

Conversion of Sugar Di-Ketals to Bio-Hydrocarbons through Catalytic Cracking over Beta Catalysts in Fixed and Fluidized Catalytic Beds
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
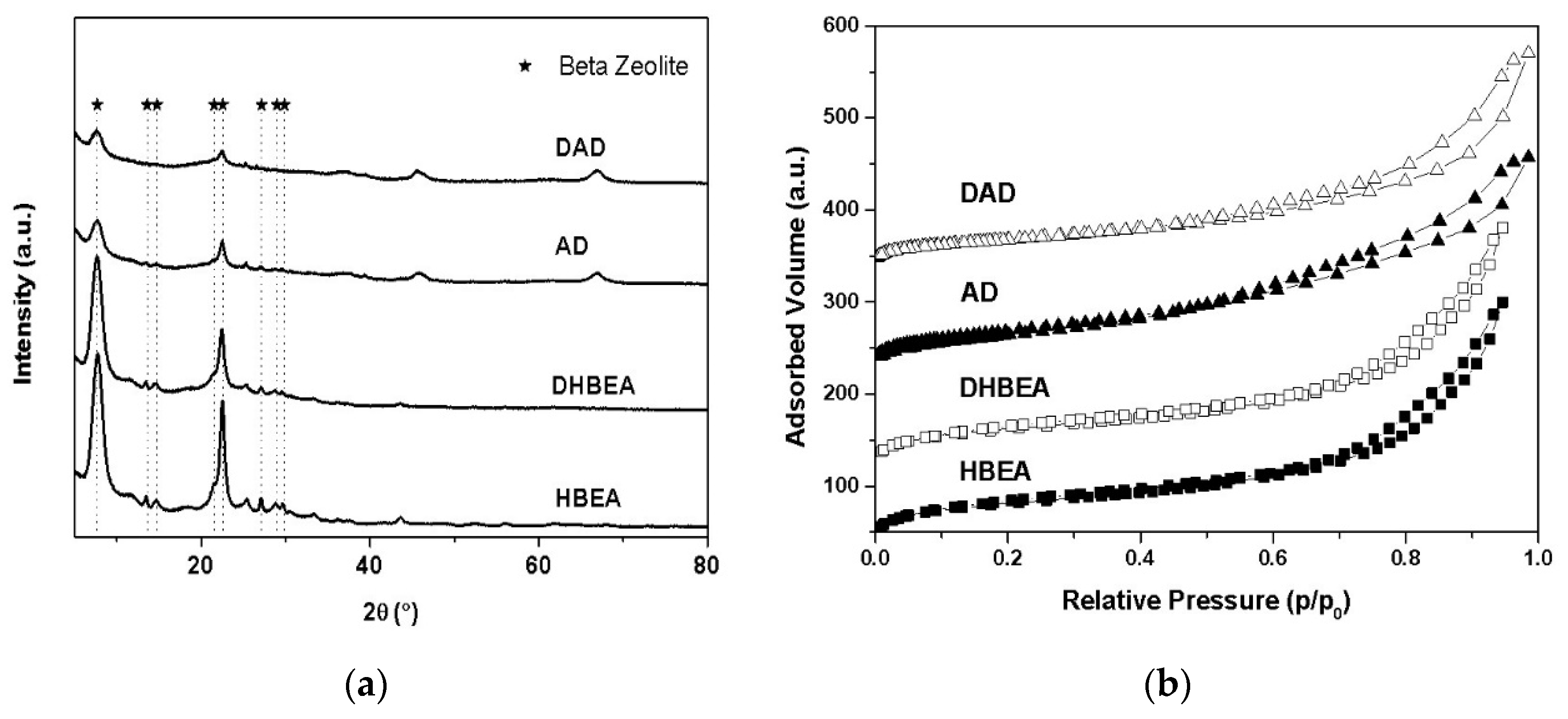
3.1. Catalyst Properties
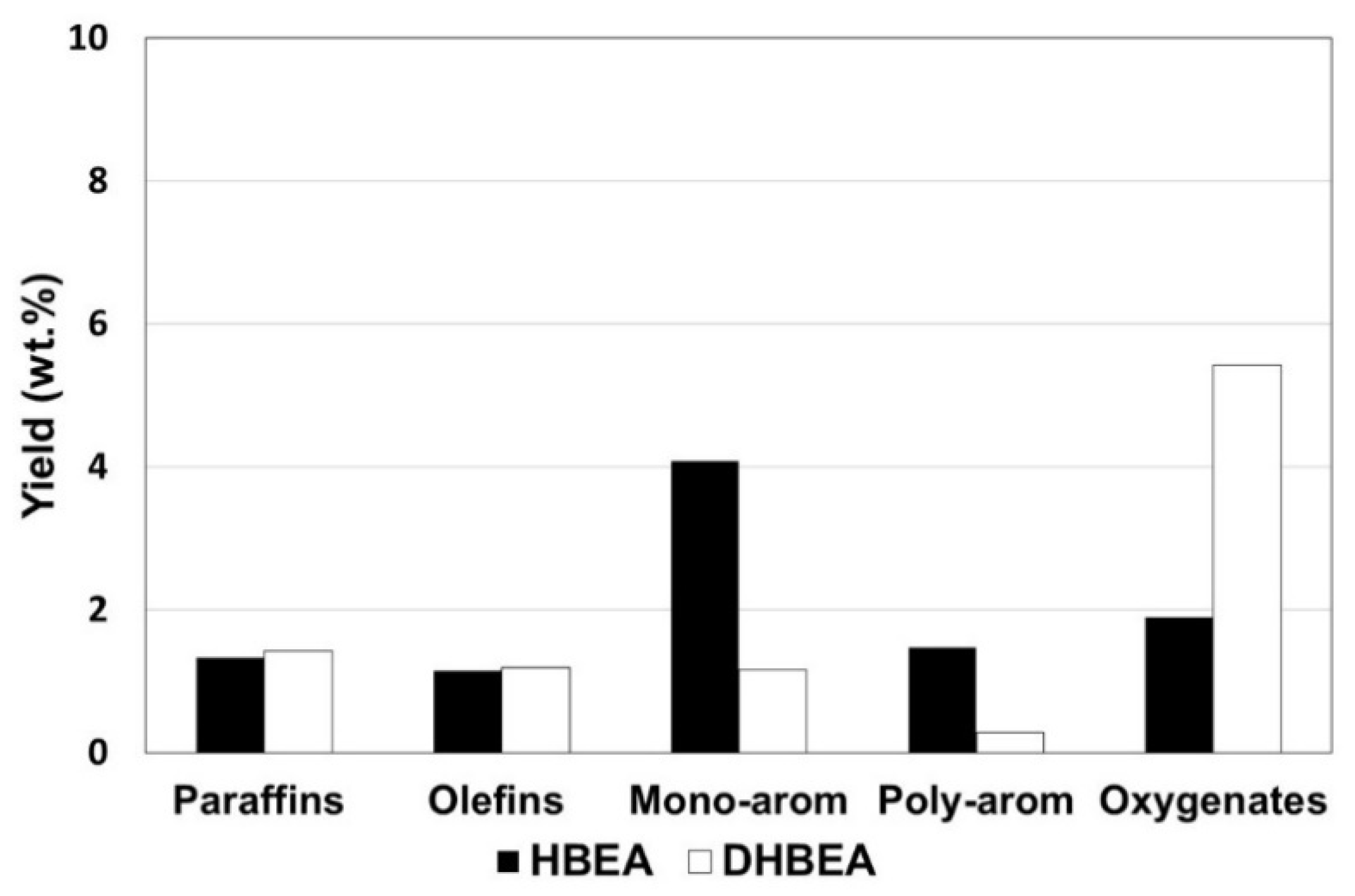
3.2. Pure Zeolite in Fixed Bed: Reducing Acid Sites by Steam Treatment
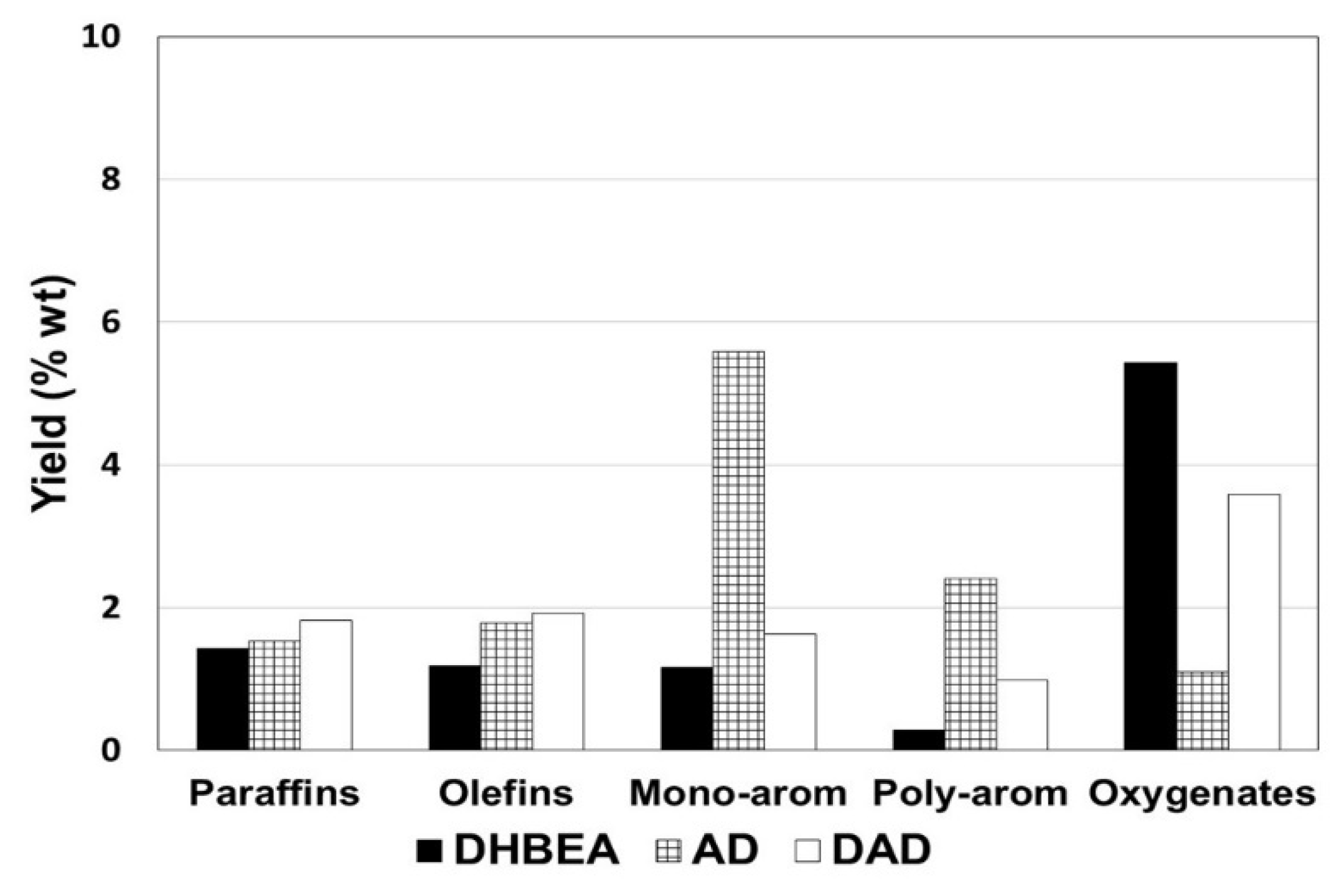
3.3. Catalysts in Fixed Bed: Comparison with Steam-Treated Beta Zeolite with Fresh and Steam-Treated Catalysts
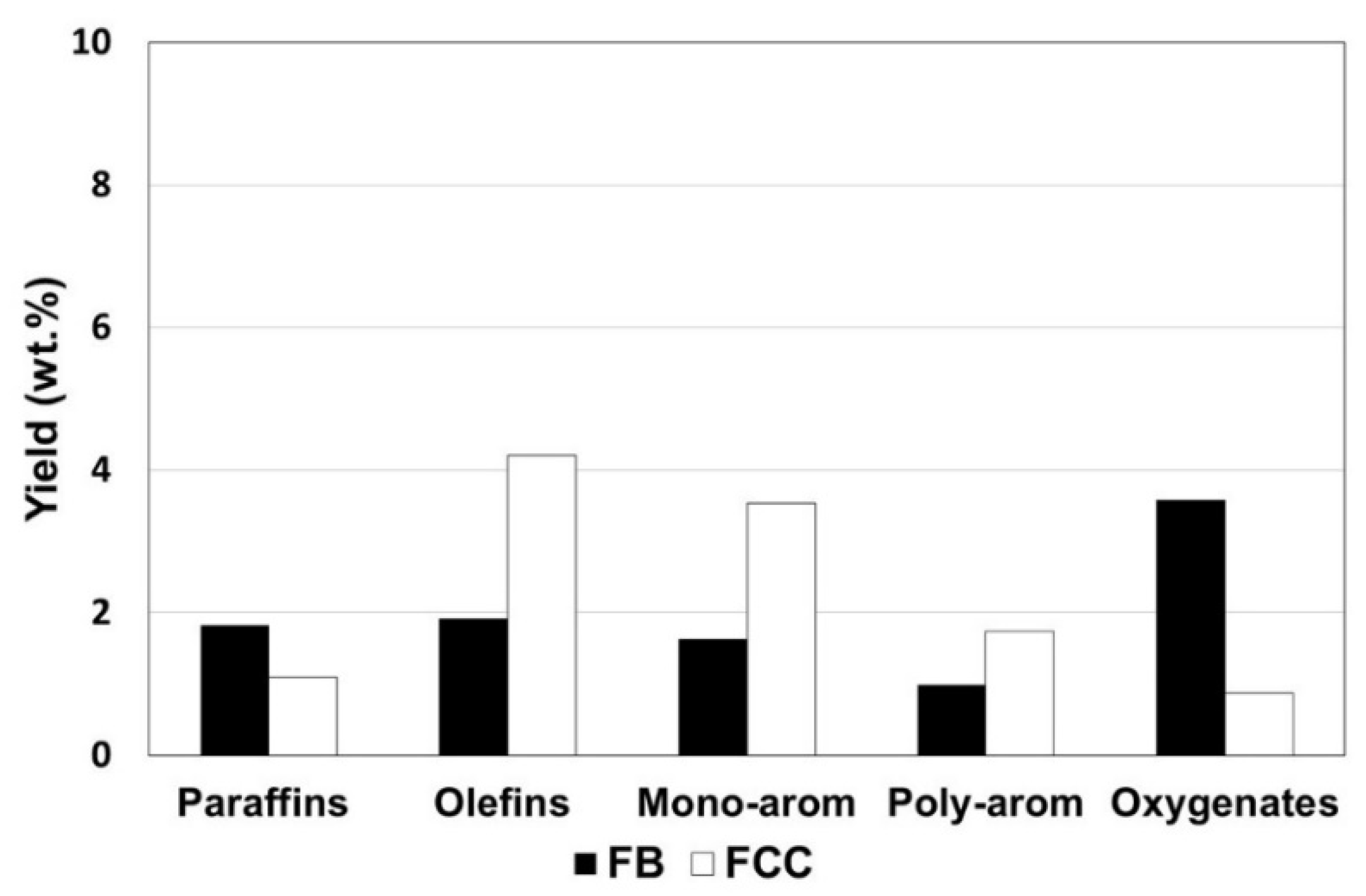
3.4. Hydrothermally Treated Catalyst in Fluidized Bed vs. in Fixed Bed
3.5. Outlook
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Pereira, M.M.; Soter de Mariz e Miranda, L.; Oliveira de Souza, M.; Brando Bezerra de Almeida, M. Processo para Produção de bio-óleo Solúvel de Hidrocarbonetos. Patent WO/2015/135046, 2015. [Google Scholar]
- dos Santos, D.N.; Pedrosa, I.V.; Fernandes, C.R.R.; Lachgar, A.; Neli, M.; Garrett, R.; Lam, Y.L.; Pereira, M.M. Catalytic sugarcane bagasse transformation into a suitable biocrude for hydrocarbon production in typical refinery processes. Sustain. Energy Fuels 2020, 4, 4158–4169. [Google Scholar] [CrossRef]
- Ibarra, Á.; Hita, I.; Arandes, J.M.; Bilbao, J. Influence of the composition of raw bio-oils on their valorization in fluid catalytic cracking (FCC) conditions. Energy Fuels 2019, 33, 7458–7465. [Google Scholar] [CrossRef]
- Mohan, D.; Pittman, C.U.; Philip, S. Pyrolysis of Wood/Biomass for Bio-oil: A Critical Review Dinesh. Prog. Energy Combust. Sci. 2017, 62, 848–889. [Google Scholar]
- Wang, G.; Dai, Y.; Yang, H.; Xiong, Q.; Wang, K.; Zhou, J.; Li, Y.; Wang, S. A review of recent advances in biomass pyrolysis. Energy Fuels 2020, 34, 15557–15578. [Google Scholar] [CrossRef]
- Durange, J.A.C.; Santos, M.R.L.; Pereira, M.M.; Fernandes, L.A.P., Jr.; Souza, M.N.; Mendes, A.N.; Mesa, L.M.; Sánchez, C.G.; Sanchez, E.M.S.; Pérez, J.M.M.; et al. Physicochemical Properties of Pyrolysis Bio-Oil from Sugarcane Straw and Sugarcane in Natura. J. Biomater. Nanobiotechnol. 2013, 4, 10–19. [Google Scholar] [CrossRef] [Green Version]
- Ibarra, Á.; Rodríguez, E.; Sedran, U.; Arandes, J.M.; Bilbao, J. Synergy in the Cracking of a Blend of Bio-oil and Vacuum Gasoil under Fluid Catalytic Cracking Conditions. Ind. Eng. Chem. Res. 2016, 55, 1872–1880. [Google Scholar] [CrossRef]
- Ibarra, Á.; Hita, I.; Azkoiti, M.J.; Arandes, J.M.; Bilbao, J. Catalytic cracking of raw bio-oil under FCC unit conditions over different zeolite-based catalysts. J. Ind. Eng. Chem. 2019, 78, 372–382. [Google Scholar] [CrossRef]
- Batalha, N.; Pinto, J.; Ferreira, H.; Baptista, D.C.; Miranda, L.S.M.; Pereira, M.M. Biohydrocarbons Production under Standard Refinery Conditions by means of a Representative Ketal Compound of Biocrude. Energy Technol. 2017, 5, 428–441. [Google Scholar] [CrossRef]
- Pinto, J.; Pedrosa, I.; Linhares, C.; Gil, R.A.S.S.; Lam, Y.L.; Pereira, M.M. Ketal Sugar Conversion Into Green Hydrocarbons by Faujasite Zeolite in a Typical Catalytic Cracking Process. Front. Chem. 2019, 7, 720. [Google Scholar] [CrossRef]
- Pinto, J.F.; Lam, Y.L.; Pereira, M.M.; Cruchade, H.; Sachse, A.; Pinard, L. Green-aromatic production in typical conditions of fluidized catalytic cracking. Fuel 2019, 254, 115684. [Google Scholar] [CrossRef]
- Pereira, S.C.; Souza, M.; Pinto, J.; Esteves, L.M.; Lam, Y.L.; Miranda, L.S.D.M.E.; Pereira, M.M. Sugar ketals as a platform molecule to overcome the limitation of converting biomass into green-hydrocarbons in a typical refinery. Sustain. Energy Fuels 2020, 4, 1312–1319. [Google Scholar] [CrossRef]
- Souza, M.O.; Pereira, S.C.; Lau, L.Y.; Soter, L.; Pereira, M.M. Hydrodeoxygenation of Xylose Isopropylidene Ketal Over Pd/HBEA Catalyst for the Production of Green Fuels. Front. Chem 2021, 9, 729787. [Google Scholar] [CrossRef]
- de Souza, M.O. Produção de Hidrocarbonetos Renováveis Através da Hidroconversão de Cetais de D-xilose Utilizando Catalisadores de Paládio em Zeólita Beta. Ph.D. Thesis, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil, 2022. [Google Scholar]
- Cardoso, C.S. Conversão do Acetal de D-Xilose em Bio-Hidrocarbonetos por Craqueamento Catalítico com Zeólita Beta. Ph.D. Thesis, Universidade Federal do Rio de Janeiro, Rio de Janeiro, Brazil, 2022. [Google Scholar]
- Cardoso, C.; Lam, Y.L.; Gil, R.A.S.; Van Bokhoven, J.A.; Pereira, M.M. Conversion of sugar diacetyls to bio-hydrocarbons by the catalytic cracking in a fixed bed with fresh and deactivated Beta zeolite. Catal. Commun. 2022, 171, 106519. [Google Scholar] [CrossRef]
- Gusev, A.A.; Psarras, A.C.; Triantafyllidis, K.S.; Lappas, A.A.; Diddams, P.A. Effect of steam deactivation severity of ZSM-5 additives on LPG olefins production in the FCC process. Molecules 2017, 22, 1784. [Google Scholar] [CrossRef] [Green Version]
- Kubicek, N.; Vaudry, F.; Chiche, B.; Hudec, P.; Di Renzo, F.; Schulz, P.; Fajula, F. Stabilization of zeolite beta for fcc application by embedding in amorphous matrix. Appl. Catal. A Gen. 1998, 175, 159–171. [Google Scholar] [CrossRef]
- Gélin, P.; Courières, T.D. Role of the amorphous matrix in the hydrothermal ageing of fluid catalytic cracking catalysts. Appl. Catal. 1991, 72, 179–192. [Google Scholar] [CrossRef]
- Etim, U.; Bai, P.; Wang, Y.; Subhan, F.; Liu, Y.; Yan, Z. Mechanistic insights into structural and surface variations in Y-type zeolites upon interaction with binders. Appl. Catal. A Gen. 2019, 571, 137–149. [Google Scholar] [CrossRef]
- Lakiss, L.; Gilson, J.-P.; Valtchev, V.; Mintova, S.; Vicente, A.; Vimont, A.; Bedard, R.; Abdo, S.; Bricker, J. Zeolites in a good shape: Catalyst forming by extrusion modifies their performances. Microporous Mesoporous Mater. 2020, 299, 110114. [Google Scholar] [CrossRef]
- Maier, S.M.; Jentys, A.; Lercher, J.A. Steaming of zeolite BEA and its effect on acidity: A comparative NMR and IR spectroscopic study. J. Phys. Chem. C 2011, 115, 8005–8013. [Google Scholar] [CrossRef]
- De Lucas, A.; Canizares, P.; Durán, A.; Carrero, A. Dealumination of HZSM-5 zeolites: Effect of steaming on acidity and aromatization activity. Appl. Catal. A Gen. 1997, 154, 221–240. [Google Scholar] [CrossRef]
- Scherzer, J. Chapter 5 Correlation Between Catalyst Formulation and Catalytic Properties. Stud. Surf. Sci. Catal. 1993, 76, 145–182. [Google Scholar] [CrossRef]
- Bonetto, L.; Corma, A.; Herrero, E. Beta Zeolite As Catalyst or Catalyst Additive for the Production of Olefins During Cracking of Gasoil. Catalysis 1992, 7, 639–646. [Google Scholar] [CrossRef]
- Cejka, J.; Morries, R.E.; Nachtigall, P. Zeolites in Catalysis, Properties and Applications; Royal Society of Chemistry: London, UK, 2017. [Google Scholar]
- Corma, A.; Sauvanaud, L. FCC testing at bench scale: New units, new processes, new feeds. Catal. Today 2013, 218, 107–114. [Google Scholar] [CrossRef]
- Varma, A.K.; Mondal, P. Pyrolysis of sugarcane bagasse in semi batch reactor: Effects of process parameters on product yields and characterization of products. Ind. Crops. Prod. 2017, 95, 704–717. [Google Scholar] [CrossRef]
- Gayubo, A.G.; Aguayo, A.T.; Atutxa, A.; Valle, B.; Bilbao, J. Undesired components in the transformation of biomass pyrolysis oil into hydrocarbons on an HZSM-5 zeolite catalyst. J. Chem. Technol. Biotechnol. 2005, 80, 1244–1251. [Google Scholar] [CrossRef]
- Pinho, A.R.; Almeida, M.D.; Mendes, F.L.; Casavechia, L.C.; Talmadge, M.; Kinchin, C.; Chum, H. Fast pyrolysis oil from pinewood chips co-processing with vacuum gas oil in an FCC unit for second generation fuel production. Fuel 2017, 188, 462–473. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bio-Oil (Thermal Pyrolysis) [4,5,6,7,8] | BP (Ketalization Ex) [2] | |
|---|---|---|
| General properties | Varied greatly as a function of source | |
| Acidity | High, pH 2 to 4 [4,5,7] | Almost neutral |
| Viscosity | High due to heavy oxygenates, examples varied: 25 to 1000 [4], 400 to 500 [6], 10 to 100 cp [7] (measured at 25 to 60 °C) | 200 cP |
| Stability (room T storage) | Unstable due to polymerization and condensation of components [4,5] | Stable after 4 months |
| Miscibility with solvents | Not miscible with most solvents | Infinitely miscible with acetone, partially with non-polar solvents and VGO |
| Composition (liq. Product) | ||
| CHO content | 55: 6: 38; typically, O 35 to 40% [5,6] | 48 to 55: 6.4 to 7: 46 to 37 [2] |
| Typical components (in wt. %) | Contains hundreds of compounds in 2 phases: aqueous phase, 15 to 30%, contains acetone, acetic acid MeOH; organic phase: 30 to 40% oxygenates, contains hydroxyl ketones/aldehydes, phenols, furans, acids, and sugars, 10 to 20% hydrocarbons [4,5,6,8] | 50% monoketals 30% higher ketals 20% lignin derivatives |
| Samples | ABET a (m2g−1) | Aext b (m2g−1) | Vmicro b (cm3g−1) | Vtotal c (cm3g−1) | Relative Cryst. XRD (%) d | Total Acid Sites (µmol/g) e | SAR f (RMN) | SiO2 g (%wt) | Al2O3 g (%wt) |
|---|---|---|---|---|---|---|---|---|---|
| AD | 250 | 193 | 0.039 | 0.38 | 100 | - | - | 45.3 | 51.9 |
| DAD | 161 | 132 | 0.013 | 0.35 | 66 | - | - | 43.6 | 54.3 |
| HBEA | 609 | 182 | 0.18 | 0.36 | 100 | 459 | 28 | 92.9 | 6.9 |
| DHBEA | 391 | 135 | 0.10 | 0.40 | 67 | 110 | 43 | 92.7 | 7.1 |
| HBEA a | DHBEA a | |
|---|---|---|
| Conversion (%) | ||
| n-Hexane conversion | 5.7 | 2.2 |
| DX conversion | 99.6 | 99.8 |
| Yield (wt. %) | ||
| Gas | 12.8 | 6.0 |
| Liquid | 84.3 | 91.2 |
| HC product b | 11.2 | 9.8 |
| Oxygenates | 2.0 | 5.4 |
| n-Hexane | 66.0 | 68.5 |
| Water c | 5.1 | 7.5 |
| Coke | 3.1 | 2.9 |
| DHBEA | AD | DAD | |
|---|---|---|---|
| Conversion (%) | |||
| n-Hexane conversion | 2.2 | 0.0 | 0.0 |
| DX conversion | 99.8 | 100 | 100 |
| Yield (wt. %) | |||
| Gas | 6.0 | 3.3 | 4.0 |
| Liquid | 91.2 | 92.8 | 93.3 |
| HC product a | 9.8 | 12.4 | 11.8 |
| Oxygenates | 5.4 | 1.1 | 3.6 |
| n-Hexane | 68.5 | 70.1 | 70.3 |
| Water b | 7.5 | 9.2 | 7.6 |
| Coke | 2.9 | 4.0 | 2.6 |
| Gas (%wt) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | CH4 | C2H4 | C2H6 | C3H8 | C3H6 | C4H10 | C4H8 | CO | CO2 | Total | |
| DHBEA | 0.1 | 2.1 | 0.2 | 0.0 | 0.0 | 0.1 | 0.0 | 0.0 | 2.9 | 0.6 | 6.0 |
| AD | 0.1 | 1.4 | 0.0 | 0.0 | 0.0 | 0.2 | 0.0 | 0.0 | 1.5 | 0.0 | 3.3 |
| DAD | 0.0 | 1.2 | 0.0 | 0.0 | 0.0 | 0.1 | 0.0 | 0.0 | 2.6 | 0.1 | 4.0 |
| Cracking Unit | FB a | FCC b |
|---|---|---|
| Ratio cat/oil | 0.3 | 3 |
| Conversion (%) | ||
| n-Hexane conversion | 0.0 | 7.1 |
| DX conversion | 100 | 100 |
| Yield (wt. %) | ||
| Gas | 4.0 | 8.0 |
| Liquid | 93.3 | 87.1 |
| Liquid product c | 11.8 | 13.6 |
| Oxygenated | 3.6 | 0.9 |
| n-Hexane | 70.3 | 65.1 |
| Water d | 7.6 | 7.5 |
| Coke | 2.6 | 4.9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cardoso, C.; Lam, Y.L.; de Almeida, M.B.B.; Pereira, M.M. Conversion of Sugar Di-Ketals to Bio-Hydrocarbons through Catalytic Cracking over Beta Catalysts in Fixed and Fluidized Catalytic Beds. Chemistry 2023, 5, 479-491. https://doi.org/10.3390/chemistry5010035
Cardoso C, Lam YL, de Almeida MBB, Pereira MM. Conversion of Sugar Di-Ketals to Bio-Hydrocarbons through Catalytic Cracking over Beta Catalysts in Fixed and Fluidized Catalytic Beds. Chemistry. 2023; 5(1):479-491. https://doi.org/10.3390/chemistry5010035
Chicago/Turabian StyleCardoso, Cristiane, Yiu L. Lam, Marlon B. B. de Almeida, and Marcelo Maciel Pereira. 2023. "Conversion of Sugar Di-Ketals to Bio-Hydrocarbons through Catalytic Cracking over Beta Catalysts in Fixed and Fluidized Catalytic Beds" Chemistry 5, no. 1: 479-491. https://doi.org/10.3390/chemistry5010035