

Synthesis and Structure of COE-11, a New Borosilicate Zeolite with a Two-Dimensional Pore System of 12-Ring Channels
 ,
,
Abstract
:
1. Introduction
2. Experimental
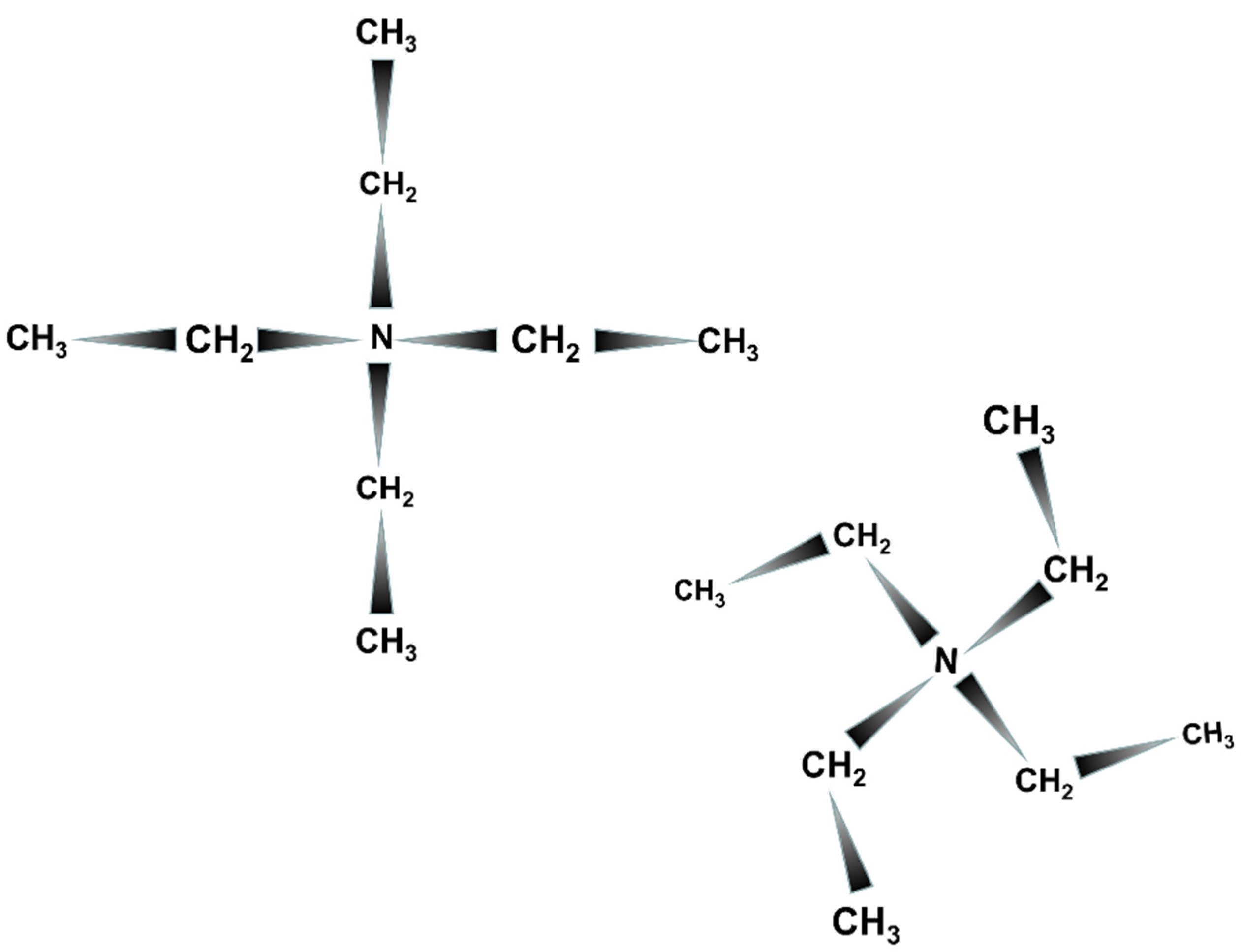
2.1. Synthesis
- -
- variable concentrations of the reaction compounds;
- -
- the type of the OSDA: TEAOH (Aldrich, 40% in water), TEABr (Sigma-Aldrich, 98%), tetraethylphosphonium bromide (Merck, 99%), tetraphenylphosphonium bromide (abcr, 98%), triethylamine (Merck, for synthesis), TEAOH + tetramethylammonium hydroxide (Merck, 25% in water);
- -
- the type of silica source: silica sol (Ludox HS40, Ludox TM50, Levasil 200A, Syton-HT50 Colloidal Silica Slurry), silica gel (Merck and homemade), borosilicate gel (homemade), fumed silica (Aerosil), mesoporous silica (calcined MCM-41, calc. MCM-48), mesoporous borosilicate (calc. B-MCM-41), crystalline sodium silicate (Na-RUB-18) and tetramethoxysilane (Sigma-Aldrich, 98%);
- -
- the type of the boron source: boric acid (J.T.Baker, p.a.), borosilicate gel (homemade), borax (Na₂B₄O₇·10H₂O) (Merck, p.a.);
- -
- the application of different heating schemes to the reaction mixture (preheated at 155 °C for different times), using preheated steel autoclaves, aged at room temperature;
- -
- and the addition of seeds.
2.2. General Characterization
2.3. Structure Analysis
3. Results and Discussion
3.1. Synthesis
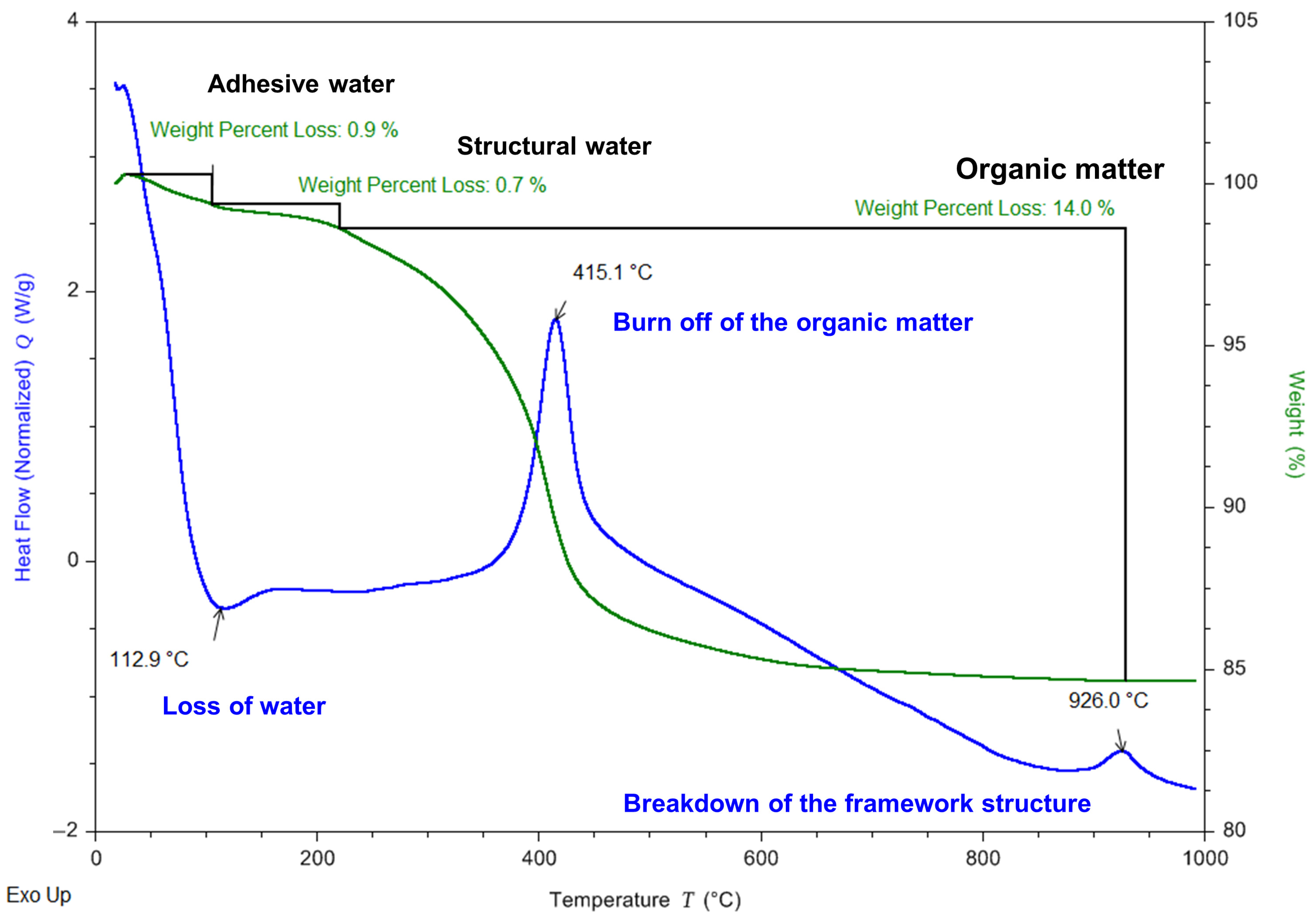
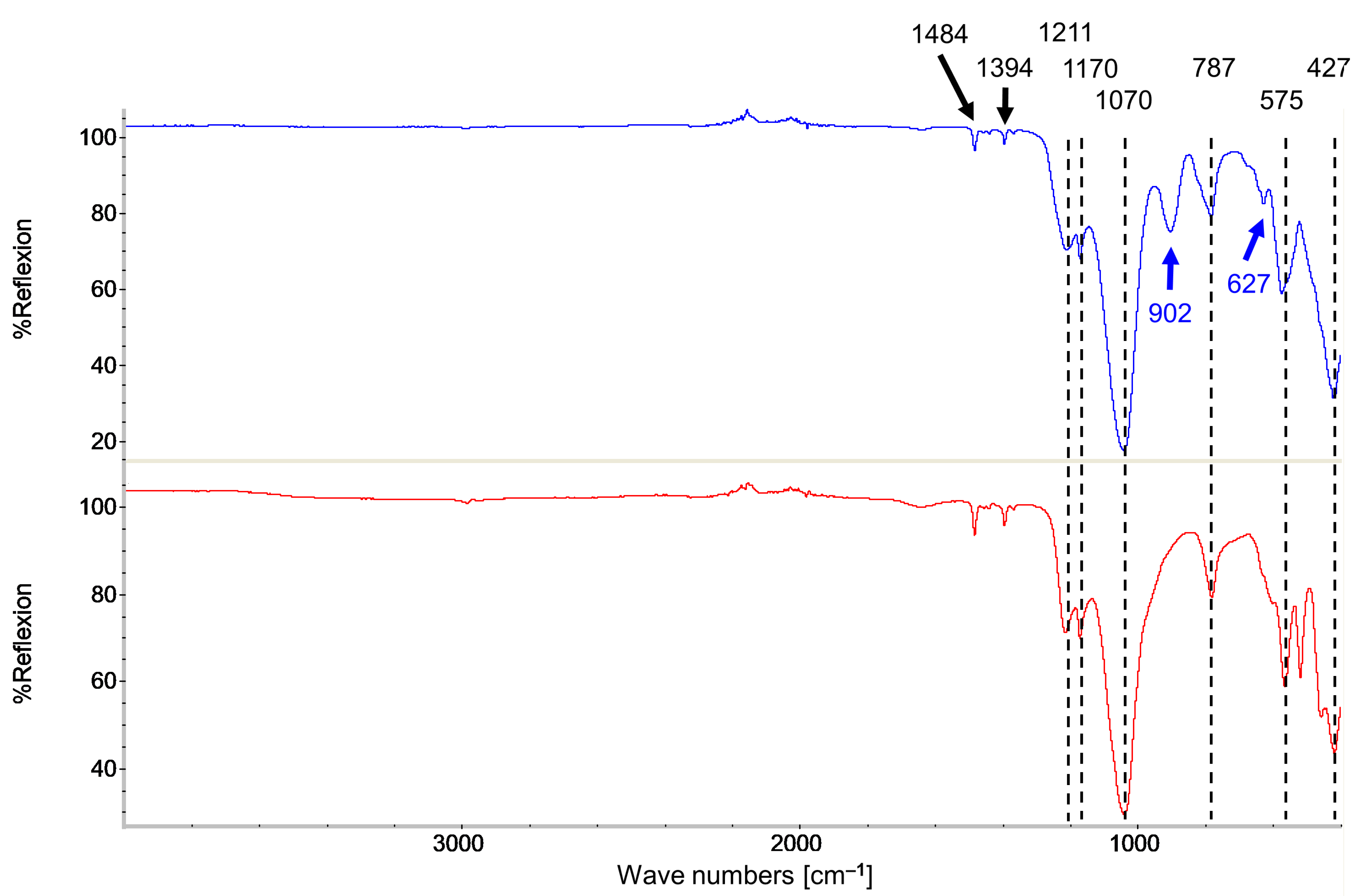
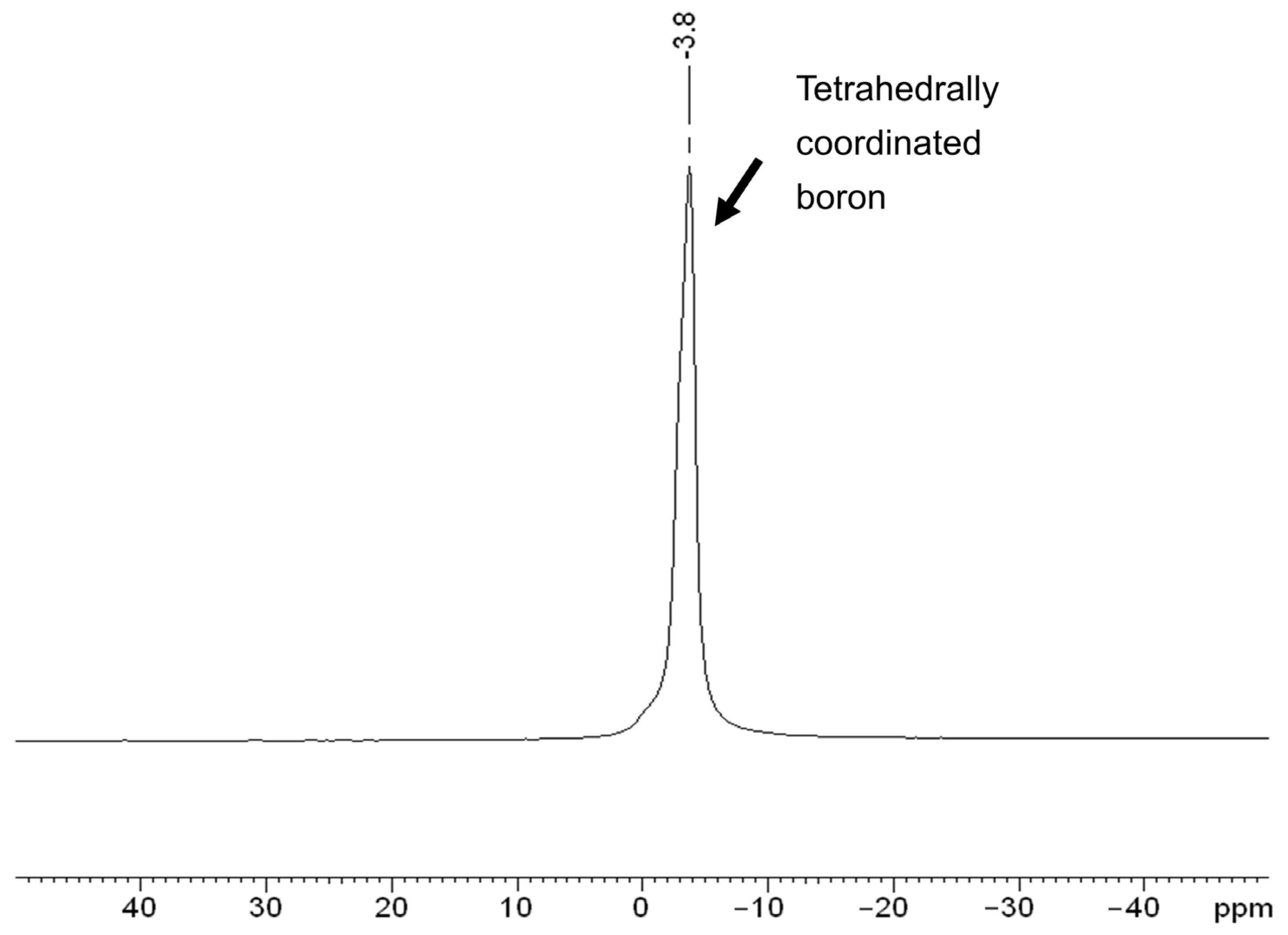
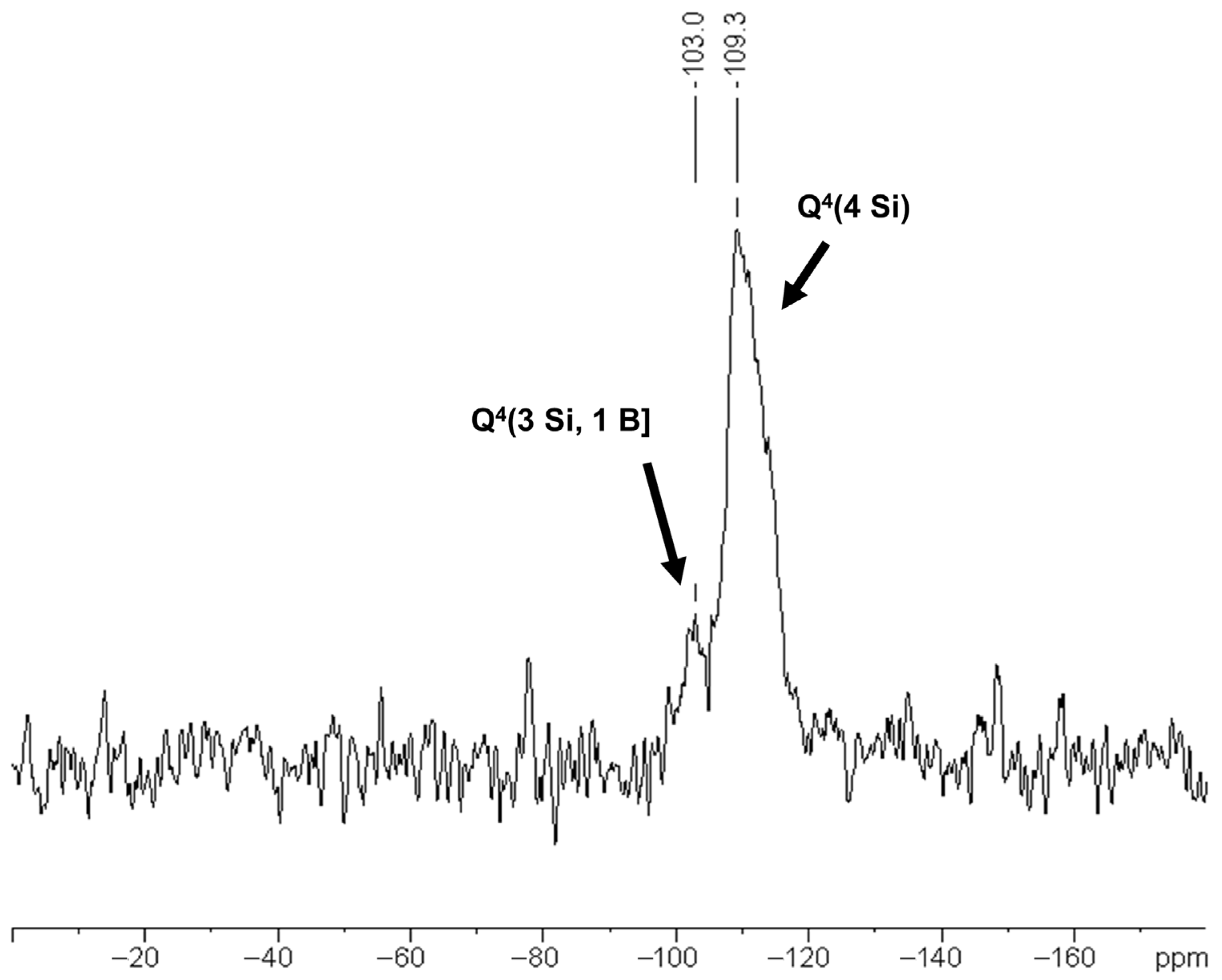
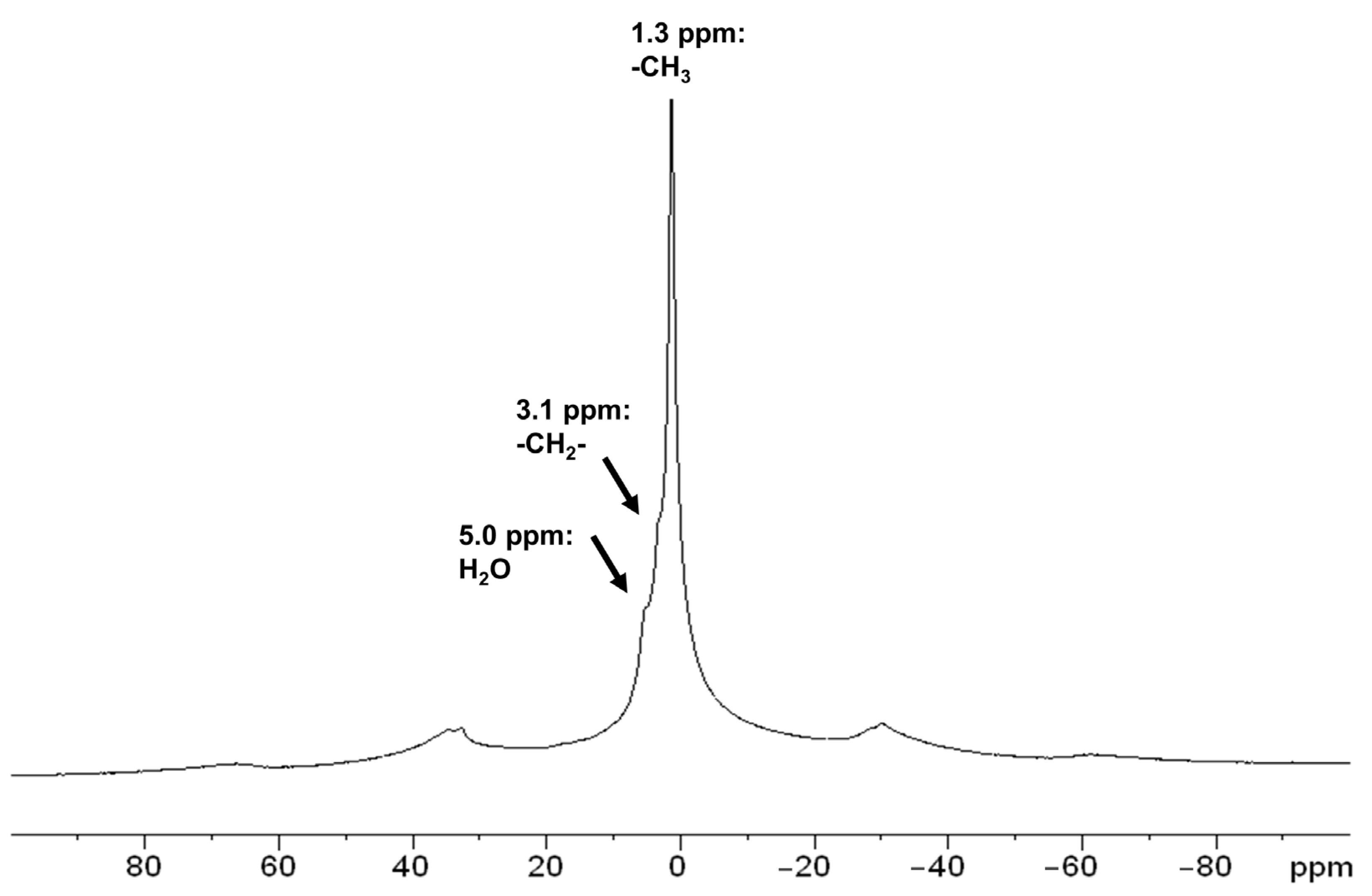
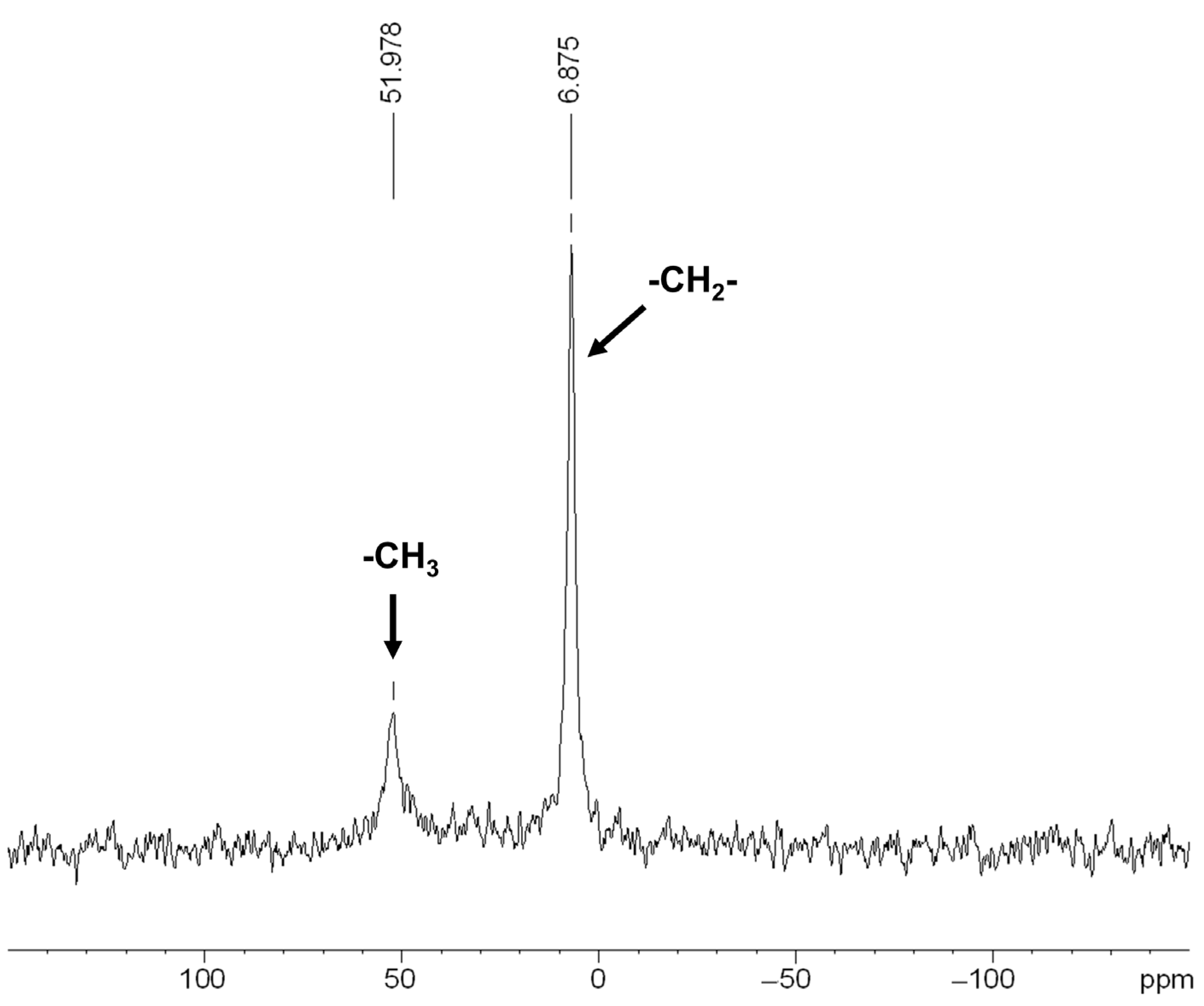
3.2. Characterization
3.3. Structure Analysis
3.3.1. Structure Determination
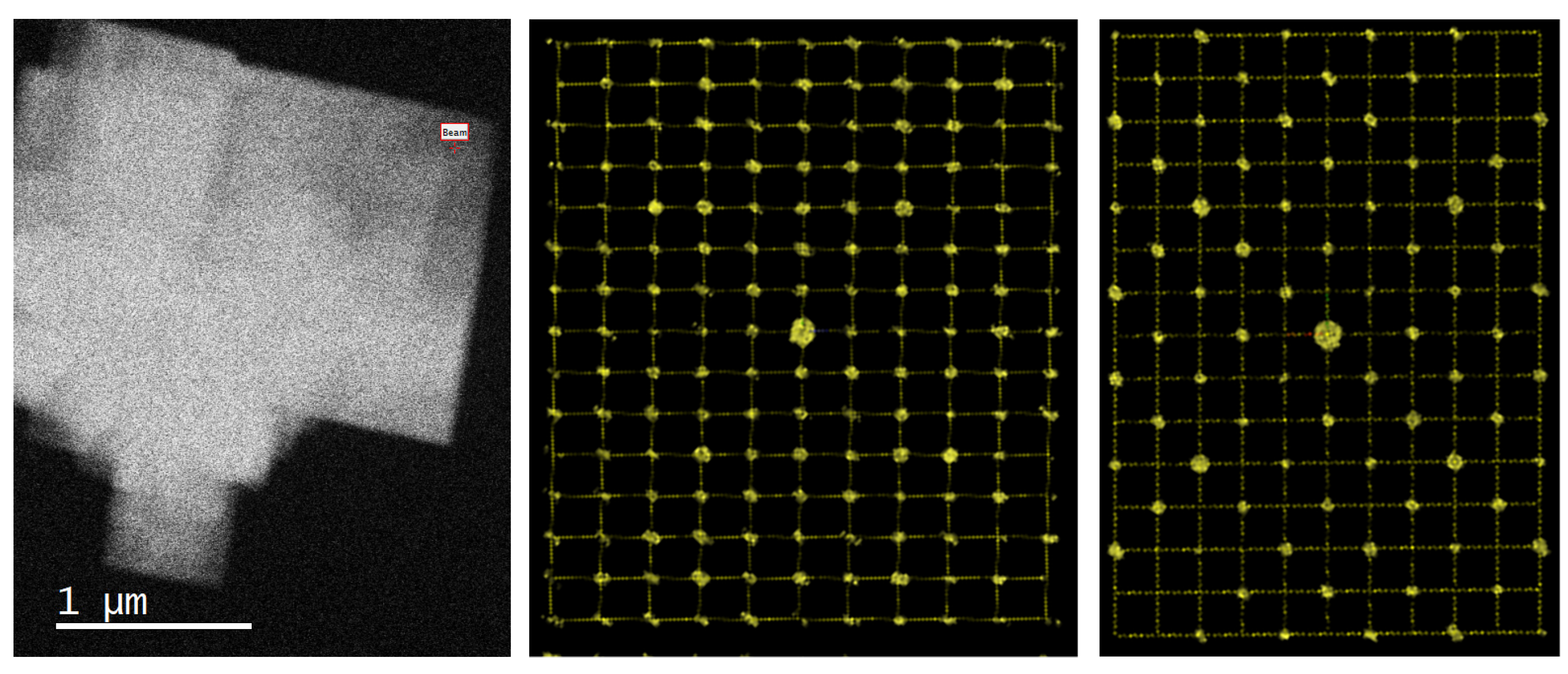
3.3.2. 3D Electron Diffraction—Structure Refinement
3.3.3. Structure Refinement Based on PXRD Data
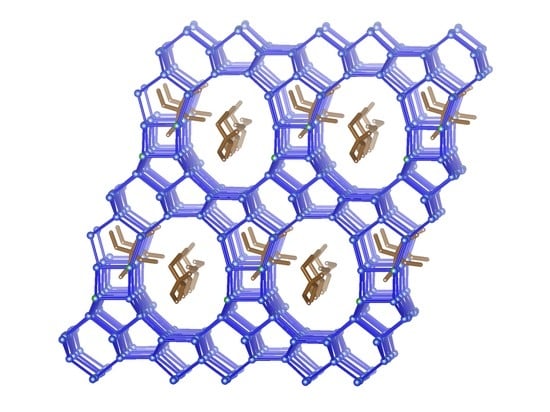
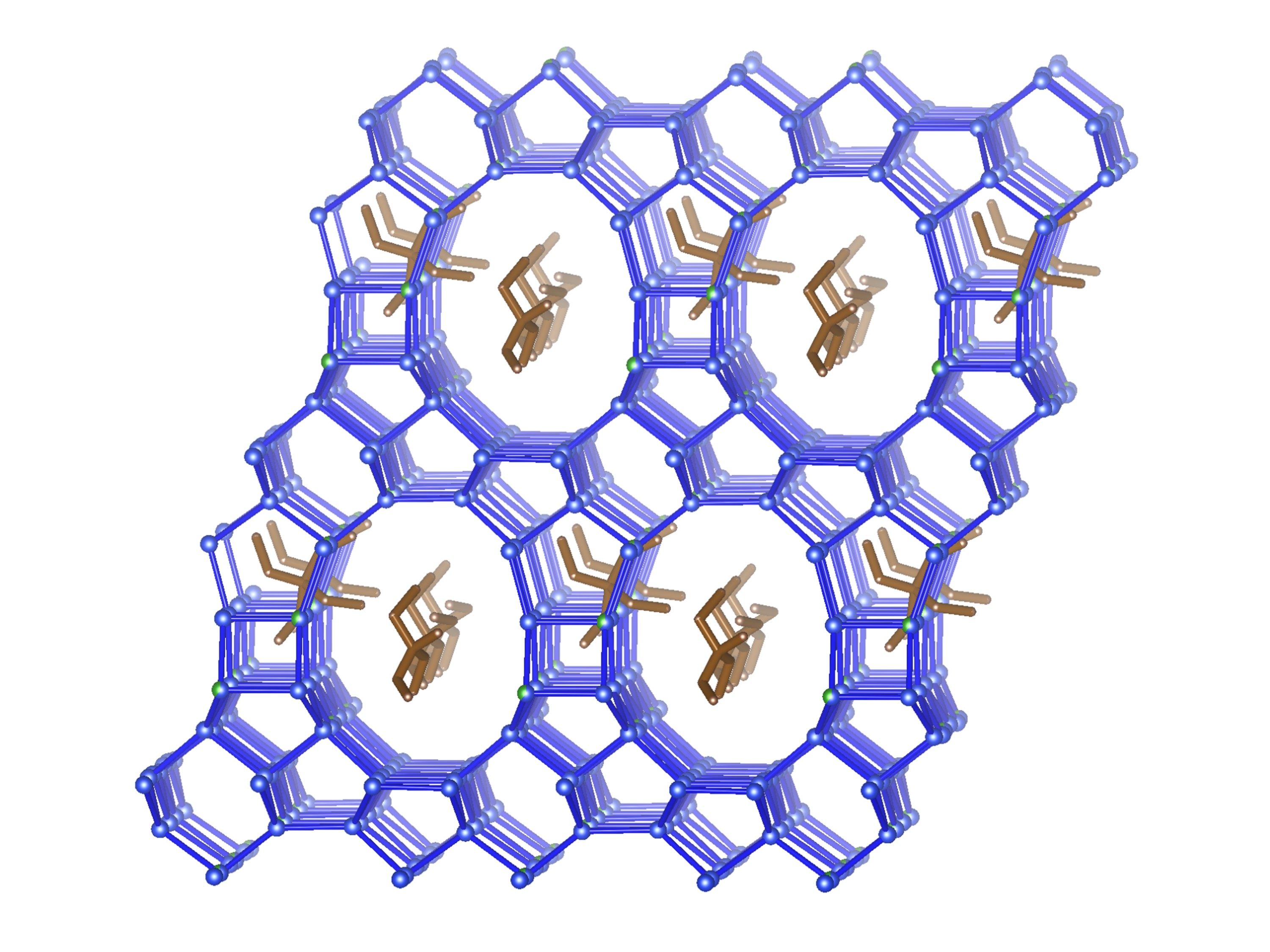
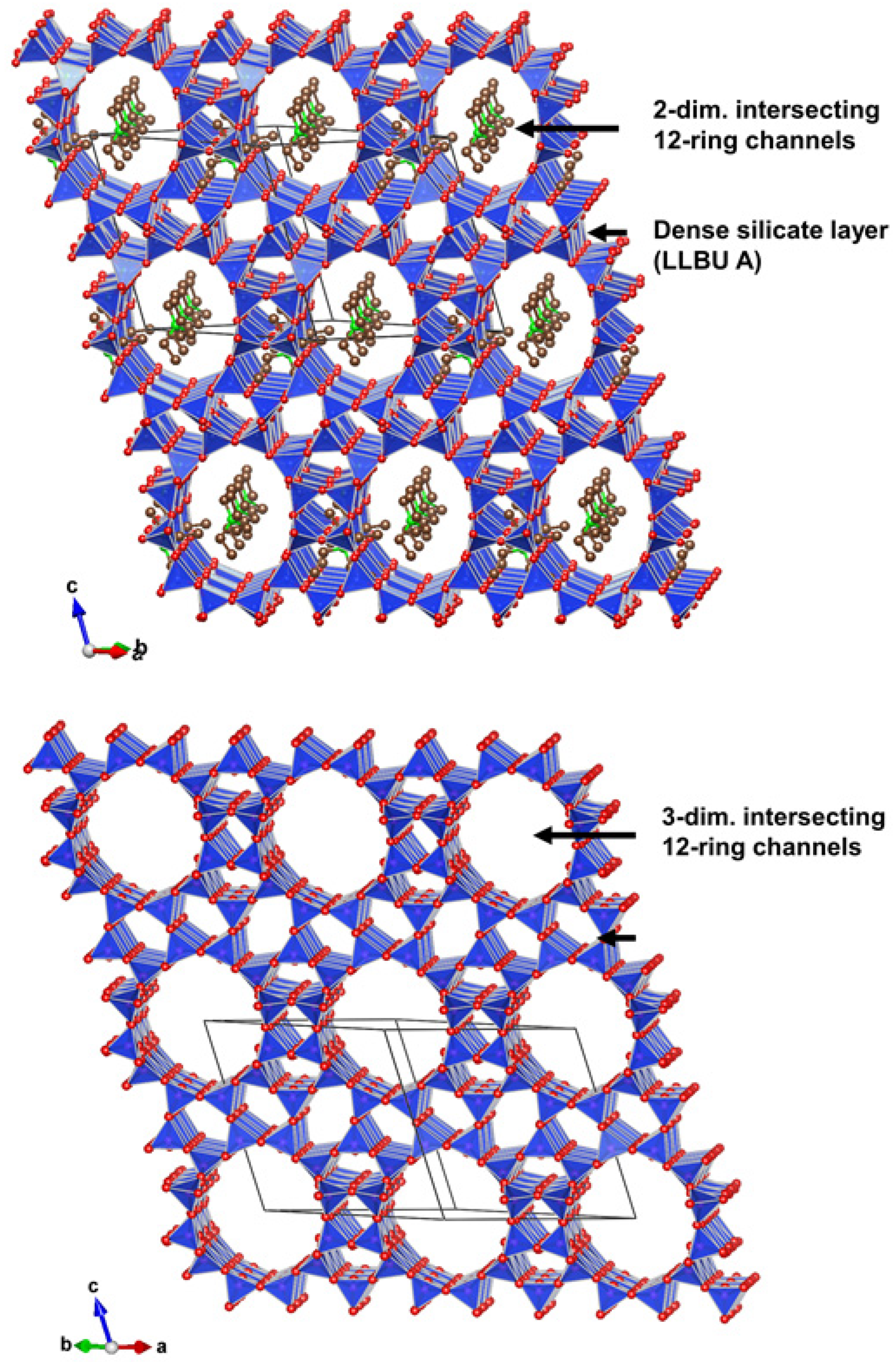
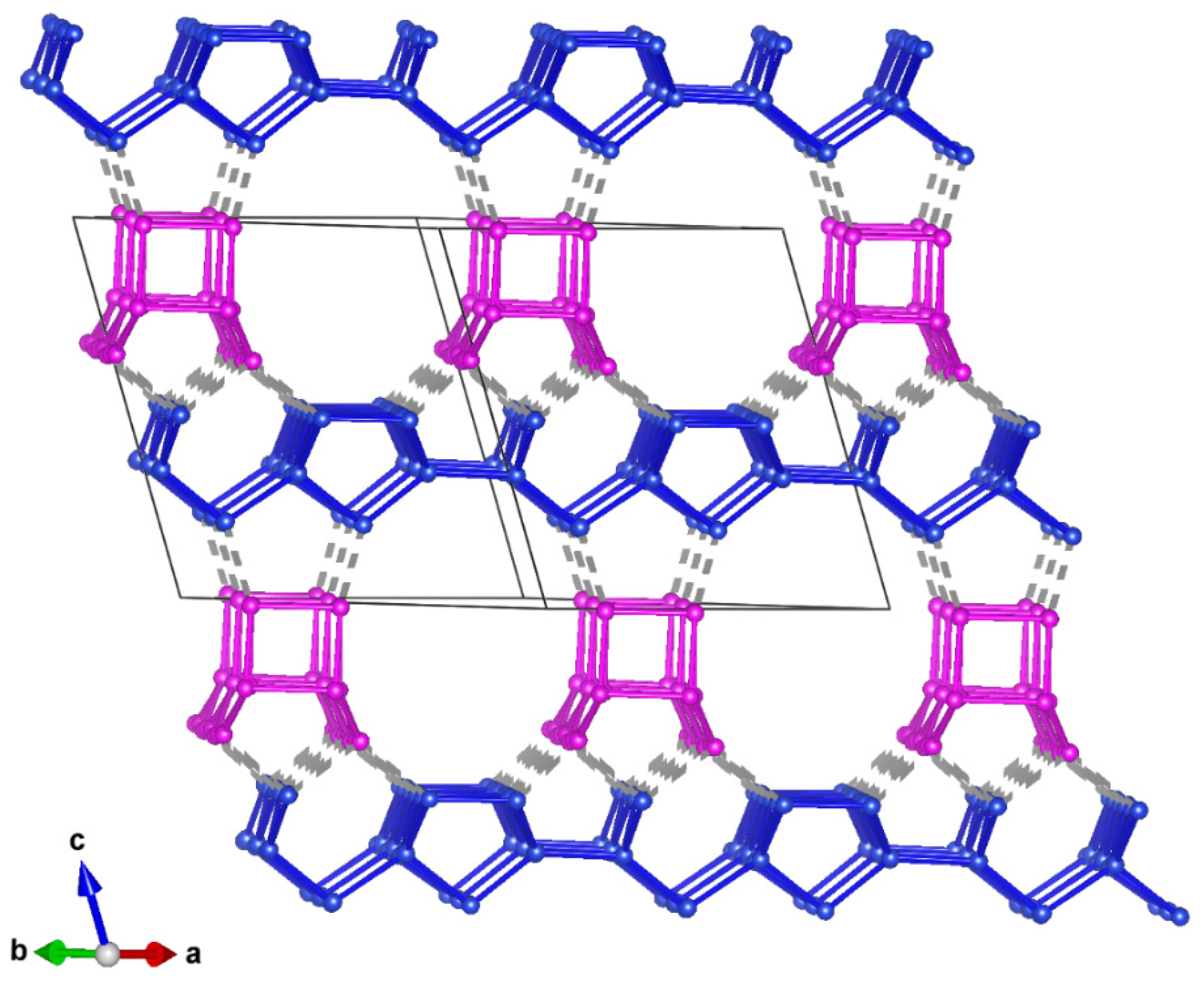
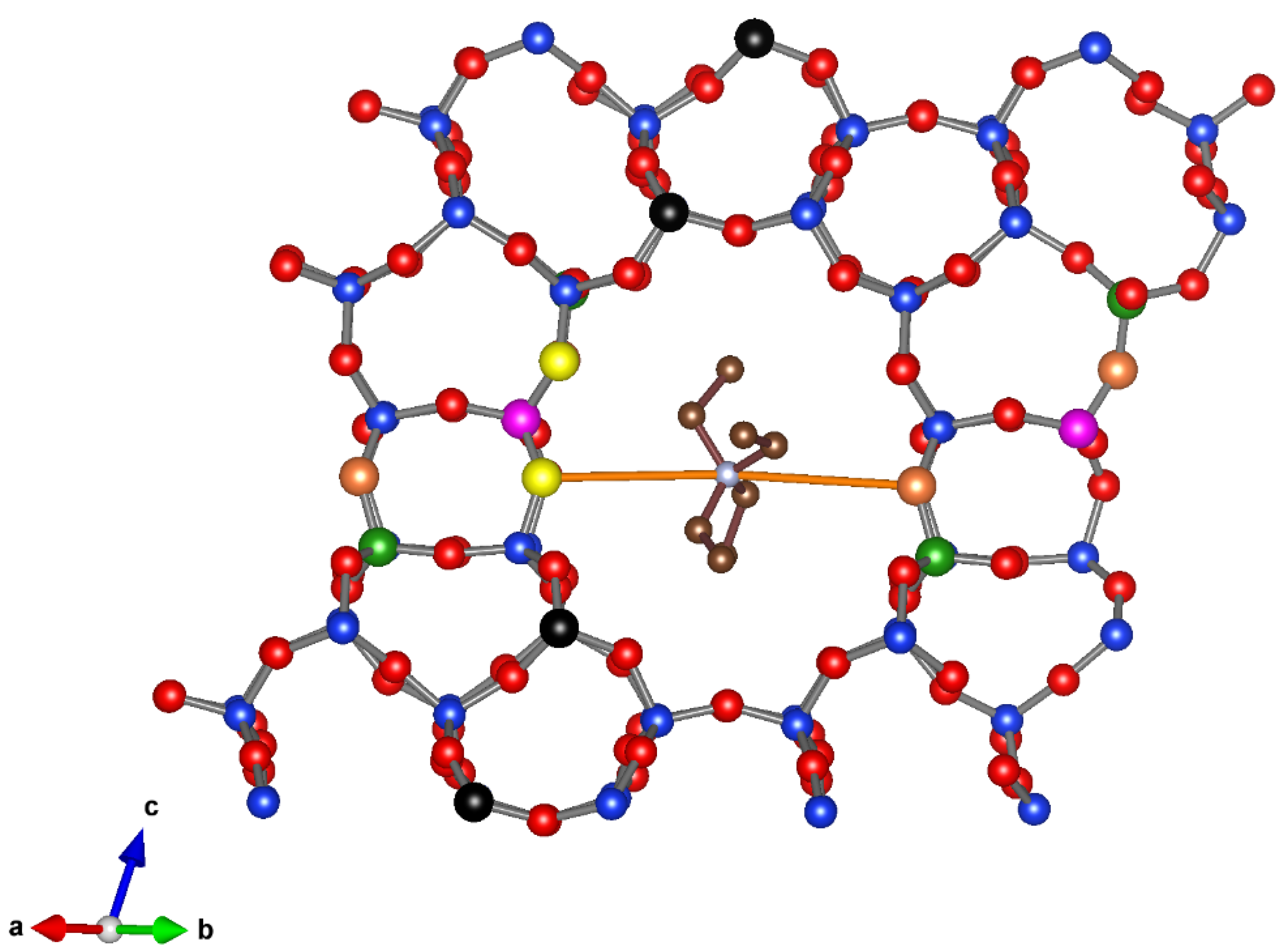
3.3.4. Description of the Structure
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Rhodes, C.J. Properties and applications of zeolites. Sci. Prog. 2010, 93, 223–284. [Google Scholar] [CrossRef] [PubMed]
- Dedecek, J.; Balgova, V.; Pashkova, V.; Klein, P.; Wichterlová, B. Synthesis of ZSM-5 Zeolites with Defined Distribution of Al Atoms in the Framework and Multinuclear MAS NMR Analysis of the Control of Al Distribution. Chem. Mater. 2012, 24, 3231–3239. [Google Scholar] [CrossRef]
- Dedecek, J.; Sobalík, Z.; Wichterlová, B. Siting and Distribution of Framework Aluminium Atoms in Silicon-Rich Zeolites and Impact on Catalysis. Catal. Rev. Sci. Eng. 2012, 54, 135–223. [Google Scholar] [CrossRef]
- Gies, H.; Marler, B. The structure controlling role of organic templates for the synthesis of porosils in the system SiO2/template/H2O. Zeolites 1992, 12, 42–49. [Google Scholar] [CrossRef]
- Lobo, R.F.; Zones, S.I.; Davis, M.E. Structure-Direction in Zeolite Synthesis. In Inclusion Chemistry with Zeolites: Nanoscale Materials by Design; Topics in Inclusion Science; Herron, N., Corbin, D.R., Eds.; Springer: Dordrecht, The Netherlands, 1995; Volume 6. [Google Scholar]
- Burton, A.W.; Zones, S.I. Organic Molecules in Zeolite synthesis: Their preparation and Structure-Directing effects. In Studies in Surface Science and Catalysis; Cejka, J., van Bekkum, H., Corma, A., Schueth, F., Eds.; Elsevier: Amsterdam, The Netherlands, 2007; Volume 168, pp. 137–179. [Google Scholar]
- Gómez-Hortigüela, L. Insights into the Chemistry of Organic Structure-Directing Agents in the Synthesis of Zeolitic Materials. In Structure and Bonding; Springer: Berlin, Germany, 2018; p. 175. [Google Scholar]
- Szostak, R. Hydrothermal Zeolite Synthesis. In Molecular Sieves, Principles of Synthesis and Identification; Springer: Dordrecht, The Netherlands, 1989; pp. 51–132. [Google Scholar]
- Marler, B.; Gies, H. Hydrous layer silicates as precursors for zeolites obtained through topotactic condensation: A review. Eur. J. Mineral. 2012, 24, 405–428. [Google Scholar] [CrossRef]
- Roth, W.J.; Nachtigall, P.; Morris, R.E.; Čejka, J. Two-Dimensional Zeolites: Current Status and Perspectives. Chem. Rev. 2014, 114, 4807−4837. [Google Scholar] [CrossRef]
- Wu, Q.; Meng, X.; Gao, X.; Xiao, F.-S. Solvent-Free Synthesis of Zeolites: Mechanism and Utility. Acc. Chem. Res. 2018, 51, 1396–1403. [Google Scholar] [CrossRef]
- Goel, S.; Zones, S.I.; Iglesia, E. Synthesis of Zeolites via Interzeolite Transformations without Organic Structure-Directing Agents. Chem. Mater. 2015, 27, 2056–2066. [Google Scholar] [CrossRef]
- Caullet, P.; Paillaud, J.-L.; Simon-Masseron, A.; Soulard, M.; Patarin, J. The fluoride route: A strategy to crystalline porous materials. C. R. Chim. 2005, 8, 245–266. [Google Scholar] [CrossRef]
- Lewis, G.J.; Miller, M.A.; Moscoso, J.G.; Wilson, B.A.; Knight, L.M.; Wilson, S.T. Experimental Charge Density Matching Approach to Zeolite Synthesis. Stud. Surf. Sci. Catal. 2004, 154, 364–372. [Google Scholar]
- Opanasenko, M.; Shamzhy, M.; Wang, Y.; Yan, W.; Nachtigall, P.; Čejka, J. Synthesis and Post-Synthesis Transformation of Germanosilicate Zeolites. Angew. Chem. Int. Ed. 2020, 59, 19380–19389. [Google Scholar] [CrossRef] [PubMed]
- Eliášová, P.; Opanasenko, M.; Wheatley, P.S.; Shamzhy, M.; Mazur, M.; Nachtigall, P.; Roth, W.J.; Morris, R.E.; Čejka, J. ADOR mechanism for the synthesis of new zeolites. Chem. Soc. Rev. 2015, 44, 7177–7206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deneyer, A.; Ke, Q.; Devos, J.; Dusselier, M. Zeolite Synthesis under Nonconventional Conditions: Reagents, Reactors, and Modi Operandi. Chem. Mater. 2020, 32, 4884–4919. [Google Scholar] [CrossRef]
- Schmidt, J.E.; Fu, D.; Deem, M.W.; Weckhuysen, B.M. Template–Framework Interactions in Tetraethylammonium-Directed Zeolite Synthesis. Angew. Chem. Int. Ed. 2016, 55, 16044–16048. [Google Scholar]
- Baerlocher, C.; Cusker, L.B.M.; Olson, D.H. Atlas of Zeolite Framework Types, 6th Revised ed.; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Database of Zeolite Structures. Available online: http://www.iza-structure.org/databases/ (accessed on 1 March 2023).
- Kolb, U.; Gorelik, T.; Kübel, C.; Otten, M.T.; Hubert, D. Towards automated diffraction tomography: Part I—Data acquisition. Ultramicroscopy 2007, 107, 507–513. [Google Scholar]
- Plana-Ruiz, S.; Krysiak, Y.; Portillo, J.; Alig, E.; Estrade, S.; Peiro, F.; Kolb, U. Fast-ADT: A fast and automated electron diffraction tomography setup for structure determination and refinement. Ultramicroscopy 2020, 211, 112951. [Google Scholar] [CrossRef]
- Kolb, U.; Mugnaioli, E.; Gorelik, T.E. Automated Electron Diffraction Tomography—A New Tool for Nano Crystal Structure Analysis. Cryst. Res. Technol. 2011, 46, 542–554. [Google Scholar] [CrossRef]
- Kolb, U.; Krysiak, Y.; Plana-Ruiz, S. Automated electron diffraction tomography—Development and applications. Acta Cryst. 2019, B75, 463–474. [Google Scholar]
- Palatinus, L. PETS: Program for Analysis of Electron Diffraction Data. Available online: http://pets.fzu.cz/ (accessed on 1 March 2023).
- Palatinus, L.; Brázda, P.; Jelínek, M.; Hrdá, J.; Steciuk, G.; Klementová, M. Specifics of the Data Processing of Precession Electron Diffraction Tomography Data and Their Implementation in the Program PETS2.0. Acta Crystallogr. 2019, B75, 512–522. [Google Scholar] [CrossRef]
- Burla, M.C.; Caliandro, R.; Carrozzini, B.; Cascarano, G.L.; Cuocci, C.; Giacovazzo, C.; Mallamo, M.; Mazzone, A.; Polidori, G. Crystal structure determination and refinement via SIR2014. J. Appl. Cryst. 2015, 48, 306–309. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELX. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hübschle, C.B.; Sheldrick, G.M.; Dittrich, B. ShelXle: A Qt graphical user interface for SHELXL. J. Appl. Cryst. 2011, 44, 1281–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Carvajal, F. A Program for Rietveld Refinement and Profile Matching Analysis of Complex Powder Diffraction Patterns-Version 7.30; ILL: Grenoble, France, 2020; Available online: http://www.ill.eu/sites/fullprof/index.html (accessed on 1 March 2023).
- Momma, K.; Izumi, F. VESTA: A Three-Dimensional Visualization System for Electronic and Structural Analysis. Available online: https://jp-minerals.org/vesta/en/ (accessed on 1 March 2023).
- Marler, B.; Krysiak, B.Y.; Großkreuz, I.; Gies, H.; Kolb, U. The crystal structure of mineral magadiite, Na2Si14O28(OH)2 × 8 H2O. Am. Mineral. 2022, 107, 2101–2110. [Google Scholar] [CrossRef]
- Marler, B.; Großkreuz, I.; Gies, H. The crystal structure of synthetic kenyaite, Na2 Si20O40(OH)2 × 8 H2O. J. Solid State Chem. 2021, 300, 122215. [Google Scholar] [CrossRef]
- Ghose, S.; Wan, C. Structural chemistry of borosilicates, part II: Searlesite, NaBSi2O5(OH): Absolute configuration, hydrogen locations, and refinement of the structure. Am. Mineral. 1976, 61, 123–129. [Google Scholar]
- Robson, H.; Lillerud, K.P. (Eds.) The sample of classical zeolite beta was synthesized according to J.P.-P. Pariente and M. Camblor. In Verified Syntheses of Zeolitic Materials, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2001; pp. 115–116. [Google Scholar]
- Karge, H.G. Characterization by IR spectroscopy. In Verified Syntheses of Zeolitic Materials, 2nd ed.; Robson, H., Lillerud, K.P., Eds.; Elsevier: Amsterdam, The Netherlands, 2001; pp. 69–71. [Google Scholar]
- Scholle, K.F.M.G.J.; Veeman, W.S. The influence of hydration on the coordination state of boron in H-Boralite studied by 11B magic angle spinning n.m.r. Zeolites 1985, 5, 118–122. [Google Scholar] [CrossRef]
- Chemical Book, Tetraethylammonium Tetrafluoroborate, CAS 429-06-1. Available online: https://www.chemicalbook.com/SpectrumEN_429-06-1_1HNMR.htmc (accessed on 1 March 2023).
- NMR Shift Database. Available online: https://nmrshiftdb.nmr.uni-koeln.de (accessed on 1 March 2023).
- Database of Disordered Zeolite Structures. Available online: https://europe.iza-structure.org/IZA-SC/DO_structures/DO_family.php?IFN=Beta (accessed on 1 March 2023).














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SiO2 | SDA | NaOH | H3BO3 | T1, Time | T2, Time | Solid Products |
|---|---|---|---|---|---|---|---|
| S01 | 1.00 | 0.33 | 0.5 | 0.1 | 130 °C, 7 days | - | Magadiite (silica source: silica gel) |
| S02 | 1.00 | 0.33 | 0.5 | 0.1 | 140 °C, 7 days | - | Amorphous, Beta, unknown phase |
| S03 | 1.00 | 0.33 | 0.5 | 0.1 | 150 °C, 7 days | - | Beta |
| S04 | 1.00 | 0.33 | 0.5 | 0.1 | 160 °C, 7 days | - | MFI, Beta, COE-11 |
| S05 | 1.00 | 0.33 | 0.5 | 0.1 | 168 °C, 7 days | - | Kenyaite, MFI, COE-11 |
| S06 | 1.00 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 160 °C, 6 days | Beta, COE-11 |
| S07 | 1.00 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | Beta, COE-11, MFI |
| S08 | 1.00 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | COE-11, Beta, MFI, MTW |
| S09 | 1.00 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 170 °C, 6 days | MFI, quartz, kenyaite |
| S10 | 1.00 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 170 °C, 6 days | MFI, quartz |
| S11 | 1.00 | 0.03 | 0.5 | 0.1 | 155 °C, 1 day | 180 °C, 6 days | Quartz |
| S12 | 1.00 | 0.03 | 0.5 | 0.1 | 155 °C, 1 day | 180 °C, 6 days | Quartz |
| Sample | SiO2 | SDA | NaOH | H3BO3 | T1, Time | T2, Time | Solid Products |
|---|---|---|---|---|---|---|---|
| S13 | 1.00 | 0.33 | 0.5 | 0.1 | - | 165 °C, 7 days | MFI, COE-11, Beta, kenyaite |
| S14 | 1.00 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | Beta, COE-11 |
| S15 | 1.00 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | Beta COE-11, MFI |
| S16 | 1.00 | 0.33 | 0.5 | 0.1 | 155 °C, 2 days | 165 °C, 5 days | Beta, COE-11, little MFI |
| S17 | 1.00 | 0.33 | 0.5 | 0.1 | 155 °C, 2 days | 165 °C, 5 days | Beta, MFI, (COE-11) |
| S18 | 1.00 | 0.33 | 0.5 | 0.1 | 155 °C, 3 days | 165 °C, 4 days | Beta, COE-11, little MFI |
| S19 | 1.00 | 0.33 | 0.5 | 0.1 | 155 °C, 3 days | 165 °C, 4 days | Beta, MFI, (COE-11) |
| S20 | 1.00 | 0.33 | 0.5 | 0.1 | 155 °C, 4 days | 165 °C, 3 days | Beta, COE-11 |
| S21 | 1.00 | 0.03 | 0.5 | 0.1 | 155 °C, 4 days | 165 °C, 3 days | COE-11, Beta, MTW |
| S22 | 1.00 | 0.33 | 0.5 | 0.1 | 155 °C, 5 days | 165 °C, 2 days | MFI |
| S23 | 1.00 | 0.33 | 0.5 | 0.1 | 155 °C, 5 days | 165 °C, 2 days | Beta, MFI, COE-11 |
| S24 | 1.00 | 0.33 | 0.5 | 0.1 | 155 °C, 7 days | - | Beta, MFI, COE-11 |
| S25 | 1.00 | 0.33 | 0.5 | 0.1 | 157 °C, 1 day | 165 °C, 9 days | MFI, Beta, COE-11 |
| S26 | 1.00 | 0.33 | 0.5 | 0.1 | 157 °C, 1 day | 165 °C, 14 days | MFI, Beta |
| S27 | 1.00 | 0.33 | 0.5 | 0.1 | 157 °C, 1 day | 165 °C, 17 days | Quartz, MFI |
| S28 | 1.00 | 0.33 | 0.5 | 0.1 | 157 °C, 1 day | 165 °C, 21 days | No product |
| Sample | SiO2 | TEAOH | NaOH | H3BO3 | T1, Time | T2, Time | Solid Products |
|---|---|---|---|---|---|---|---|
| S29 | Ludox HS40 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | MFI, COE-11, Kenyaite |
| S30 | Ludox HS40 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | COE-11, Beta, MFI, kenyaite |
| S31 | Ludox HS40 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | Beta, COE-11, (MFI) |
| S32 | Ludox HS40 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | COE-11, beta, MFI |
| S33 | Gel, Merck | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | Unidentified phases |
| S34 | Gel, Merck | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | Kenyaite, MFI, COE-11, unknown phase |
| S35 | Gel, lab made | 0.33 | 0.5 | 0.1 | 155 °C, 4 days | 165 °C, 3 days | COE-11, Searlsite, MFI, (Beta) |
| S36 | Gel, lab made | 0.33 | 0.5 | 0.1 | 155 °C, 4 days | 165 °C, 3 days | Searlsite, MFI, Beta, COE-11 |
| S37 * | B-Si-Gel, lab made | 0.33 | 0.5 | 0.1 | 155 °C, 4 days | 165 °C, 3 days | Amorphous, quartz, MFI |
| S38 * | B-Si-Gel, lab made | 0.33 | 0.5 | 0.1 | 155 °C, 4 days | 165 °C, 3 days | Amorphous, Beta, MFI |
| S39 | Levasil 150 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | Beta, MFI |
| S40 | Levasil 150 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | MFI, beta, kenyaite, COE-11 |
| S41 § | Levasil 500 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | MOR |
| S42 § | Levasil 500 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | MOR |
| S43 | Syton HT50 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | MFI, COE-11, Beta |
| S44 | Syton HT50 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | MFI |
| S45 | Ludox SM30 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | MFI, Beta, COE-11 |
| S46 | Ludox SM30 | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | MFI, Beta, COE-11 |
| S47 | Tetramethoxisilane | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | Quartz, amorphous |
| S48 | Tetramethoxisilane | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | Quartz, amorphous |
| S49 | Na-RUB-18 | 0.33 | 0.5 | 0.1 | 155 °C, 4 days | 165 °C, 3 days | Quartz, Beta, searlsite, MOR |
| S50 | Na-RUB-18 | 0.33 | 0.5 | 0.1 | 155 °C, 4 days | 165 °C, 3 days | Amorphous, Beta, MOR, quartz, searlsite |
| S51 | Calc. MCM-48 | 0.33 | 0.5 | 0.1 | 155 °C, 4 days | 165 °C, 3 days | Beta, MFI, COE-11, kenyaite |
| S52 | Calc. MCM-48 | 0.33 | 0.5 | 0.1 | 155 °C, 4 days | 165 °C, 3 days | Beta, COE-11, CHA |
| S53 | Calc. MCM-41 | 0.33 | 0.5 | 0.1 | 155 °C, 4 days | 165 °C, 3 days | Amorphous, Beta |
| S54 | Calc. MCM-41 | 0.33 | 0.5 | 0.1 | 155 °C, 4 days | 165 °C, 3 days | Beta |
| S55 | Calc. B-MCM-41 | 0.33 | 0.5 | 0.1 | 155 °C, 4 days | 165 °C, 3 days | MFI, MTW, magadiite, kenyaite |
| S56 | Calc. B-MCM-41 | 0.33 | 0.5 | 0.1 | 155 °C, 4 days | 165 °C, 3 days | Amorphous, Beta |
| S57 | Fumed silica | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | MFI, COE-11, kenyaite |
| S58 | Fumed silica | 0.33 | 0.5 | 0.1 | 155 °C, 1 day | 165 °C, 6 days | MFI, kenyaite, COE-11 |
| H | K | L | 2theta | Iobs | d-hkl | H | K | L | 2theta | Iobs | d-hkl |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 0 | 1 | 6.758 | 2198.1 | 13.068 | 2 | 2 | 2 | 23.197 | 117.6 | 3.831 |
| 1 | 1 | 0 | 7.543 | 2399.4 | 11.711 | 2 | 4 | 0 | 23.360 | 99.7 | 3.805 |
| −1 | 1 | 1 | 8.503 | 1454.5 | 10.390 | −4 | 2 | 2 | 23.367 | 86.1 | 3.804 |
| 0 | 2 | 0 | 10.194 | 102.9 | 8.670 | −4 | 0 | 3 | 23.443 | 3.3 | 3.792 |
| −2 | 0 | 1 | 10.441 | 7.4 | 8.466 | 1 | 1 | 3 | 23.797 | 64.4 | 3.736 |
| 2 | 0 | 0 | 11.136 | 33.4 | 7.939 | −1 | 3 | 3 | 24.347 | 46.5 | 3.653 |
| 1 | 1 | 1 | 11.539 | 175.7 | 7.663 | 0 | 4 | 2 | 24.624 | 12.8 | 3.612 |
| 0 | 2 | 1 | 12.241 | 152.5 | 7.225 | 4 | 2 | 0 | 24.645 | 36.2 | 3.609 |
| −1 | 1 | 2 | 13.401 | 102.9 | 6.602 | −2 | 4 | 2 | 24.678 | 50.9 | 3.605 |
| 0 | 0 | 2 | 13.540 | 74.5 | 6.534 | −2 | 0 | 4 | 24.924 | 3.7 | 3.570 |
| −2 | 0 | 2 | 13.636 | 52.5 | 6.489 | 2 | 4 | 1 | 25.596 | 9.9 | 3.477 |
| −2 | 2 | 1 | 14.612 | 53.4 | 6.057 | −4 | 2 | 3 | 25.621 | 19.7 | 3.474 |
| 2 | 2 | 0 | 15.119 | 68.3 | 5.855 | 3 | 3 | 1 | 25.664 | 483.7 | 3.468 |
| 2 | 0 | 1 | 15.202 | 8.4 | 5.823 | −3 | 3 | 3 | 25.702 | 486.1 | 3.463 |
| −3 | 1 | 1 | 16.143 | 30.1 | 5.486 | 4 | 0 | 1 | 25.933 | 3.2 | 3.433 |
| 1 | 3 | 0 | 16.306 | 22.2 | 5.432 | −1 | 1 | 4 | 26.025 | 13.5 | 3.421 |
| −1 | 3 | 1 | 16.777 | 55.9 | 5.280 | 3 | 1 | 2 | 26.071 | 22.2 | 3.415 |
| 0 | 2 | 2 | 16.977 | 7.7 | 5.218 | −3 | 1 | 4 | 26.127 | 20.5 | 3.408 |
| −2 | 2 | 2 | 17.054 | 74.3 | 5.195 | −5 | 1 | 2 | 26.275 | 0.4 | 3.389 |
| 1 | 1 | 2 | 17.382 | 163.4 | 5.098 | 1 | 5 | 0 | 26.281 | 3.2 | 3.388 |
| 3 | 1 | 0 | 17.505 | 1.9 | 5.062 | −5 | 1 | 1 | 26.558 | 9.2 | 3.354 |
| −3 | 1 | 2 | 17.532 | 171.0 | 5.055 | −1 | 5 | 1 | 26.582 | 2.3 | 3.351 |
| 2 | 2 | 1 | 18.337 | 385.3 | 4.834 | −2 | 2 | 4 | 26.991 | 300.3 | 3.301 |
| 1 | 3 | 1 | 18.519 | 56.3 | 4.787 | 2 | 0 | 3 | 27.003 | 11.7 | 3.299 |
| −2 | 0 | 3 | 18.864 | 0.0 | 4.700 | 0 | 0 | 4 | 27.274 | 136.1 | 3.267 |
| −1 | 1 | 3 | 19.501 | 48.3 | 4.548 | −4 | 0 | 4 | 27.470 | 16.8 | 3.244 |
| −1 | 3 | 2 | 19.746 | 51.1 | 4.492 | 1 | 5 | 1 | 27.739 | 108.8 | 3.213 |
| 0 | 0 | 3 | 20.370 | 4.4 | 4.356 | −5 | 1 | 3 | 27.751 | 77.1 | 3.212 |
| 0 | 4 | 0 | 20.470 | 16.2 | 4.335 | 4 | 2 | 1 | 27.930 | 5.2 | 3.192 |
| −4 | 0 | 1 | 20.581 | 22.1 | 4.312 | 1 | 3 | 3 | 27.943 | 11.3 | 3.190 |
| 2 | 0 | 2 | 20.781 | 0.0 | 4.271 | −2 | 4 | 3 | 27.976 | 16.4 | 3.187 |
| −4 | 0 | 2 | 20.970 | 3.7 | 4.233 | 5 | 1 | 0 | 28.552 | 76.7 | 3.124 |
| 3 | 1 | 1 | 21.106 | 15.8 | 4.206 | −1 | 5 | 2 | 28.591 | 52.6 | 3.120 |
| −3 | 1 | 3 | 21.151 | 32.1 | 4.197 | 2 | 2 | 3 | 28.932 | 255.1 | 3.084 |
| −2 | 2 | 3 | 21.487 | 402.8 | 4.132 | 0 | 4 | 3 | 29.035 | 26.6 | 3.073 |
| 0 | 4 | 1 | 21.579 | 64.9 | 4.115 | 0 | 2 | 4 | 29.186 | 27.3 | 3.057 |
| −3 | 3 | 1 | 21.721 | 62.0 | 4.088 | −4 | 4 | 1 | 29.187 | 69.4 | 3.057 |
| 4 | 0 | 0 | 22.378 | 36.2 | 3.970 | 2 | 4 | 2 | 29.331 | 31.5 | 3.043 |
| 1 | 3 | 2 | 22.666 | 23.1 | 3.920 | −4 | 2 | 4 | 29.370 | 10.8 | 3.039 |
| 3 | 3 | 0 | 22.762 | 1186.7 | 3.904 | −4 | 4 | 2 | 29.469 | 11.4 | 3.029 |
| −3 | 3 | 2 | 22.783 | 1016.4 | 3.900 | −1 | 3 | 4 | 29.884 | 26.1 | 2.987 |
| 0 | 2 | 3 | 22.827 | 23.8 | 3.892 | 3 | 3 | 2 | 29.925 | 58.1 | 2.983 |
| −4 | 2 | 1 | 23.016 | 115.0 | 3.861 | −3 | 3 | 4 | 29.974 | 108.3 | 2.979 |
| −2 | 4 | 1 | 23.030 | 160.9 | 3.859 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marler, B.; Gies, H.; De Baerdemaeker, T.; Müller, U.; Parvulescu, A.-N.; Zhang, W.; Yokoi, T.; Xiao, F.-S.; Meng, X.; De Vos, D.; et al. Synthesis and Structure of COE-11, a New Borosilicate Zeolite with a Two-Dimensional Pore System of 12-Ring Channels. Chemistry 2023, 5, 730-752. https://doi.org/10.3390/chemistry5020052
Marler B, Gies H, De Baerdemaeker T, Müller U, Parvulescu A-N, Zhang W, Yokoi T, Xiao F-S, Meng X, De Vos D, et al. Synthesis and Structure of COE-11, a New Borosilicate Zeolite with a Two-Dimensional Pore System of 12-Ring Channels. Chemistry. 2023; 5(2):730-752. https://doi.org/10.3390/chemistry5020052
Chicago/Turabian StyleMarler, Bernd, Hermann Gies, Trees De Baerdemaeker, Ulrich Müller, Andrei-Nicolae Parvulescu, Weiping Zhang, Toshiyuki Yokoi, Feng-Shou Xiao, Xiangju Meng, Dirk De Vos, and et al. 2023. "Synthesis and Structure of COE-11, a New Borosilicate Zeolite with a Two-Dimensional Pore System of 12-Ring Channels" Chemistry 5, no. 2: 730-752. https://doi.org/10.3390/chemistry5020052