] [PubMed]
- Umar, M.; Sabir, Z.; Raja, M.A.Z.; Baskonus, H.M.; Yao, S.W.; Ilhan, E. A novel study of Morlet neural networks to solve the nonlinear HIV infection system of latently infected cells. Results Phys. 2021, 25, 104235. [Google Scholar] [CrossRef]
- Umar, M.; Sabir, Z.; Raja, M.A.Z.; Shoaib, M.; Gupta, M.; Sanchez, Y.G. A Stochastic Intelligent Computing with Neuro-Evolution Heuristics for Nonlinear SITR System of Novel COVID-19 Dynamics. Symmetry 2020, 12, 1628. [Google Scholar] [CrossRef]
- Yang, J.H.; Wright, S.N.; Hamblin, M.; McCloskey, D.; Alcantar, M.A.; Schrubbers, L.; Lopatkin, A.J.; Satish, S.; Nili, A.; Palsson, B.O.; et al. A White-Box Machine Learning Approach for Revealing Antibiotic Mechanisms of Action. Cell 2019, 177, 1649–1661.e9. [Google Scholar] [CrossRef]
- Lewis, J.E.; Kemp, M.L. Integration of machine learning and genome-scale metabolic modeling identifies multi-omics biomarkers for radiation resistance. Nat. Commun. 2021, 12, 2700. [Google Scholar] [CrossRef]
- Vijayakumar, S.; Rahman, P.K.S.M.; Angione, C. A Hybrid Flux Balance Analysis and Machine Learning Pipeline Elucidates Metabolic Adaptation in Cyanobacteria. Iscience 2020, 23, 101818. [Google Scholar] [CrossRef]
- Ramos, J.R.C.; Oliveira, G.P.; Dumas, P.; Oliveira, R. Genome-scale modeling of Chinese hamster ovary cells by hybrid semi-parametric flux balance analysis. Bioprocess Biosyst. Eng. 2022, 45, 1889–1904. [Google Scholar] [CrossRef]
- Le Novere, N.; Bornstein, B.; Broicher, A.; Courtot, M.; Donizelli, M.; Dharuri, H.; Li, L.; Sauro, H.; Schilstra, M.; Shapiro, B.; et al. BioModels Database: A free, centralized database of curated, published, quantitative kinetic models of biochemical and cellular systems. Nucleic Acids Res. 2006, 34, D689–D691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olivier, B.G.; Snoep, J.L. Web-based kinetic modelling using JWS Online. Bioinformatics 2004, 20, 2143–2144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mochao, H.; Barahona, P.; Costa, R.S. KiMoSys 2.0: An upgraded database for submitting, storing and accessing experimental data for kinetic modeling. Database J. Biol. Databases Curation 2020, 2020, baaa093. [Google Scholar] [CrossRef] [PubMed]
- Hucka, M.; Fineey, A.; Sauro, H.M.; Bolouri, H.; Doyle, J.C.; Kitano, H.; Arkin, A.P.; Bornstein, B.J.; Bray, D.; Cornish-Bowden, A.; et al. The systems biology markup language (SBML): A medium for representation and exchange of biochemical network models. Bioinformatics 2003, 19, 524–531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, J.; Costa, R.S.; Alexandre, L.; Ramos, J.; Oliveira, R. SBML2HYB: A Python interface for SBML compatible hybrid modelling. Bioinformatics 2023, 39, btad044. [Google Scholar] [CrossRef] [PubMed]
- Pinto, J.; Mestre, M.; Ramos, J.; Costa, R.S.; Striedner, G.; Oliveira, R. A general deep hybrid model for bioreactor systems: Combining first principles with deep neural networks. Comput. Chem. Eng. 2022, 165, 107952. [Google Scholar] [CrossRef]
- Chassagnole, C.; Fell, D.A.; Rais, B.; Kudla, B.; Mazat, J.P. Control of the threonine-synthesis pathway in Escherichia coli: A theoretical and experimental approach. Biochem. J. 2001, 356, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.G.; Tanner, B.C.W.; Chang, S.T.; Esteban, M.; Katze, M.G. Virus infection rapidly activates the P58(IPK) pathway, delaying peak kinase activation to enhance viral replication. Virology 2011, 417, 27–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dano, S.; Madsen, M.F.; Schmidt, H.; Cedersund, G. Reduction of a biochemical model with preservation of its basic dynamic properties. Febs J. 2006, 273, 4862–4877. [Google Scholar] [CrossRef]
- Kingma, D.P.; Ba, J. Adam: A method for stochastic optimization. arxiv 2014, arXiv:1412.6980. [Google Scholar]
- Hoops, S.; Sahle, S.; Gauges, R.; Lee, C.; Pahle, J.; Simus, N.; Singhhal, M.; Xu, L.; Mendes, P.; Kummer, U. COPASI—A COmplex PAthway SImulator. Bioinformatics 2006, 22, 3067–3074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konig, M.; Drager, A.; Holzhutter, H.G. CySBML: A Cytoscape plugin for SBML. Bioinformatics 2012, 28, 2402–2403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, B.B.; Morris, J.; Martin, E.B. Model selection for partial least squares regression. Chemom. Intell. Lab. 2002, 64, 79–89. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case Study | Number of Species | Number of Reactions | Number of Parameters | JWS Online ID | Reference |
|---|---|---|---|---|---|
| E. coli threonine synthesis pathway | 11 | 7 | 47 | chassagnole1 | [38] |
| P58IPK signal transduction pathway | 9 (4 fixed) | 9 | 10 | goodman | [39] |
| Yeast glycolytic oscillations | 7 (1 fixed) | 11 | 31 | dano1 | [40] |
| Hybrid Model | WMSE Train | WMSE Test | WMSE Test (Noise Free) | AICc | CPU Time (h:m:s) | Number of Weights |
|---|---|---|---|---|---|---|
| 11 × 5 × 5 × 7 | 1.03 | 0.99 | 0.07 | 838 | 00:31:00 | 132 |
| 11 × 10 × 10 × 7 | 1.07 | 1.00 | 0.08 | 2510 | 00:29:00 | 307 |
| 11 × 15 × 15 × 7 | 1.04 | 0.99 | 0.08 | 2102 | 00:35:00 | 532 |
| 11 × 20 × 20 × 7 | 1.03 | 0.98 | 0.07 | 2400 | 00:33:00 | 807 |
| 11 × 5 × 5 × 5 × 7 | 1.03 | 0.99 | 0.07 | 918 | 00:32:00 | 162 |
| 11 × 10 × 10 × 10 × 7 | 1.05 | 0.98 | 0.07 | 1890 | 00:40:00 | 417 |
| 11 × 15 × 15 × 15 × 7 | 1.04 | 1.01 | 0.08 | 2659 | 00:36:00 | 772 |
| 11 × 20 × 20 × 20 × 7 | 1.04 | 1.00 | 0.07 | 3684 | 00:35:00 | 1227 |
| Hybrid Model | WMSE Train | WMSE Test | WMSE Test (Noise Free) | AICc | CPU Time (h:m:s) | Number of Weights |
|---|---|---|---|---|---|---|
| 5 × 5 × 5 × 9 | 1.60 | 1.51 | 0.54 | 1916 | 00:12:10 | 114 |
| 5 × 10 × 10 × 9 | 1.59 | 1.48 | 0.53 | 2181 | 00:11:54 | 269 |
| 5 × 15 × 15 × 9 | 1.61 | 1.50 | 0.56 | 2810 | 00:15:15 | 474 |
| 5 × 20 × 20 × 9 | 1.58 | 1.49 | 0.51 | 3480 | 00:20:48 | 729 |
| 5 × 5 × 5 × 5 × 9 | 1.45 | 1.50 | 0.48 | 1890 | 00:13:15 | 144 |
| 5 × 10 × 10 × 10 × 9 | 1.23 | 1.28 | 0.12 | 1430 | 00:16:10 | 379 |
| 5 × 15 × 15 × 15 × 9 | 1.35 | 1.36 | 0.31 | 2140 | 00:19:30 | 714 |
| 5 × 20 × 20 × 20 × 9 | 1.34 | 1.40 | 0.36 | 4150 | 00:27:12 | 1149 |
| Hybrid Model | WMSE Train | WMSE Test | WMSE Test (Noise Free) | AICc | CPU Time (h:m:s) | Number of Weights |
|---|---|---|---|---|---|---|
| 7 × 5 × 5 × 11 | 20.12 | 21.05 | 20.14 | 5730 | 01:05:00 | 136 |
| 7 × 10 × 10 × 11 | 1.87 | 1.99 | 1.67 | 3818 | 01:20:00 | 311 |
| 7 × 15 × 15 × 11 | 1.74 | 1.78 | 1.56 | 4120 | 01:15:00 | 536 |
| 7 × 20 × 20 × 11 | 1.16 | 1.43 | 0.98 | 2740 | 01:24:00 | 811 |
| 7 × 5 × 5 × 5 × 11 | 5.33 | 5.84 | 5.14 | 3930 | 01:33:00 | 166 |
| 7 × 10 × 10 × 10 × 11 | 0.93 | 0.94 | 0.11 | −41 | 01:31:00 | 421 |
| 7 × 15 × 15 × 15 × 11 | 0.98 | 0.97 | 0.21 | 784 | 01:20:00 | 776 |
| 7 × 20 × 20 × 20 × 11 | 0.97 | 0.97 | 0.17 | 2213 | 01:40:00 | 1231 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, J.; Ramos, J.R.C.; Costa, R.S.; Oliveira, R. A General Hybrid Modeling Framework for Systems Biology Applications: Combining Mechanistic Knowledge with Deep Neural Networks under the SBML Standard. AI 2023, 4, 303-318. https://doi.org/10.3390/ai4010014
Pinto J, Ramos JRC, Costa RS, Oliveira R. A General Hybrid Modeling Framework for Systems Biology Applications: Combining Mechanistic Knowledge with Deep Neural Networks under the SBML Standard. AI. 2023; 4(1):303-318. https://doi.org/10.3390/ai4010014
Chicago/Turabian StylePinto, José, João R. C. Ramos, Rafael S. Costa, and Rui Oliveira. 2023. "A General Hybrid Modeling Framework for Systems Biology Applications: Combining Mechanistic Knowledge with Deep Neural Networks under the SBML Standard" AI 4, no. 1: 303-318. https://doi.org/10.3390/ai4010014