
Multiple Intramolecular Hydrogen Bonding in Large Biomolecules: DFT Calculations and Deuterium Isotope Effects on 13C Chemical Shifts as a Tool in Structural Studies



Abstract
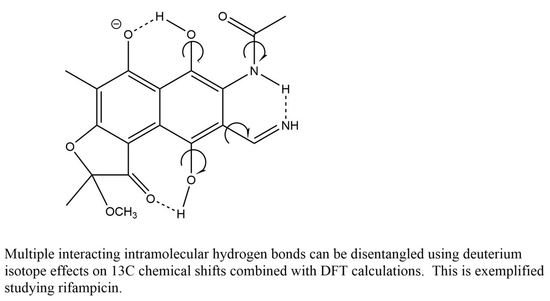
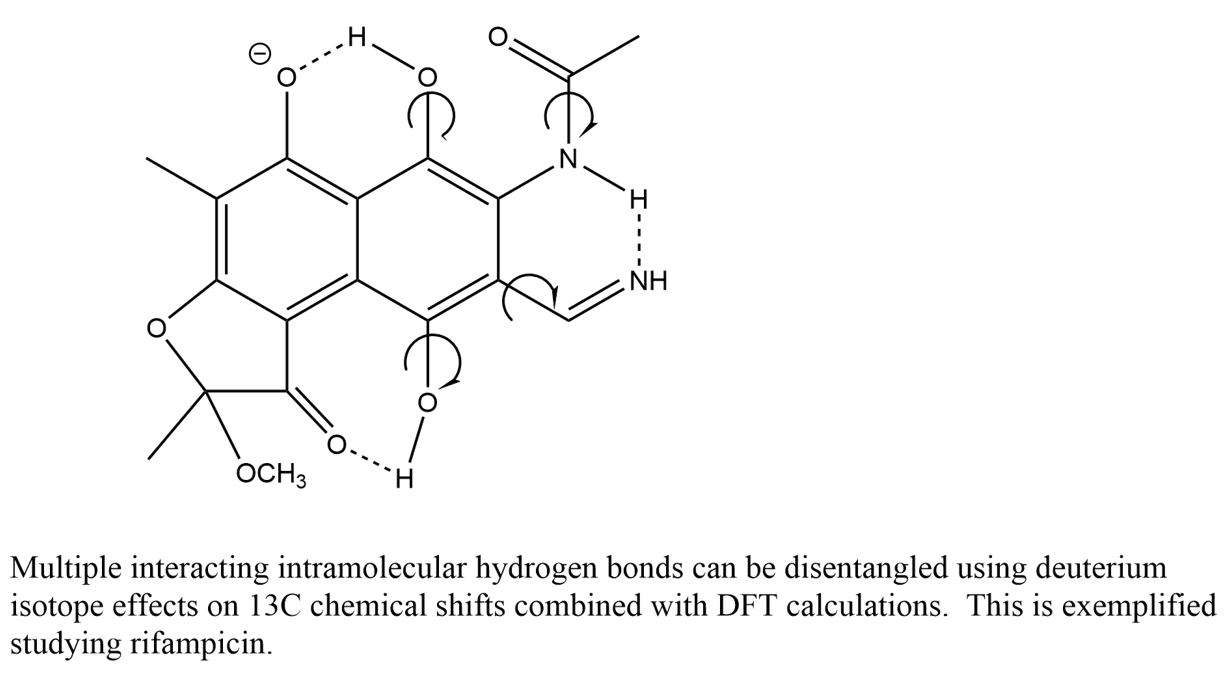
:
1. Introduction
2. Experimental
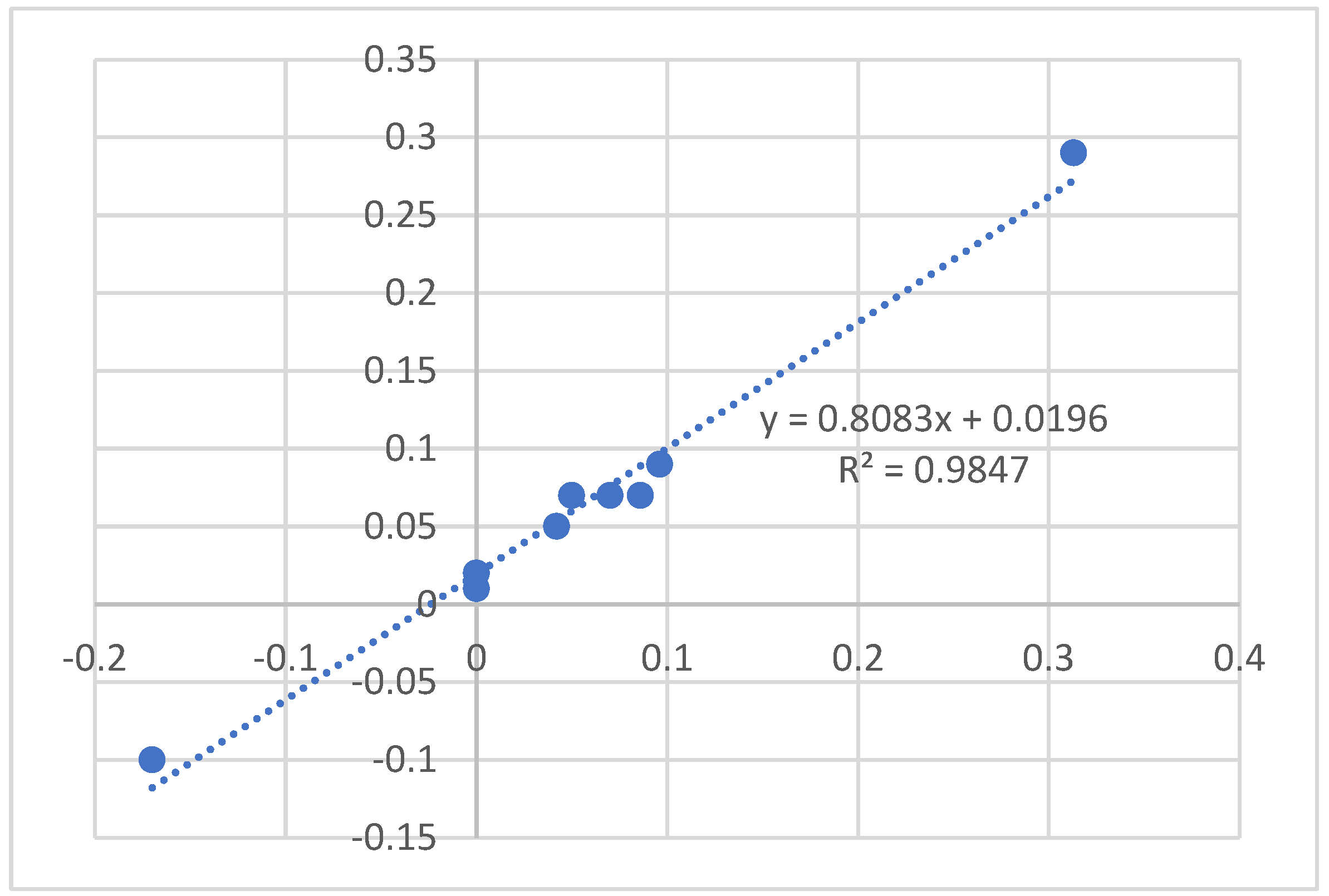
2.1. Calculations
2.2. NMR
2.3. Deuteriation
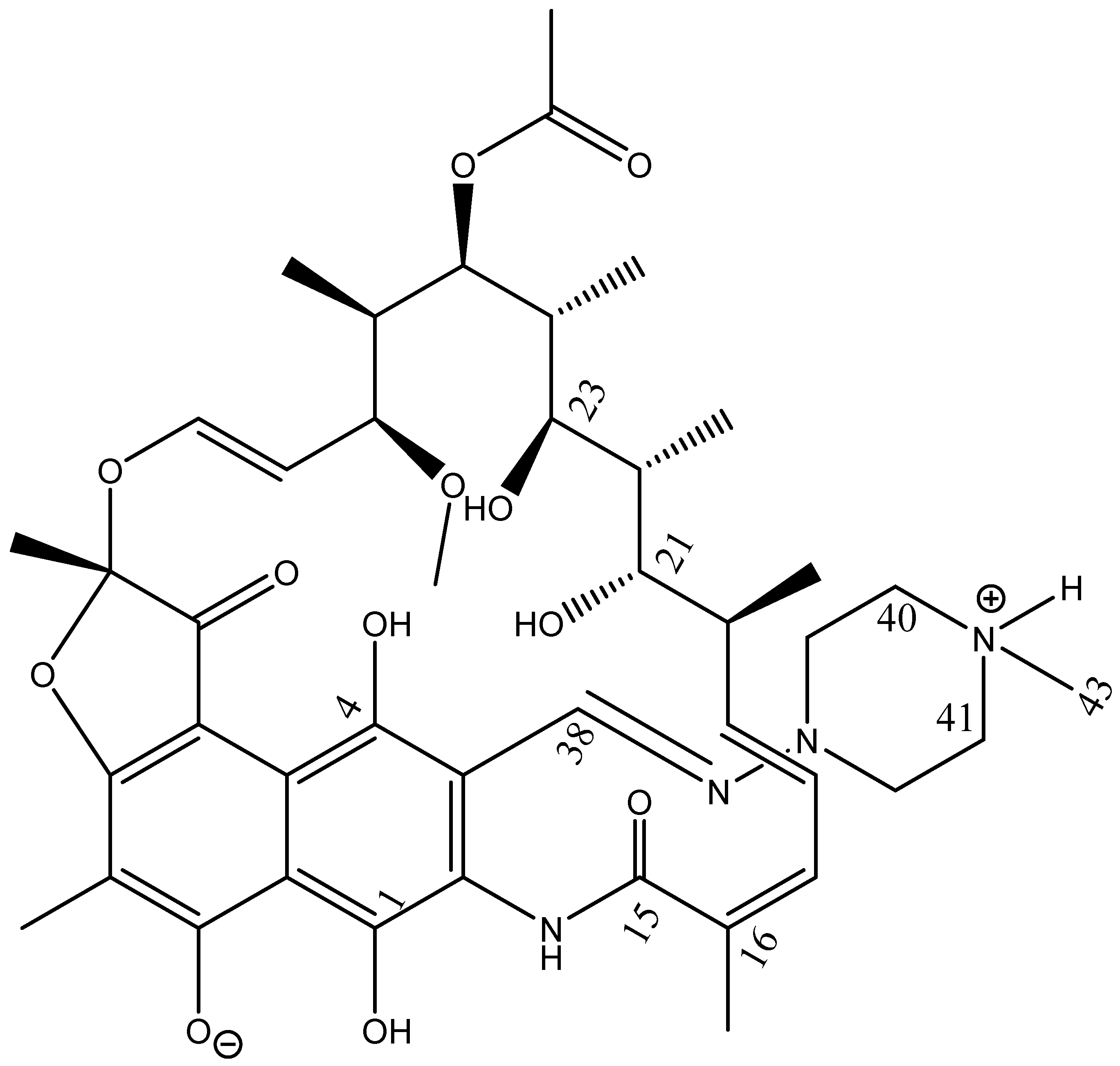
3. Results
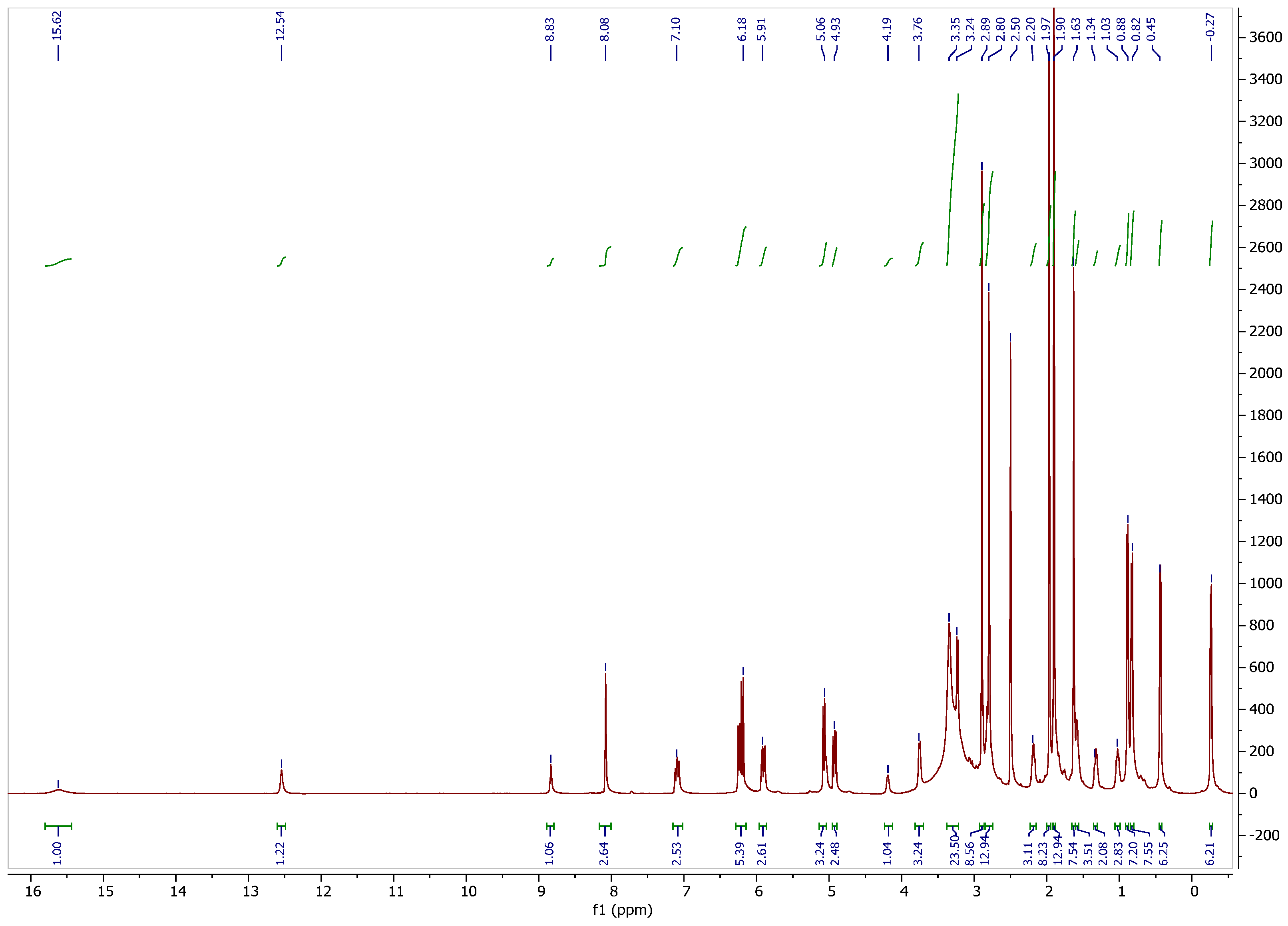
3.1. 1H NMR Spectrum
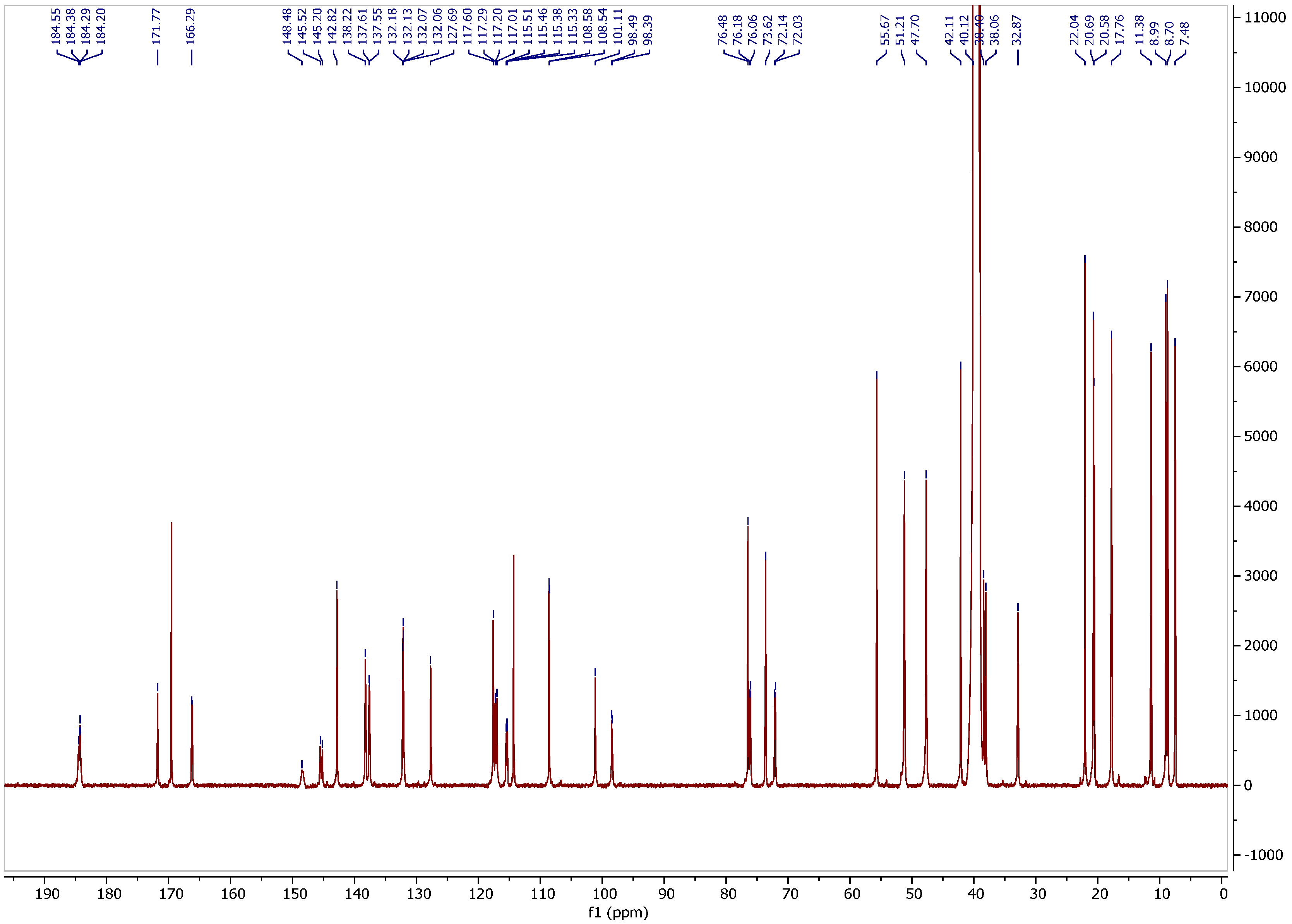
3.2. 13C NMR Spectrum
3.3. Assignment of Isotope Effects on 13C Chemical Shifts
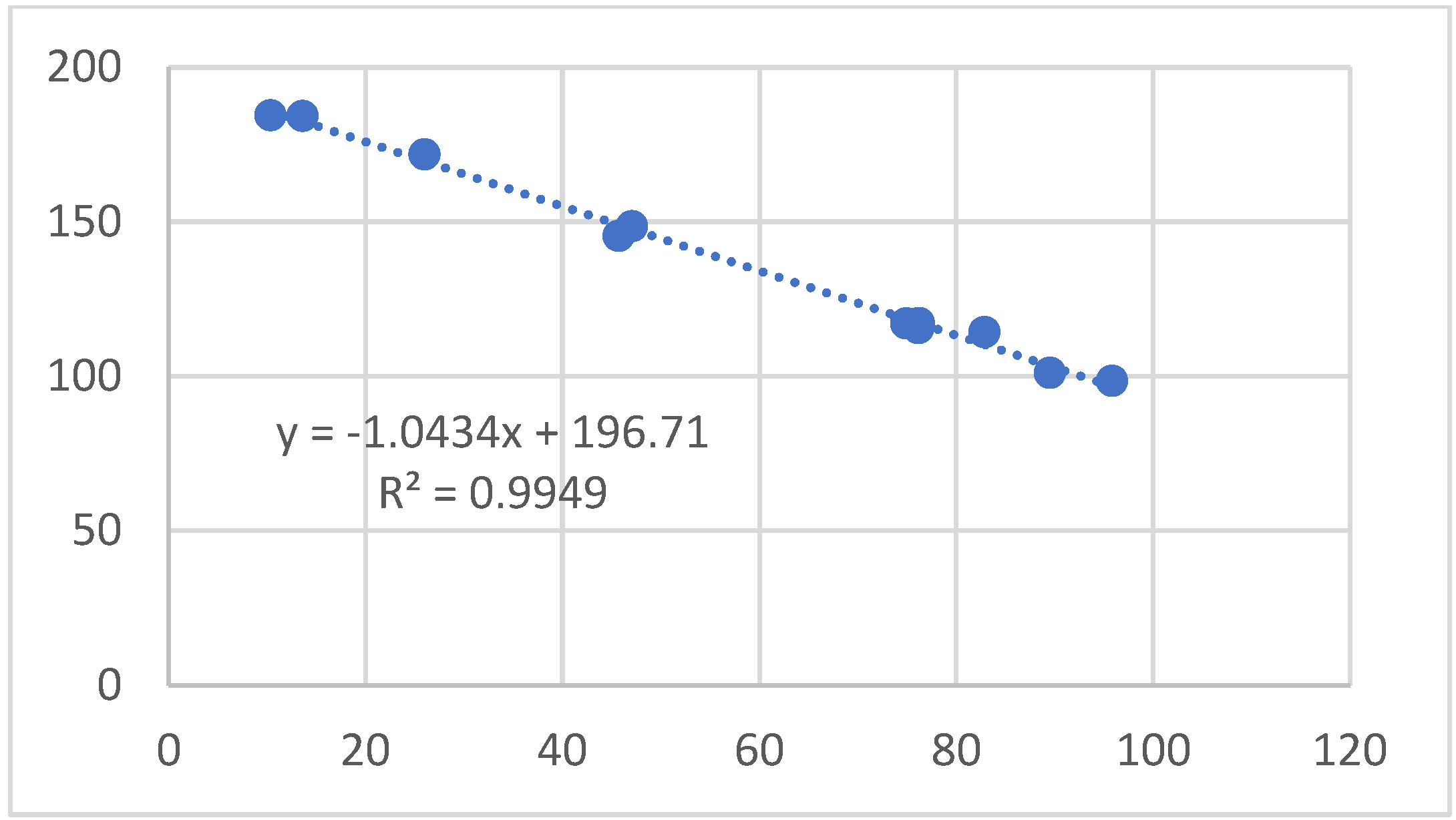
3.4. Isotope Effects
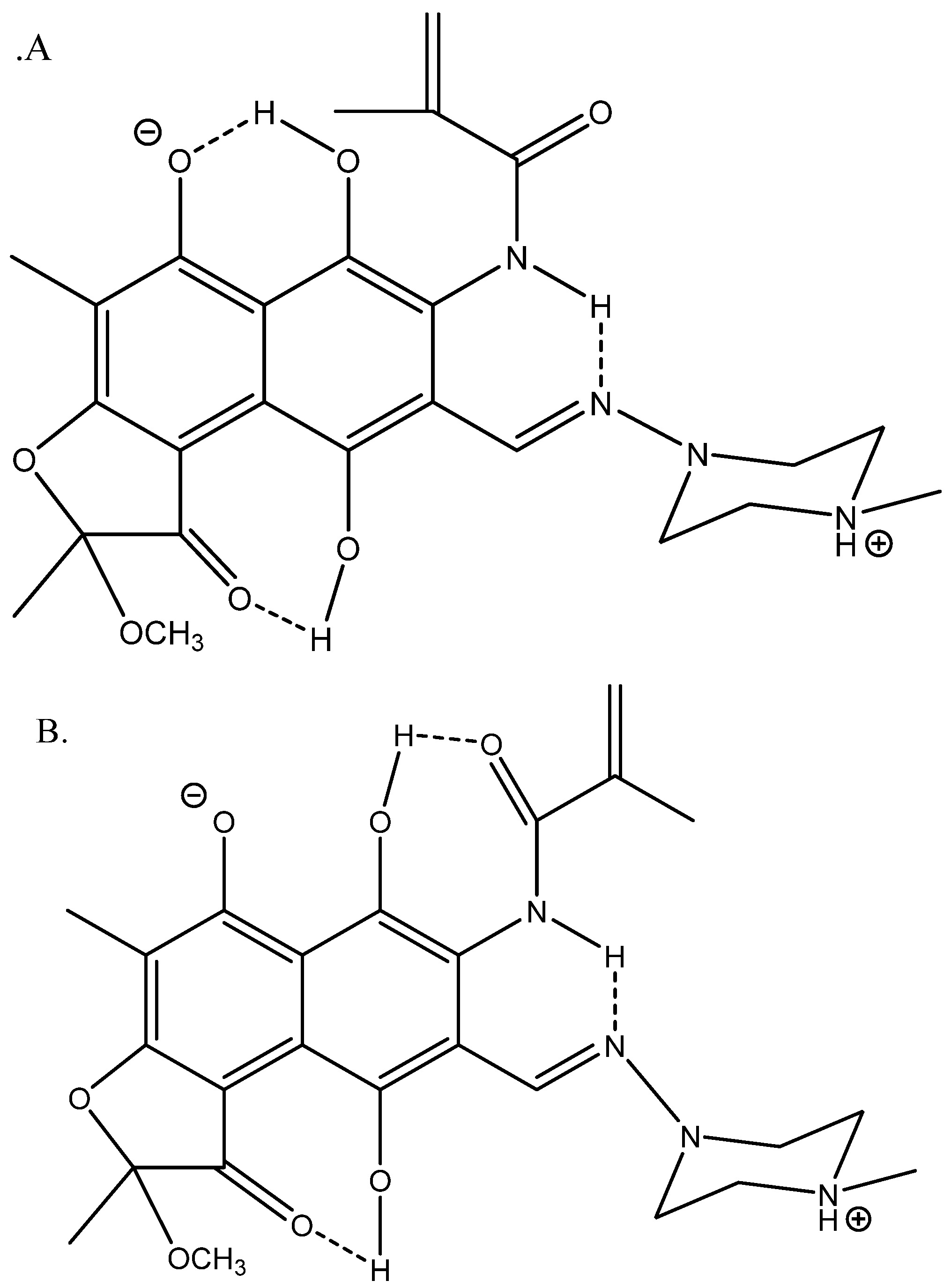
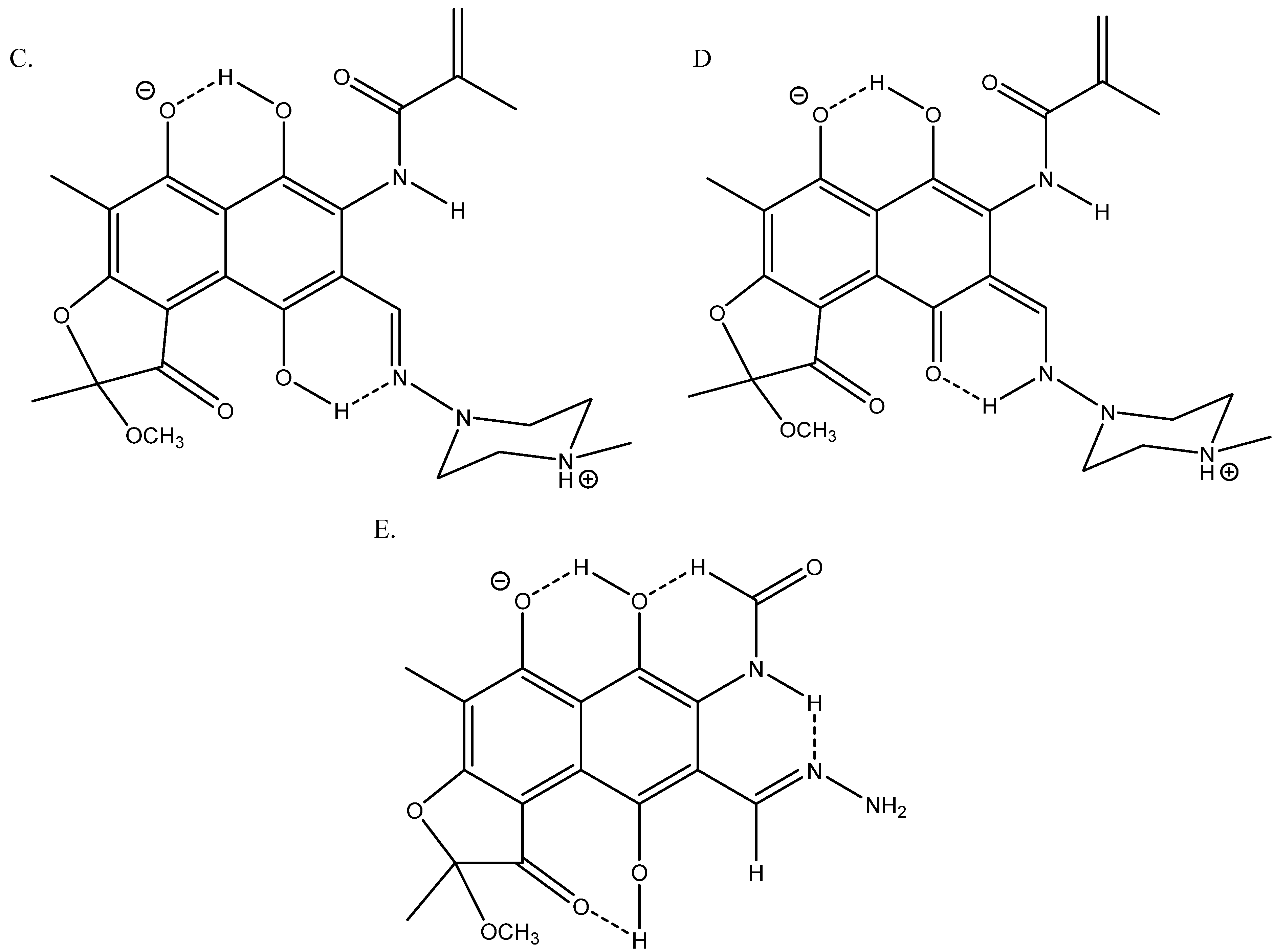
3.5. Calculations
3.5.1. 13C Nuclear Shieldings
3.5.2. Isotope Effects on Chemical Shifts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pyta, K.; Janas, A.; Skrzypczak, N.; Schilf, W.; Wicher, B.; Gdaniec, M.; Bartl, F.; Przybylski, P. Specific Interactions between Rifamycin Antibiotics and Water Influencing Ability to Overcome Natural Cell Barriers and the Range of Antibacterial Potency. ACS Infect. Dis. 2019, 5, 1754–1763. [Google Scholar] [CrossRef] [PubMed]
- Kozyra, P.; Kaczor, A.; Karczmarzyk, Z.; Wysocki, W.; Pitucha, M. Experimental and computational studies of tautomerism pyridine carbonyl thiosemicarbazide derivatives. Struct. Chem. 2023. [Google Scholar] [CrossRef]
- Pyta, K.; Przybylski, P.; Klich, K.; Stefańska, J. A new model of binding of rifampicin and its amino analogues as zwitterionsto bacterial RNA polymerase. Org. Biomol. Chem. 2012, 10, 8283–8297. [Google Scholar] [CrossRef] [PubMed]
- Hansen, P.E. Isotope Effects on Chemical shifts of small molecules. Molecules 2022, 27, 2405. [Google Scholar] [CrossRef]
- Hansen, P.E. Isotope Effects on Chemical Shifts of Proteins and Peptides. Magn. Reson. Chem. 2000, 38, 1–10. [Google Scholar] [CrossRef]
- Hansen, P.E. Isotope Effects on Chemical Shifts in the Study of Hydrogen Bonded Biological Systems. Progress NMR 2020, 120–121, 109–117. [Google Scholar] [CrossRef]
- Kim, Y.-I.; Manalo, M.N.; Perés, M.L.; LiWang, A. Computational and empirical trans-hydrogen bond deuterium isotope shifts suggest that N1–N3 A: U hydrogen bonds of RNA are shorter than those of A:T hydrogen bonds of DNA. J. Biomol. NMR 2006, 34, 229–236. [Google Scholar] [CrossRef]
- Manalo, M.N.; Perés, L.M.; LiWang, A. Hydrogen-bonding and base-stacking interactions are coupled in DNA, as suggested by Calculated and experimental trans-Hbond deuterium isotope effects. J. Am. Chem. Soc. 2007, 129, 11298–11299. [Google Scholar] [CrossRef]
- Hansen, P.E. Methods to distinguish tautomeric cases from static ones. In Tautomerism: Ideas, Compounds, Applications; Antonov, L., Ed.; Wiley-VCH: Weinheim, Germany, 2016. [Google Scholar]
- Pyta, K.; Przybylski, P.; Wicker, B.; Gdaniec, M.; Stefańska, J. Intramolecular proton transfer impact on antibacterial properties of ansamycin antibiotic rifampicin and its new amino analogues. Org. Biomol. Chem. 2012, 10, 2385–2388. [Google Scholar] [CrossRef]
- Morgante, P.; Peverat, R. The devil in the details: A tutorial review on some undervalued aspects of density functional theory calculations. Quantum Chem. 2020, 120, e26332. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Ireta, J.; Neugebauer, J.; Scheffler, M. On the Accuracy of DFT for Describing Hydrogen Bonds: Dependence on the Bond Directionality. J. Phys. Chem. A 2004, 108, 5692–5698. [Google Scholar] [CrossRef]
- Afonin, A.V.; Ushakov, I.A.; Simonenko, D.E.; Shmidt, E.Y.; Zorina, N.V.; Mikhaleva, A.I.; Trofimov, B.A. 1H and 13C NMR Study of Bifurcated Intramolecular Hydrogen Bonds in 2,6-Bis(2-pyrrolyl)pyridine and 2,6-Bis(1-vinyl-2-pyrrolyl)pyridine. Russ. J. Org. Chem. 2005, 41, 1516–1521, Translated from Zhurnal Organicheskoi Khimii, Vol. 41, No. 10, 2005. [Google Scholar] [CrossRef]
- Afonin, A.V.; Vashchenko, A.V.; Ushakov, I.A.; Zorina, N.V.; Schmidt, E.Y. Comparative analysis of hydrogen bondingwith participation of the nitrogen, oxygen andsulfur atoms in the 2(2-heteroaryl)pyrroles andtheir trifluoroacetyl derivatives based on the 1H,13C,15N spectroscopy and DFT calculations. Magn. Reson. Chem. 2008, 46, 441–447. [Google Scholar] [CrossRef]
- Afonin, A.V.; Vashchenko, A.V.; Sigalov, M.V. Estimating the energy of intramolecular hydrogenbonds from 1H NMR and QTAIM calculations. Org. Biomol. Chem. 2016, 14, 11199. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Ditchfield, R.; Hehre, W.J.; Pople, J.A. Self-consistent Molecular-orbital methods. 9. Extended Gaussian-type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. [Google Scholar] [CrossRef]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilization of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Scalmani, G.; Frisch, M.J. Continuous surface charge polarizable continuum models of solvation. I. General formalism. J. Chem. Phys. 2010, 132, 114110. [Google Scholar] [CrossRef]
- Ditchfield, R. Self-consistent perturbation theory of diamagnetism. I. A gauge-invariant LCAO method for N.M.R. chemical shifts. Mol. Phys. 1974, 27, 789–807. [Google Scholar] [CrossRef]
- Wolinski, K.; Hilton, F.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Jameson, C.J.; Osten, H.-J. The NMR isotope shift in polyatomic molecules. Estimation of the dynamic factors. J. Chem. Phys. 1984, 81, 4300–4305. [Google Scholar] [CrossRef]
- Jameson, C.J. Isotopes in the Physical and Biomedical Sciences. Isotopic Applications in NMR Studies; Buncel, E., Jones, J.R., Eds.; Elsevier: Amsterdam, The Netherlands, 1991. [Google Scholar]
- Dziembowska, T.; Rozwadowski, Z.; Filarowski, A.; Hansen, P.E. A multinuclear NMR study of proton transfer equilibrium in Schiff bases derived from 2-hydroxy-1-naphtaldehyde. Deuterium isotope effects on 13C and 15N chemical shifts. Magn. Reson. Chem. 2001, 39, S67–S80. [Google Scholar] [CrossRef]
- Filarowski, A.; Koll, A.; Rospenk, M.; Krol-Starzomska, I.; Hansen, P.E. Tautomerism of sterically hindered Schiff bases. Deuterium Isotope Effects on 13C Chemical Shifts. J. Phys. Chem. A 2005, 109, 4464–4473. [Google Scholar] [CrossRef] [PubMed]
- Jarret, M.; Sin, N.; Dintzner, M. Deuterium Isotope Effects on13C NMR Chemical Shifts of Amides. Microchem. J. 1997, 56, 19–21. [Google Scholar] [CrossRef]
- Newmark, R.A.; Hill, J.R. Assignment of Primary and Secondary Amide Carbonyl Resonances in Carbon-13 NMR. J. Magn. Reson. 1976, 21, 1–7. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Carbon | 13C CS a | IE OH-1 b | IE H-4 c | IE NHCO c | IE NH+ d | IE OH-21,23 c |
|---|---|---|---|---|---|---|
| C-1 | 148.52 | 0.33 (0.3) | ||||
| C-2 d | 115.51 | 0.05 (0.07) d | 0.13 | |||
| C-3 | 114.24 | |||||
| C-4 | 145.40 | −0.10 (−0.15) | 0.31 (0.29) | |||
| C-5 | 98.51 | 0.07 (0.08) | 0.05 (0.09) | |||
| C-6 | 171.77 | |||||
| C-7 | 101.02 | −0.09 (0.01) | ||||
| C-8 | 184.21 | 0.08 (0.09) | ||||
| C-9 d | 117.21 | −0.09 (−0.14) | 0.0 (0.02) | |||
| C-10 d | 117.05 | 0.07 (−0.11) | 0.07 (0.07) | |||
| C-11 | 184.43 | 0.11 (0.07) | −0.17 (−0.10) | |||
| C-12 | 108.55 | 0.042 | ||||
| C-13 | ||||||
| C-14 | ||||||
| C-15 | 166.22 | 0.099 | ||||
| C-16 | 132.17 | 0.046 | ||||
| C-21 | 0.12 | |||||
| C-23 | 0.12 | |||||
| C-38 | 137.59 | 0.07 (0.03) | ||||
| C-40,41 | 51.21 | 0.07 | ||||
| C-43 | 42.11 | 0.04 e |
| 0 μL | 2 μL | 4 μL | 9 μL | 20 μL | 30 μL | 10 μL a | |
|---|---|---|---|---|---|---|---|
| C11 | 184.43 | 184.37(H) b 184.57(D) | 184.29(H) 184.51(D) | 184.31(H) 184.47(D) | 184.35(H) 184.55(D) | 184.40(H) 184.60(D) | 184.44(5) |
| C8 | 184.21 | 184.21(H) 184.13(D) | 184.29 (H) 184.20 (D) | 184.31 (D) 184.23(H) | 184.30(D) | 184.35(D) | 184.25 |
| C6 | 171.77 | 171.77 | 171.78 | 171.77 | 171.83 | 171.89 | 171.80 |
| C35 | 169.48 | 169.49 | 169.51 | 169.54 | 169.64 | 169.72 | 169.53 |
| C15 | 166.22 | 166.25 | 125.26 | 166.29 | 166.42 | 166.52 | 166.28 |
| C1 | 148.52 | 148.45 | 148.34 | 148.28 | 148.28 | 148.32 | 148.57 |
| C4 | 145.40 | 145.48(H) 145.17(D) | 145.50(H) 145.19(D) | 145.54(D) 145.21(H) | 145.63(D) 145.32(H) | 145.67(D) 145.36(H) | 145.40 |
| C29 | 142.79 | 142.79 | 142.81 | 142.82 | 142.89 | 142.94 | 142.80 |
| C19 | 138.13 | 138.16 | 138.18 | 138.19 | 138.30 | 138.37 | 138.17 |
| C38 | 137.59 | 137.60 | 137.54 | 137.49 | 137.54 | 137.55 | 137.55 |
| C16 | 132.17 | 132.15 | 132.13 | 132.17 | 132.18 | 132.20 | 132.18 |
| C17 | 131.99 | 132.02 | 132.05 | 132.06 | - | - | 132.03 |
| C18 | 127.72 | 127.70 | 127.71 | 127.68 | 127.73 | 127.76 | 127.70 |
| C28 | 117.59 | 117.58 | 117.58 | 117.56 | 117.63 | 117.67 | 117.61 |
| C9 | 117.21 | 117.26(D) 117.17(H) | 117.28(D) 117.20(H) | 117.30(D) 117.21(H) | 117.30(D) w c (H) | 117.35 w(H) | 117.23 |
| C10 | 117.05 | 117.00 | 117.01 | 116.98 | 117.04 | 117.09 | 117.08 |
| C2 e | 115.51 | 115.48(HH) 115.44(DH) 115.34(HD) 115.31(DD) | 115.48(HH) 115.44(DH) 115.36(DH) 115.31(DD) | 115.46(HH) 115.42(DH) 115.33(HD) 115.29(DD) | 115.35(DD) | 115.51 d 115.38(DD) | 115.51 |
| C3 | 114.24 | 114.24 | 114.27 | 114.27 | 114.34 | 114.38 | 114.25 |
| C12 | 108.55 | 108.55 | 108.57 | 108.57 | 108.62 | 108.67 | 108.57 |
| C7 | 101.02 | 101.07 | 101.08 | 101.11 | 101.23 | 101.31 | 101.06 |
| C5 | 98.51 | 98.48 | 98.46 | 98.44 | 98.42 | 98.48 | 98.54 |
| C37 | 55.63 | 55.64 | 55.65 | 55.65 | 55.72 | 55.77 | 55.65 |
| C40 | 51.21 | 51.17 | 51.19 | 51.18 | 51.24 | 51.27 | 51.25 |
| C39 | 47.68 | 47.66 | 47.69 | 47.68 | 47.72 | 47.76 | 47.71 |
| N-CH3 | 42.11 | 42.07 | 42.09 | 42.09 | 42.14 | 42.21 | 42.16 |
| Carbon | NS a of Structure C | NS of Structure D | Difference b |
|---|---|---|---|
| C-1 | 86.46 | 79.79 | 6.67 |
| C-2 | 18.06 | 18.62 | −0.56 |
| C-3 | 73.73 | 77.66 | −3.93 |
| C-4 | 73.35 | 64.57 | −21.22 |
| C-5 | 94.86 | 90.29 | 4.57 |
| C-6 | 24.91 | 26.62 | −1.71 |
| C-7 | 47.25 | 41.61 | 5.64 |
| C-8 | 42.11 | 12.06 | 30.05 |
| C-9 | 91.15 | 74.96 | 16.19 |
| C-10 | 83.39 | 92.13 | −8.74 |
| C-14 | 11.64 | 10.14 | 1.50 |
| C-19 | 36.79 | 37.93 | −1.14 |
| C-21 | 27.40 | 54.74 | −27.34 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hansen, P.E.; Kamounah, F.S. Multiple Intramolecular Hydrogen Bonding in Large Biomolecules: DFT Calculations and Deuterium Isotope Effects on 13C Chemical Shifts as a Tool in Structural Studies. Chemistry 2023, 5, 1317-1328. https://doi.org/10.3390/chemistry5020089
Hansen PE, Kamounah FS. Multiple Intramolecular Hydrogen Bonding in Large Biomolecules: DFT Calculations and Deuterium Isotope Effects on 13C Chemical Shifts as a Tool in Structural Studies. Chemistry. 2023; 5(2):1317-1328. https://doi.org/10.3390/chemistry5020089
Chicago/Turabian StyleHansen, Poul Erik, and Fadhil S. Kamounah. 2023. "Multiple Intramolecular Hydrogen Bonding in Large Biomolecules: DFT Calculations and Deuterium Isotope Effects on 13C Chemical Shifts as a Tool in Structural Studies" Chemistry 5, no. 2: 1317-1328. https://doi.org/10.3390/chemistry5020089