
Aluminum-Catalyzed Cross Selective C3–N1′ Coupling Reactions of N-Methoxyindoles with Indoles



Graduate School of Medicine, Dentistry and Pharmaceutical Sciences, Okayama University, Okayama City 700-8530, Japan
*
Author to whom correspondence should be addressed.
Chemistry 2023, 5(1), 452-462; https://doi.org/10.3390/chemistry5010033
Submission received: 13 February 2023
/
Revised: 27 February 2023
/
Accepted: 2 March 2023
/
Published: 3 March 2023
(This article belongs to the Section Molecular Organics)
Abstract
:C3–N1′ bond formation of bisindoles has been a great challenge due to the intrinsic reactivity of indoles as both C3 and N1-nucleophilic character. Herein, we demonstrate an C3–N1′ cross-coupling reaction of indoles using N-methoxyindoles as N-electrophilic indole reagents in the presence of Lewis acid. The bisindoles generated in this transformation are latent C3-nucleophile, allowing them to be used as strategic intermediates in sequential C3–N1′–C3′–N1″ triindole formations. The potential synthetic usefulness of this sequential transformation was highlighted upon application to the construction of C3–N1 looped polyindoles.
Keywords:
1′H-1,3′-biindole; N-electrophilic; N-methoxyindoles; bisindoles; aluminum; cross-coupling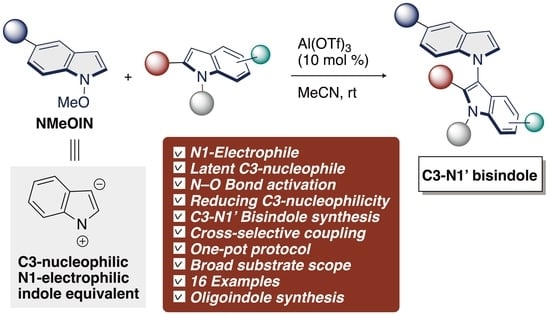
1. Introduction
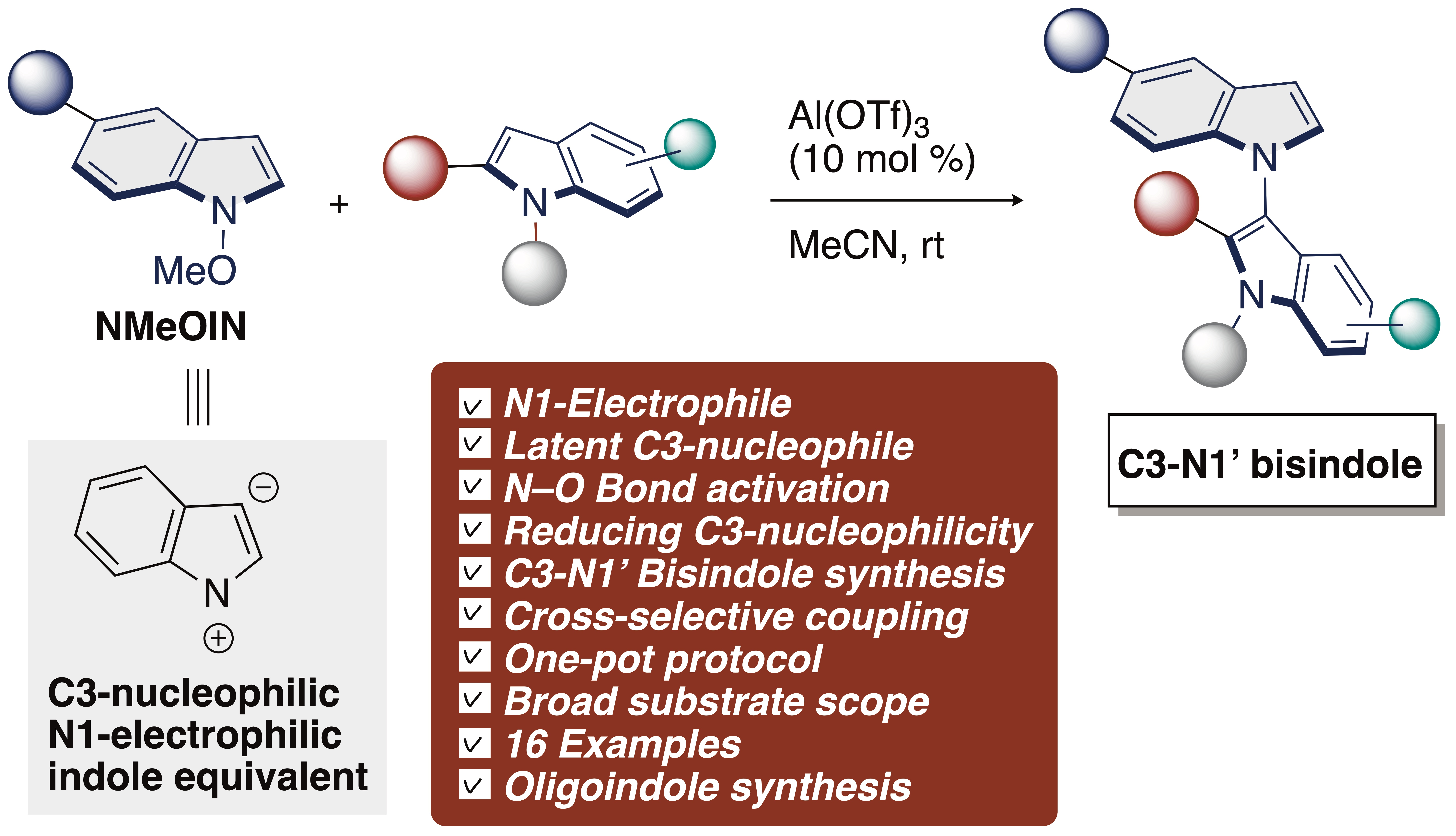
C3–N1′ Heterodimeric tryptophan or tryptamine dimers comprising a pyrroloindoline skeleton are ubiquitous in biologically active alkaloids and form a class of privileged components in medicinal chemistry [1,2,3,4,5,6,7,8,9,10,11,12]. In sharp contrast, construction of C3–N1′ heterodimeric indole skeletons have proven more challenging due to the difficulties associated with introduction of the indole nitrogen (N1′) in the C3-position of indoles, and no approaches have been reported to date (Figure 1) [13,14]. In general, a C3–N1′ cross-coupling reaction between two indole derivatives is one of the most difficult challenges because the most nucleophilic position is the C3-position of the indole nucleus and the most electrophilic site is the C2-position [15,16,17,18,19]. Consequently, cross-coupling reactions take place largely at C2–C3′ due to the intrinsic property. Therefore, in contrast to the well-established C2–C3′ cross-coupling reactions, the C3–N1′ cross-coupling reactions of indoles has received much less attention [20,21,22,23,24,25].
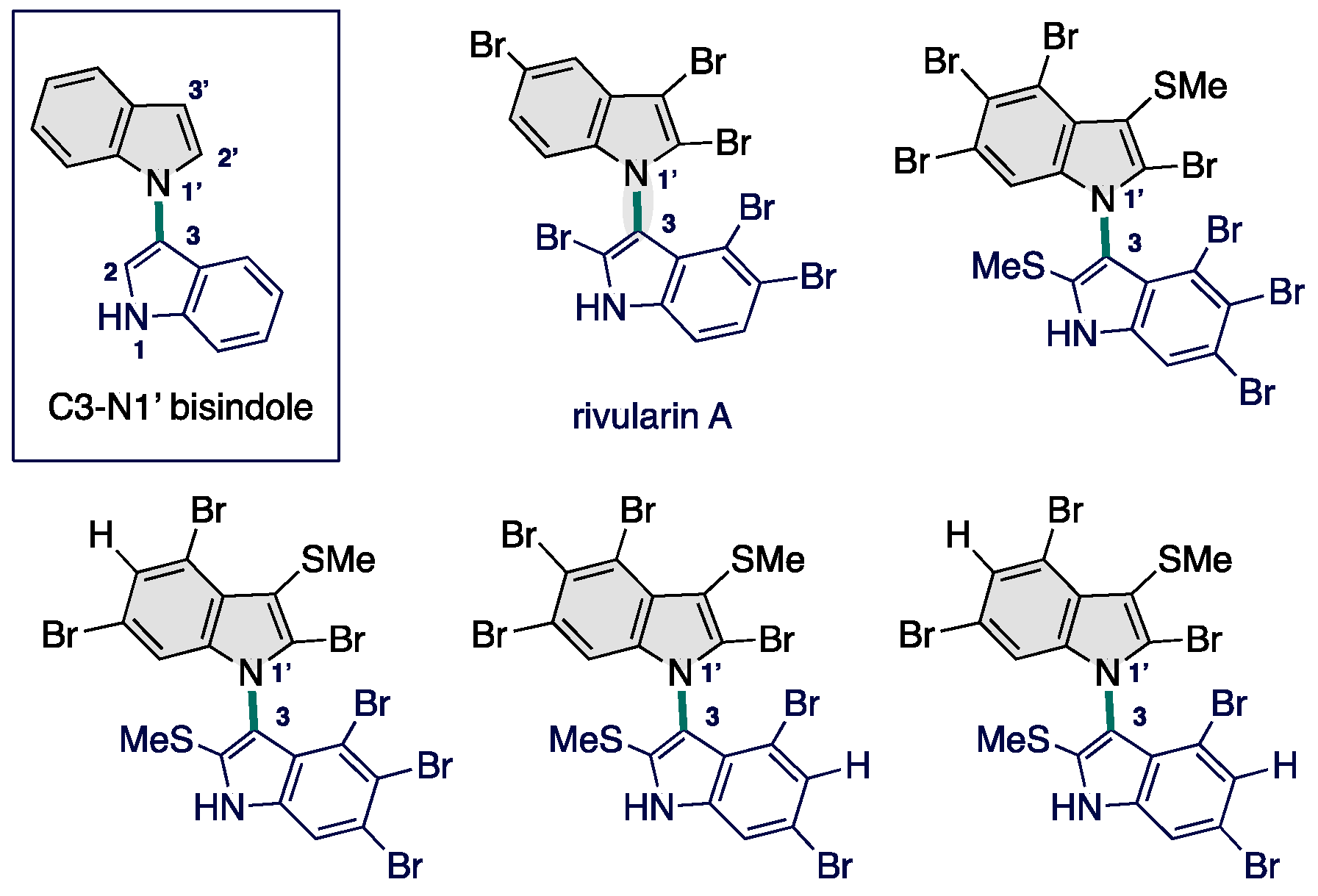
Somei has reported on C3–N1′ bond-forming reactions of N-hydroxy tryptamines in the presence of excess amounts of strong acids to form C3–N1′ heterodimers in 84% yield (Scheme 1a) [26,27,28,29,30]. Although it is necessary to use C3-substituted indoles such as a tryptamine, this strategy contrasts the many indole coupling efforts motivated by the intrinsic C3- and N1-nucleophilicity. However, umpolung of indole nitrogen constitutes a rarely developed latent alternative for direct C3–N1′ bond-forming reactions, whereas electrophilic nitrogen chemistry is well-developed with the leaving group placed at the amine nitrogen atom [31]. Nonetheless, underlying cross-selectivity challenges using C3-unsubstituted indoles remain for development (Scheme 1b). Recently, Buchwald and co-workers described a CuH-catalyzed N-alkylation of C3-unsubstituted N-benzyloxyindoles via hydroamination, which relies on the polarity reversal strategy triggered by the Cu catalyst [32]. To date, other than their use as electrophilic indole nitrogen surrogates toward site-selective alkylation, no general and useful synthetic methods of construction of C3–N1′ bisindoles have been exploited.
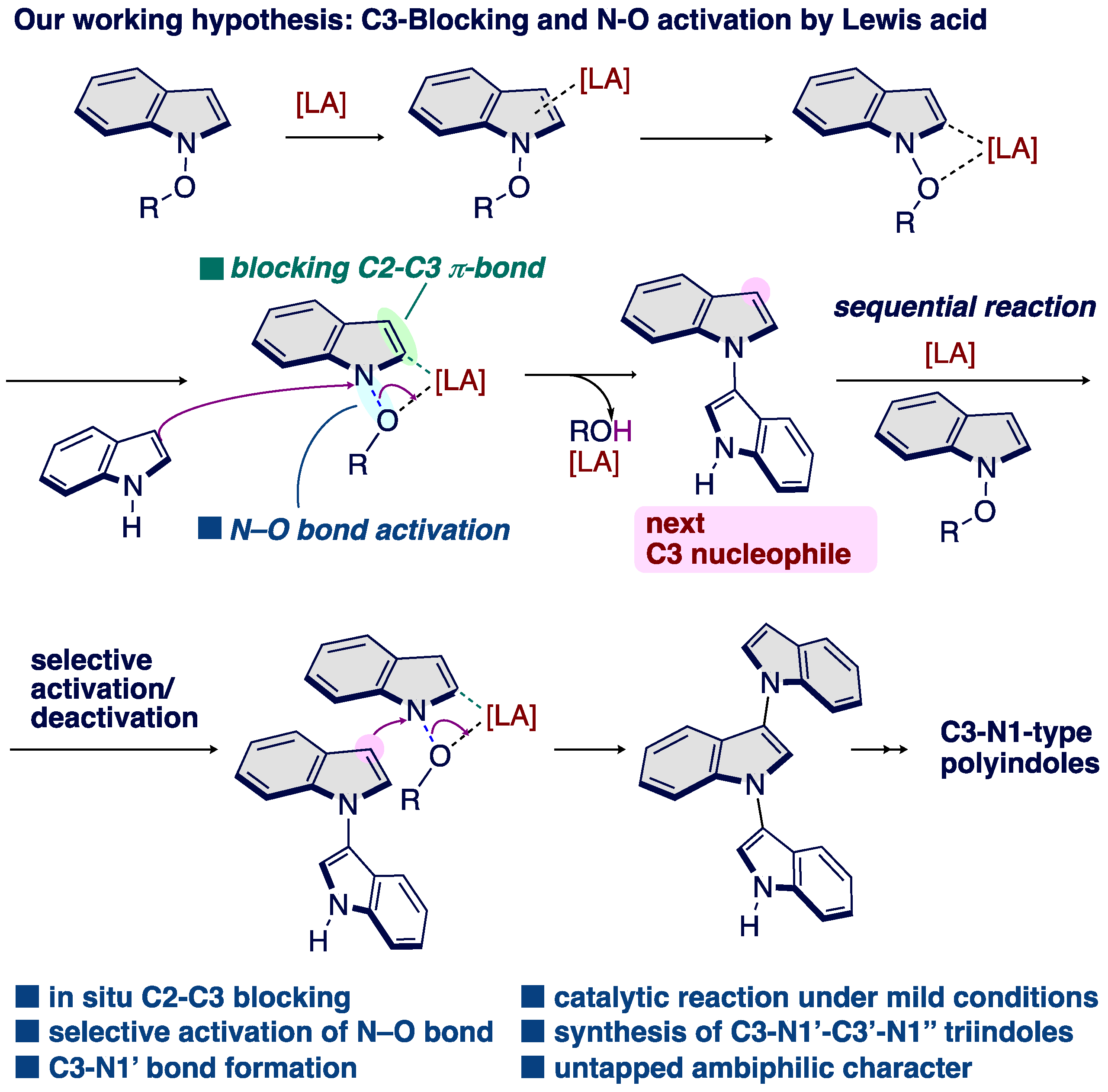
Over the past five years, our group has had an intensive focus on the development and application of umpoled indole surrogates [33,34,35,36,37,38,39,40,41,42,43,44]. These results led us to find that in situ generated 3-methoxyindoles act as a C3-electrophilic reagent that can be harnessed for C–N, C–O, and C–C bond-forming SNAr reactions under indium catalysts [45,46]. In this context, our group has successfully established indium-mediated C–O bond activation for the SNAr reaction with a release of MeOH as a leaving group. By analogy to our indium-catalyzed SNAr reaction, we hypothesized that N-alkoxy indoles might be suitable competent substrate as a N1-electrophilic indole precursor by a Lewis acid activation of alkoxy group through an elimination of ROH, thereby producing a C3–N1′ bisindole (Scheme 2). In this hypothesis, N-alkoxyindole is first combined with Lewis acids (LA) to form an LA–indole complex, which shows an N-electrophilic character by N–O bond activation along with reducing C3-nucleophilicity by coordinating at the C2–C3 π-bond [47,48,49]. Thus, the use of LA could potentially enhance the rate of C3–N1′ cross-coupling in the use of indoles as a nucleophile [50,51], thus altering the balance between homo- and cross-coupling process. This bisindole can serve as re-birthed nucleophiles in a sequential protocol to multiple C3–N1′ bond formation that are otherwise incompatible with Lewis acid-mediated methods. We therefore decided to focus on umpolung of N-alkoxy indoles [52,53]. Herein, we report the successful execution of this hypothesis to enable the construction of C3–N1′ heterodimeric indole skeletons from simple indoles and N-methoxy indoles. The resulting investigations offer most concise catalytic protocol for constructing C3-N1′ heterodimeric indole skeletons developed to date, and shed light on the “old and new” N-methoxyindoles. Notably, this is the first example of a catalytic SNAr reaction at the nitrogen center of C3-unsubstituted N-methoxyindoles is performed in the C–N bond formations [26,27,28,29,30,32].
2. Materials and Methods
High-resolution MS spectra were recorded with a Brucker micrOTOF mass spectrometers (ESI-TOF-MS). The NMR experiments were performed with JEOL JNM-ECZ600R (1H NMR: 600 MHz, 13C NMR: 151 MHz) spectrometer, Varian 600-MR ASW (1H NMR: 600 MHz, 13C NMR: 151 MHz) spectrometer and Varian 400-MR ASW (1H NMR: 400 MHz, 13C NMR: 100 MHz) spectrometer, and chemical shifts are expressed in ppm (δ) using residual undeuterated solvent as an internal reference (CDCl3, 1H NMR: δ 7.25, 13C NMR: δ 77.1). The following abbreviations were used to explain NMR peak multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, sep = septet, m = multiplet, dd = doublet of doublets, ddd = doublet of doublet of doublets, br = broad; coupling constants in Hz; integration. Reactions were monitored by thin layer chromatography (TLC) carried out on a silica gel plates (60F-254) and visualized under UV illumination at 254 or 365 nm depending on the compounds. Flash column chromatography was performed on silica gel (WAKO Gel 75–150 mesh, WAKO Co., Ltd., Tokyo, Japan).
2.1. General Procedure for Synthesis of NMeOINs [54,55]
A solution with the indoline (2 mmol) and Na2WO4·2H2O (0.1 mmol, 0.05 eq) in MeOH (6 mL) and H2O (0.6 mL) was cooled to 0 °C. A total of 30% H2O2 (2.24 mL, 20 mmol) was added dropwise. The mixture was stirred for 5–10 min at room temperature. Then, (MeO)2SO2 (6 mmol, 3 eq) and K2CO3 (10 mmol, 5 eq) was added to the reaction mixture and stirred until the complete disappearance of N-hydroxyindolines indicated by TLC. After H2O (20 mL) was added to the mixture, the whole was extracted with AcOEt (3 × 20 mL), washed with brine (20 mL). The combined organic layer was dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (AcOEt/hexane = 1/20–1/5) to give 1.
5-Methyl-1-methoxyindole (1b): 167 mg, 52% yield. colorless oil; 1H NMR (600 MHz, CDCl3) δ: 7.43 (d, J = 6.0 Hz, 1H), 7.40–7.37 (m, 1H), 7.25 (d, J = 3.0 Hz, 1H), 7.14–7.11 (m, 1H), 6.31 (d, J = 3.0 Hz, 1H), 4.09 (s, 3H), 2.49 (s, 3H); 13C NMR (151 MHz, CDCl3) δ: 130.4, 129.3, 124.7, 124.1, 123.2, 120.9, 108.1, 97.6, 65.8, 21.5; HRMS (ESI) m/z: 162.0920 (Calcd for C10H12NO [M + H]+: 162.0919).
5-Chloro-1-methoxyindole (1c): 184 mg, 51% yield. colorless oil; 1H NMR (400 MHz, CDCl3) δ: 7.56 (d, J = 2.0 Hz, 1H), 7.37 (d, J = 8.4 Hz, 1H), 7.29 (d, J = 3.6 Hz, 1H), 7.20 (dd, J = 8.4, 2.0 Hz, 1H), 6.31 (d, J = 3.6 Hz, 1H), 4.08 (s, 3H); 13C NMR (100 MHz, CDCl3) δ: 130.2, 125.6, 125.1, 124.2, 122.6, 120.5, 109.3, 97.6, 65.9; HRMS (ESI) m/z: 182.0373, 184.0344 (Calcd for C9H9ClNO [M + H]+: 182.0373, 184.0343).
5-Bromo-1-methoxyindole (1d): 230 mg, 51% yield. colorless oil; 1H NMR (600 MHz, CDCl3) δ: 7.71 (s, 1H), 7.31–7.31 (m, 2H), 7.25–7.25 (m, 1H), 6.29 (d, J = 3.6 Hz, 1H), 4.07 (s, 3H); 13C NMR (151 MHz, CDCl3) δ:130.5, 125.9, 125.3, 124.1, 123.7, 113.2, 109.7, 97.6, 66.2; HRMS (ESI) m/z: 225.9867, 227.9847 (Calcd for C9H9rNO [M + H+: 225.9868, 227.9847).
2.2. General Procedure for Synthesis of 1′H-1,3′-Biindole Derivatives (Scheme 3)
To a solution of 1a (1 mmol) and 2 (1 mmol, 1 eq) in MeCN (10 mL, 0.1 M) was added Al(OTf)3 (0.1 mmol, 10 mol%) at room temperature. The mixture was stirred until the complete disappearance of starting material indicated by TLC. After H2O (20 mL) was added to the mixture, the whole was extracted with AcOEt (3 × 20 mL), washed with brine (20 mL). The combined organic layer was dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (AcOEt/hexane = 1/20–1/5) to give 3.
1,3′-Bisindole (3ab): 105 mg, 45% yield. colorless solid; 1H NMR (400 MHz, CDCl3) δ: 8.17 (br s, 1H), 7.73–7.71 (m, 1H), 7.49–7.45 (m, 2H), 7.37–7.28 (m, 4H), 7.21–7.13 (m, 3H), 6.71–6.70 (m, 1H); 13C NMR (100 MHz, CDCl3) δ: 137.5, 134.7, 129.6, 128.6, 123.7, 123.1, 122.0, 120.9, 120.5, 120.0, 119.2, 118.6, 117.6, 111.7, 110.9, 102.5; HRMS (ESI) m/z: 233.1079 (Calcd for C16H13N2 [M + H]+: 233.1079).
2.3. General Procedure for Synthesis of Oligoindoles (Scheme 4)
To a solution of 1a (53.0 mg, 0.36 mmol) and 3ah (73.9 mg, 0.3 mmol) in MeCN (3 mL, 0.1 M) was added Al(OTf)3 (28.5 mg, 0.06 mmol) under reflux. The mixture was stirred until the complete disappearance of starting material indicated by TLC. After H2O (10 mL) was added to the mixture, the whole was extracted with AcOEt (3 × 10 mL), washed with brine (10 mL). The combined organic layer was dried over Na2SO4, filtered and concentrated in vacuo. The residue was purified by silica gel column chromatography (AcOEt/hexane = 1/20–1/5) and PTLC (acetone/hexane = 1/5) to give 3ah (24.4 mg, 33% yield), 4 (16.3 mg, 15% yield), 5 (7.2 mg, 5% yield) and 6 (1.0 mg, 1% yield).
1″-Methyl-1,3′:1′,3″-terindole (4): 16.3 mg, 15% yield. colorless oil; 1H NMR (600 MHz, CDCl3) δ: 7.73 (dd, J = 7.8, 1.2 Hz, 1H), 7.57–7.53 (m, 3H), 7.48–7.42 (m, 4H), 7.37–7.34 (m, 2H), 7.26 (ddd, J = 7.8, 6.6, 1.2 Hz, 1H), 7.22–7.16 (m, 4H), 6.73 (dd, J = 3.0, 1.2 Hz, 1H), 3.92 (s, 3H); 13C NMR (151 MHz, CDCl3) δ:137.5, 136.6, 135.7, 129.7, 128.7, 124.5, 124.2, 124.1, 124.1, 123.1, 123.0, 122.1, 120.9, 120.6, 120.4, 120.0, 118.8, 118.6, 117.5, 115.3, 111.5, 111.0, 109.9, 102.6, 33.3; HRMS (ESI) m/z: 362.1658 (Calcd for C25H20N3 [M + H]+: 362.1657).
1‴-Methyl-1,3′:1′,3″:1″,3‴-quaterindole (5): 7.2 mg, 5% yield. colorless oil; 1H NMR (600 MHz, CDCl3) δ: 7.73 (d, J = 7.8 Hz, 1H), 7.66 (s, 1H), 7.63 (s, 1H), 7.62 (d, J = 7.8 Hz, 1H), 7.58–7.53 (m, 3H), 7.50–7.44 (m, 4H), 7.37–7.34 (m, 2H), 7.31–7.27 (m, 2H), 7.24–7.16 (m, 5H), 6.73 (dd, J = 3.0, 0.6 Hz, 1H), 3.93 (s, 3H); 13C NMR (151 MHz, CDCl3) δ: 137.5, 136.6, 136.5, 135.7, 129.7, 128.7, 124.7, 124.3, 124.1, 124.1, 123.4, 123.2, 123.1, 122.1, 120.9, 120.7, 120.4, 120.1, 118.8, 118.6, 117.6, 116.7, 115.2, 111.6, 111.5, 111.0, 110.0, 102.6, 33.3; HRMS (ESI) m/z: 477.2075 (Calcd for C33H25N4 [M + H]+: 477.2079).
1″″-Methyl-1,3′:1′,3″:1″,3‴:1‴:3″″-quinqueindole (6): 1.0 mg, 1% yield. colorless oil; 1H NMR (600 MHz, CDCl3) δ: 7.76 (s, 1H), 7.74 (d, J = 7.8 Hz, 1H), 7.69 (s, 1H), 7.66–7.64 (m, 3H), 7.60–7.59 (m, 3H), 7.56–7.51 (m, 2H), 7.49–7.46 (m, 3H), 7.39–7.29 (m, 6H), 7.24–7.17 (m, 5H), 6.74 (d, J = 3.0 Hz, 1H), 3.95 (s, 3H); HRMS (ESI) m/z: 592.2505 (Calcd for C41H30N5 [M + H]+: 592.2501).
Detailed synthetic procedure and corresponding analytic data can be found in the Supplementary Materials.
3. Results and Discussion
3.1. Optimization of Reaction Conditions
To investigate the feasibility of the envisaged SNAr reaction, we select N-methoxyindole (NMeOIN, 1a) and 2-methylindole (2a) as model substrates for optimization. Initially, 1a and 2a were reacted in the presence of In(OTf)3 [47] in MeCN at room temperature for 1.5 h (Table 1, run 1). We were gratified to observe that the use of indium catalyst enabled our proposed reactivity, leading to C3-N1′ bisindole 3aa in 72% yield. From the catalysts tested (InF·3H2O, InBr3, InCl3·4H2O, Ga(OTf)3, La(OTf)3, Bi(OTf)3, AgOTf, Yb(OTf)3, Cu(OTf)2, Zn(OTf)2, and Al(OTf)3) (runs 2–12), In(OTf)3, Ga(OTf)3, Bi(OTf)3, Cu(OTf)2, and Al(OTf)3) [50,51] were found to promote the reaction quite well, affording the C3-N1′ bisindole 3aa in 72%, 79%, 60%, 72%, and 83 % yields, respectively. The highest isolated yield 87% was obtained from the reaction with Al(OTf)3 (run 12). Among the aluminum catalysts (Al(OTf)3, AlCl3, and Al(OiPr)3), Al(OTf)3 proved to be the best catalyst (runs 12–14). Next, to investigate the effect of the solvent with Al(OTf)3, additional optimization was performed (runs 15–17). To our surprise, different solvents showed a notable effect on the Al(OTf)3-catalyzed reaction. Chlorobenzene (PhCl) showed the same effects as CHCl3 (runs 15 and 17), while 1,4-dioxane led to low conversion (run 16). When performed in the presence of TfOH, the reaction gave 3aa in 54% yield (run 18). Finally, the reaction failed to proceed in the absence of catalyst or solvent (runs 19 and 20). In our cases, Al(OTf)3 could not be recovered after the reactions [51]. Based on the above results, the optimized reaction conditions were determined (10 mol% of Al(OTf)3, MeCN, and room temperature).
3.2. Scope and Limitations
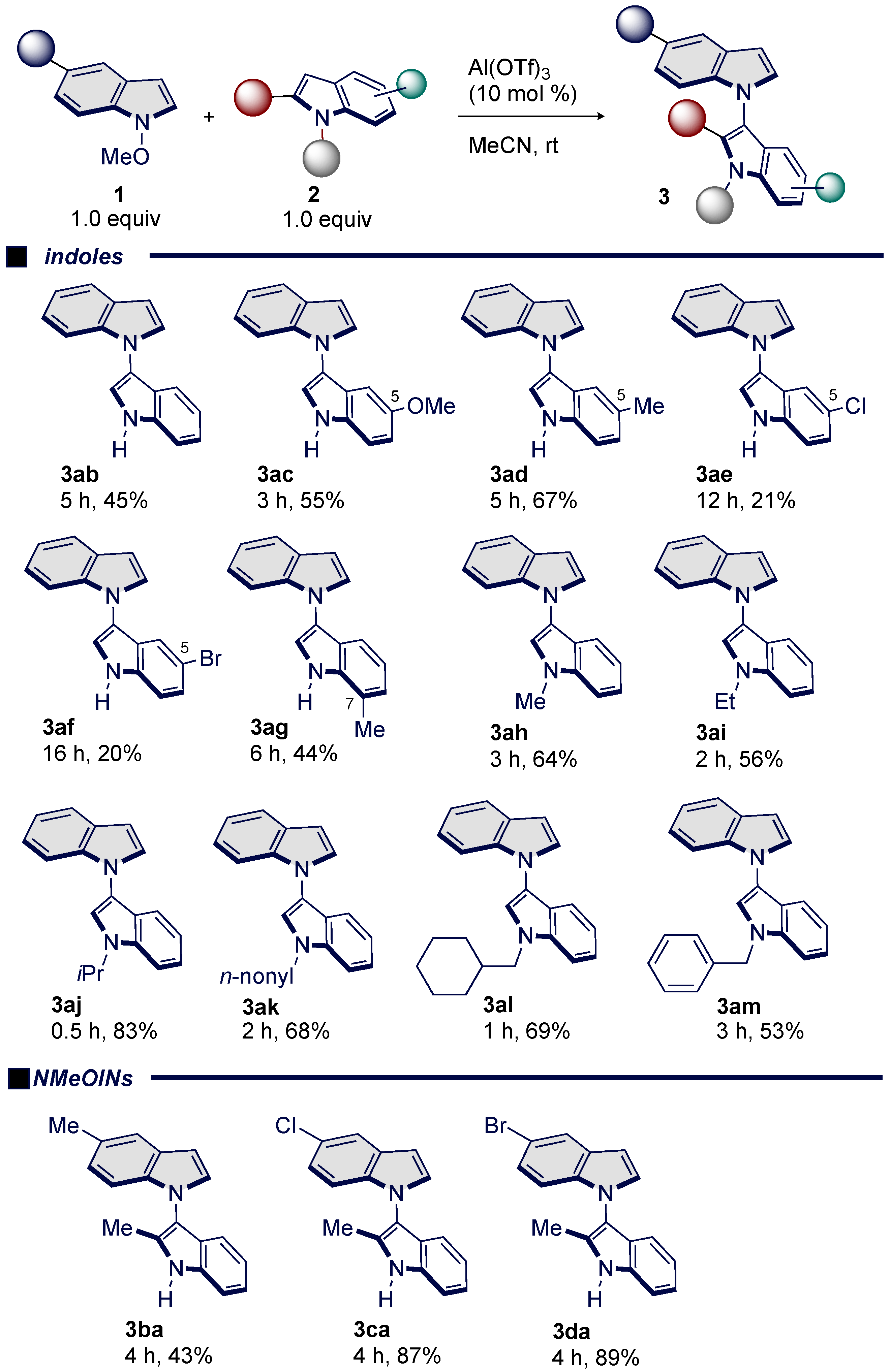
With the optimized reaction conditions in hand, we investigated a range of indoles 2 and NMeOIN 1a to assess the generality of this transformation (Scheme 3). Unsubstituted indole afforded bisindole 3ab in 45% yield. The presence of electron-withdrawing group was found to have a negative influence on the reaction (3ac, 3ad, 3ag vs. 3ae, 3af), which might be due to the lack of nucleophilicity. Next, we focused on the reactivity of N-substituted indoles. N-Methylindole reacted well with NMeOIN 1a yielding product 3ah in 64% yield. Further investigations revealed that some N-alkylindoles were applicable to deliver the N-alkylated bisindoles bearing the ethyl (3ai), isopropyl (3aj), n-nonyl (3ak), and cyclohexylmethylene (3al) groups. Additionally, the reaction of benzyl-substituted indole afforded 3am in 53% yield. However, the reaction of Ts-indole with 1a resulted in no reaction due to its low nucleophilicity.
The scope of the NMeOIN 1 was also investigated. With the electro-donating group attached to the indole-ring, the reaction proceeded smoothly, leading to 3ba in 45% yield. Interestingly, in contrast to 3ba, the presence of electron-withdrawing group attached to the indole-ring was found to have a positive effect, increasing in yields (3ca: 87%, 3da: 89%). From the scope and limitation experiments, we conclude that this transformation is quite sensitive to substituents on the indole-ring. In addition, the preferential C3–N1′ reactivity of NMeOIN in all cases can be rationalized based on the both N-activated and C2–C3 deactivated abilities of Al(OTf)3 toward 1a. This observed selectivity can prove helpful in synthetic application such as C3–N1′-type bisindole alkaloids and polyindoles [13,14].
3.3. Synthesis of Oligoindoles
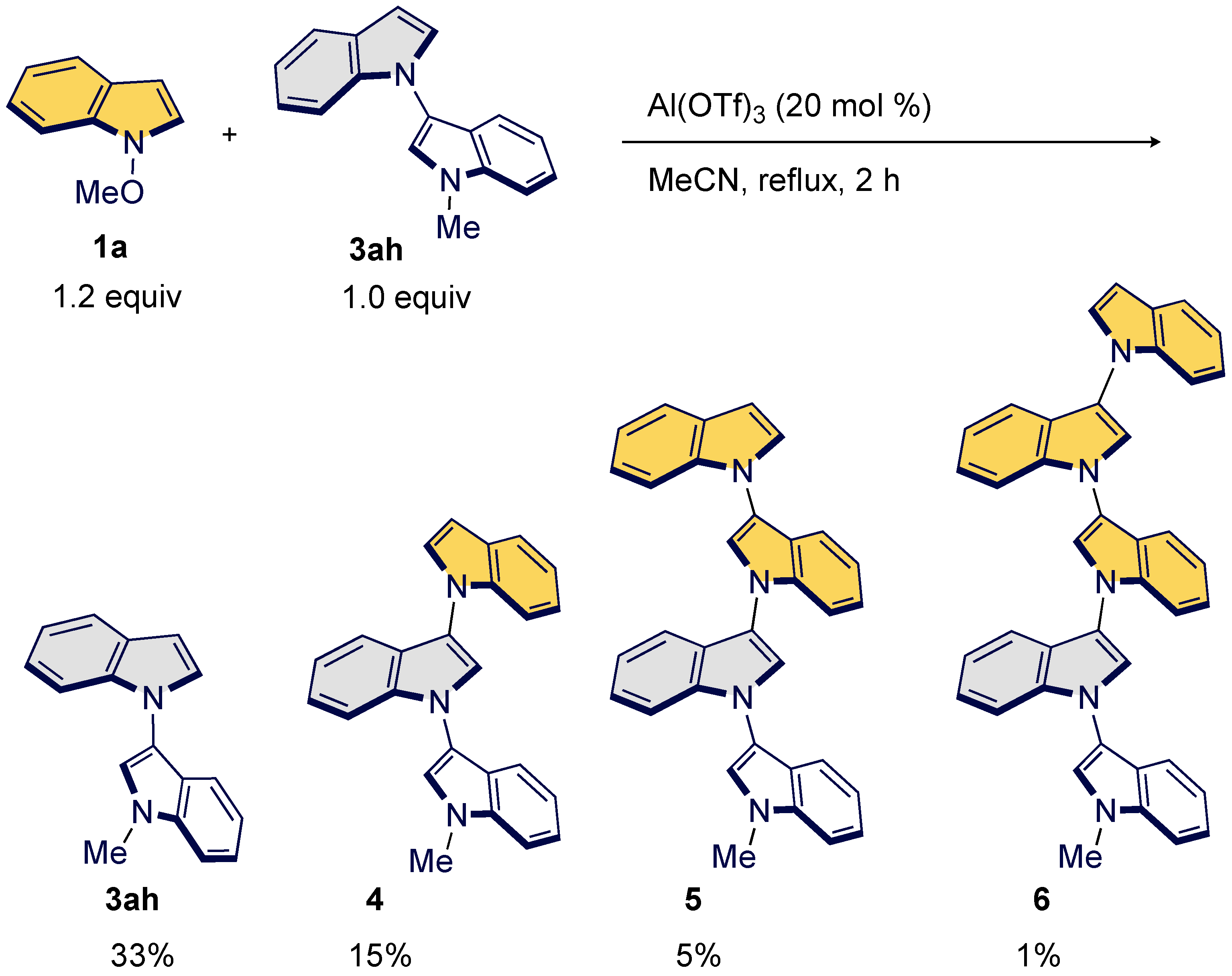
To probe the feasibility of a formation of oligoindoles, we tested the reaction of bisindole 3ah with 1a (Scheme 4). As construction of oligoindoles through C3–N1′ bond formation is unprecedented [56,57,58,59]; we hope this transformation will promote further progress in the material sciences [60,61,62,63,64]. After intensive investigations, we found that a reaction using 20 mol% of Al(OTf)3 under reflux conditions plays a crucial role in delivering previously untapped C3–N1′ homologs such as trimer 4, tetramer 5, and pentamer 6 in one-pot protocol.
3.4. Scalability of the Aluminum-Catalyzed Cross Selective C3–N1′Cross-Coupling Reaction
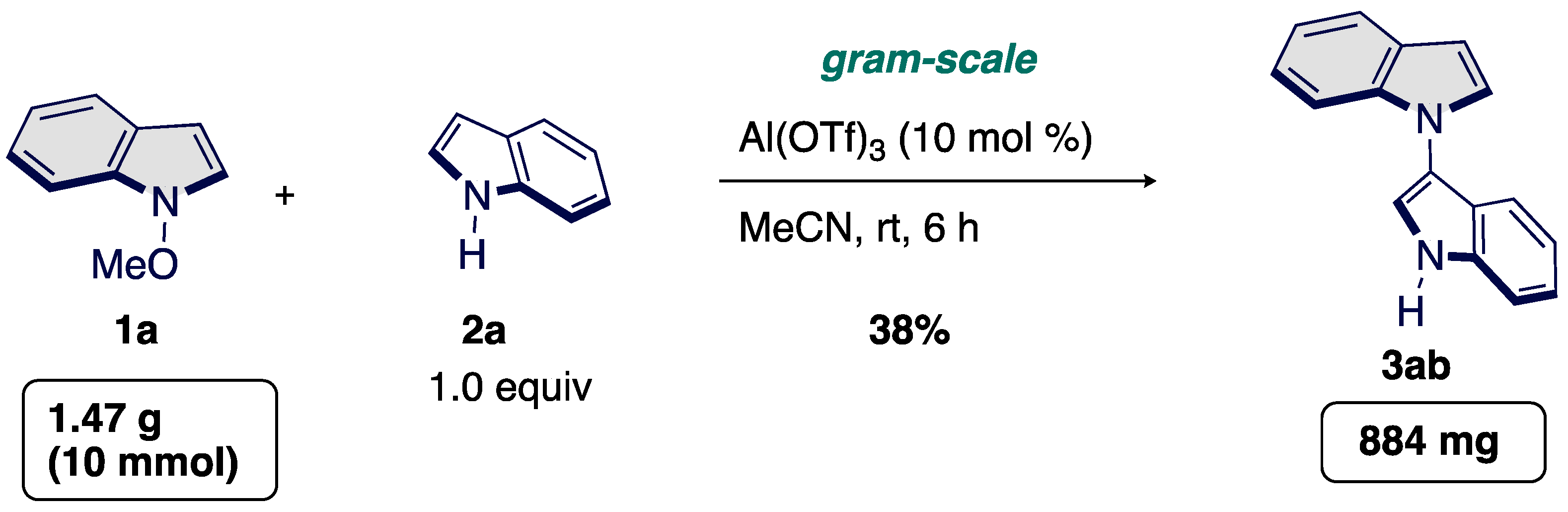
Considering the potential synthetic utility, we next scaled-up synthesis of 3. The synthesis of bisindole 3 could be scalable; as shown in Scheme 5, we efficiently prepared large quantities of a representative bisindole 3ab from NMeOIN (10 mmol) with indole. Notably, our transformation could be scaled up to 10 mmol with an acceptable loss of efficiency for 3ab (38% yield vs. 45% yield).
As mentioned above, this is the first example that N-methoxyindoles showed unprecedented ambiphilic reactivity of N1-electrophile and C3-nucleophile triggered by σ-activation/π-deactivation [47,48,49]. Our protocol also expands the umpolung chemistry of indoles [52,53], thereby affording unprecedented construction of C3–N1′ polyindoles.
4. Conclusions
In conclusion, we have successfully developed a novel strategy that addresses the latent N-electrophilicity and intrinsic C3-nucleophilicity of N-methoxyindoles toward less well-developed C3–N1′ bond formation of bisindoles in the presence of Lewis acid. Given the C2–C3 deactivation/N–O activation protocol, our transformation was applicable to site-selective synthesis of C3–N1′ bisindoles and C3–N1′–C3′–N1″ triindoles. Importantly, these transformations could be achieved only with the aid of Lewis acid. Additionally, the C3–N1 oligomerization paves the way for further application in material sciences.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/chemistry5010033/s1, The Supplementary Materials contain detailed procedures for synthesis of compounds and analytical data including 1H- and 13C-NMR spectra.
Author Contributions
Conceptualization, T.A.; investigation, T.A.; resources, T.A.; visualization, T.A.; structures, T.A.; experiments, K.T., T.Y. and S.H.; writing—original draft preparation, T.A.; writing—review and editing, T.A. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partly supported by JSPS KAKENHI (22K06503). This work was also supported by JST SPRING, Grant Number JPMJSP2126.
Data Availability Statement
Not applicable.
Acknowledgments
T.Y. thanks Nagai Memorial Research Scholarship from the Pharmaceutical Society of Japan.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Espejo, V.R.; Rainier, J.D. An expeditious synthesis of C(3)-N(1′) heterodimeric indolines. J. Am. Chem. Soc. 2008, 130, 12894–12895. [Google Scholar] [CrossRef]
- Newhouse, T.; Baran, P.S. Total synthesis of (±)-psychotriamine. J. Am. Chem. Soc. 2008, 130, 10886–10887. [Google Scholar] [CrossRef]
- Espejo, V.R.; Rainier, J.D. Total synthesis of kapakahine E and F. Org. Lett. 2010, 12, 2154–2157. [Google Scholar] [CrossRef]
- Pérez-Balado, C.; de Lera, Á.R. Concise total synthesis and structural revision of (+)-pestalazine B. Org. Biomol. Chem. 2010, 8, 5179–5186. [Google Scholar] [CrossRef]
- Foo, K.; Newhouse, T.; Mori, I.; Takayama, H.; Baran, P.S. Total synthesis of guided structure elucidation of (+)-psychotetramine. Angew. Chem. Int. Ed. 2011, 50, 2716–2719. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Xia, T.; Yao, L.; Deng, H.; Liao, X. Enantioselective and diastereoselective azo-coupling/iminium-cyclizations: A unified strategy for the total syntheses of (–)-psychotriasine and (+)-pestalazine B. Chem. Sci. 2015, 6, 3599–3605. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, A.A.; Chisholm, J.D. Lewis acid catalyzed displacement of trichloroacetamidates in the synthesis of functionalized pyrroloindolines. Org. Lett. 2016, 18, 4100–4103. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Yi, J.-C.; Xheng, Z.-B.; Tang, Y.; Dai, L.-X.; You, S.-L. Enantioselective synthesis of 3a-amino-pyrroloindolines by copper-catalyzed direct asymmetric dearomative amination of tryptamines. Angew. Chem. Int. Ed. 2016, 55, 751–754. [Google Scholar] [CrossRef]
- Dai, J.; Xiong, D.; Yuan, T.; Liu, J.; Chen, T.; Shao, Z. Chiral primary amine catalysis for asymmetric Mannich reactions of aldehydes with ketimines: Stereoselectivity and reactivity. Angew. Chem. Int. Ed. 2017, 56, 12697–12701. [Google Scholar] [CrossRef]
- Nelson, B.M.; Loach, R.P.; Schiesser, S.; Movassaghi, M. Concise total synthesis of (+)-asperazine A and (+)-pestalazine B. Org. Biomol. Chem. 2018, 16, 202–207. [Google Scholar] [CrossRef]
- Gentry, E.C.; Rono, L.J.; Hale, M.E.; Matsuura, R.; Knowles, R.R. Enantioselective synthesis of pyrroloindolines via noncovalent stabilization of indole radical cations to the synthesis of alkaloid natural products. J. Am. Chem. Soc. 2018, 140, 3394–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakamata, H.; Ueda, H.; Tokuyama, H. Construction of indole structure on pyrroloindolines via AgNTf2-mediated amination/cyclization cascade: Application to total synthesis of (+)-pestazine B. Org. Lett. 2019, 21, 4205–4209. [Google Scholar] [CrossRef] [PubMed]
- Norton, R.S.; Wells, R.J. A series of chiral polybrominated biindoles from the marine blue-green alga Rivularia firma. Application of carbon-13 NMR spin-lattice relaxation data and carbon-13-proton coupling constants to structure elucidation. J. Am. Chem. Soc. 1982, 104, 3628–3635. [Google Scholar] [CrossRef]
- Kubota, N.K.; Iwamoto, H.; Fukuzawa, Y.; Uchio, Y. Five new sulfur-containing polybrominated bisindoles from the red alga Laurencia brongniartii. Heterocycles 2005, 65, 2675–2682. [Google Scholar]
- Liang, Z.; Zhao, J.; Zhang, Y. Palladium-catalyzed regioselective oxidative coupling of indoles and one-pot synthesis of acetoxylated biindolyls. J. Org. Chem. 2010, 75, 170–177. [Google Scholar] [CrossRef]
- Li, Y.-X.; Ji, K.-G.; Wang, H.-X.; Ali, S.; Liang, Y.-M. Iodine-induced regioseletive C–C and C–N bonds formation of N-protected indoles. J. Org. Chem. 2011, 76, 744–747. [Google Scholar] [CrossRef]
- Guo, T.; Han, S.-L.; Liu, Y.-C.; Liu, Y.; Liu, H.-M. Convenient synthesis of antiproliferative 2,3-dihydro-2,3′-bisindoles via dimerization of N–H indole derivatives. Tetrahedron Lett. 2016, 57, 1097–1099. [Google Scholar] [CrossRef]
- Huang, P.; Peng, X.; Hu, D.; Liao, H.; Tang, S.; Liu, L. Regioselective synthesis of 2,3′-biindoles mediated by an NBS-induced homo-coupling of indoles. Org. Biomol. Chem. 2017, 15, 9622–9629. [Google Scholar] [CrossRef]
- Yin, B.; Huang, P.; Lu, Y.; Liu, L. TEMPO-catalyzed oxidative homocoupling route to 3,2′-biindolin-2-ones via an indolin-3-one intermediate. RSC Adv. 2017, 7, 606–610. [Google Scholar] [CrossRef] [Green Version]
- Benkovics, T.; Guzei, I.A.; Yoon, T.P. Oxaziridine-mediated oxyamination of indoles: An approach to 3-aminoindoles and enantiomerically enriched 3-aminopyrroloindolines. Angew. Chem. Int. Ed. 2010, 49, 9153–9157. [Google Scholar] [CrossRef]
- Lee, D.J.; Yoo, E.J. Efficient synthesis of C–N-couped heterobiaryls by sequential N–H functionalization reactions. Org. Lett. 2015, 17, 1830–1833. [Google Scholar] [CrossRef] [PubMed]
- Li, T.-R.; Cheng, B.-Y.; Wang, Y.-N.; Zhang, M.-M.; Lu, L.-Q.; Xiao, W.-J. A copper-catalyzed decarboxylative amination/hydroamination sequence: Switchable synthesis of functionalized indoles. Angew. Chem. Int. Ed. 2016, 55, 12422–12426. [Google Scholar] [CrossRef] [PubMed]
- Yonekura, K.; Yoshimura, Y.; Akehi, M.; Tsuchimoto, T. A heteroarylamine library: Indium-catalyzed nucleophilic aromatic substitution of alkoxyheteroarenes with amines. Adv. Synth. Catal. 2018, 360, 1159–1181. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Tian, J.-S.; Tan, P.-W.; Cao, Q.; Zhang, X.-X.; Cao, Z.-Y.; Zhou, F.; Wang, X.; Zhou, J. Ragiodivergent intramolecular nucleophilic addition of ketimines for the diverse synthesis of azacycles. Angew. Chem. Int. Ed. 2020, 59, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- Shan, X.-H.; Zheng, H.-X.; Yang, B.; Tie, L.; Fu, J.-L.; Qu, J.-P.; Kang, Y.-B. Copper-catalyzed oxidative benzylic C–H cyclization via iminyl radical from intermolecular anion-radical redox relay. Nat. Commun. 2019, 10, 908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somei, M. 1-Hydroxyindoles. Heterocycles 1999, 50, 1157–1211. [Google Scholar] [CrossRef]
- Somei, M. A frontier in indole chemistry: 1-Hydroxyindoles, 1-hydroxytryptamines, and 1-hydrxytryptophans. Top. Heterocycl. Chem. 2006, 6, 77–111. [Google Scholar]
- Somei, M.; Yamada, F.; Hayashi, T.; Goto, A.; Saga, Y. Nucleophilic substitution reaction on the nitrogen of indole nucleus: Formation of 1-(indol-3-yl)indoles upon reaction of 1-hydroxyindoles with indole in formic acid. Heterocycles 2001, 55, 457–460. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Peng, W.; Nakai, Y.; Yamada, K.; Somei, M. Nucleophilic substitution reaction on the nitrogen of indole nucleus: A novel synthesis of 1-aryltryptamines. Heterocycles 2002, 57, 421–424. [Google Scholar]
- Yamada, F.; Goto, A.; Peng, W.; Hayashi, T.; Saga, Y.; Somei, M. Nucleophilic substitution reaction at the 1-position of 1-hydroxytryptamine and -tryptophan derivatives. Heterocycles 2003, 61, 163–172. [Google Scholar]
- O’Neil, L.G.; Bower, J.F. Electrophilic aminating agents in total synthesis. Angew. Chem. Int. Ed. 2021, 60, 25640–25666. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Kim, S.-T.; Jeong, J.; Baik, M.-H.; Buchwald, S.L. CuH-Catalyzed enantioselective alkylation of indole derivatives with ligand-controlled regiodivergence. J. Am. Chem. Soc. 2019, 141, 3901–3909. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Suzuki, T.; Anada, M.; Matsunaga, S.; Yamada, K. 2-Hydroxyindoline-3-triethylammonium bromide: A reagent for formal C3-electrophilic reactions of indoles. Org. Lett. 2017, 19, 4275–4278. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Yamada, K. Dehydrative Mannich-type reaction for the synthesis of azepinobisindole alkaloid iheyamine A. Org. Lett. 2018, 20, 1469–1472. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Shimizu, H.; Takada, S.; Tanaka, T.; Yoshikawa, M.; Yamada, K. Double “open and shut” transformation of g-carbolines triggered by ammonium salts: One-pot synthesis of multiheterocyclic compounds. Org. Lett. 2018, 20, 1589–1592. [Google Scholar] [CrossRef]
- Abe, T.; Satake, S.; Yamada, K. Biomimetic synthesis of iheyamine A from spirocyclic oxindoles. Heterocycles 2019, 99, 379–388. [Google Scholar] [CrossRef] [Green Version]
- Abe, T.; Aoyama, S.; Ohmura, M.; Taniguchi, M.; Yamada, K. Revisiting furodiindolines: One-pot synthesis of furodiindolines using indole 2,3-epoxide surrogates and their synthetic applications. Org. Lett. 2019, 21, 3367–3371. [Google Scholar] [CrossRef]
- Abe, T.; Yamada, K.; Nishi, T. Development and application of indole-2,3-epoxide surrogates. J. Synth. Org. Chem. 2020, 78, 597–607. [Google Scholar] [CrossRef]
- Abe, T.; Yamashiro, T.; Hirao, S. A metal-, oxidant-, and fluorous solvent-free synthesis of a-indolylketones enabled by an umpolung strategy. Chem. Commun. 2020, 56, 10183–10186. [Google Scholar] [CrossRef]
- Abe, T.; Noda, K.; Sawada, D. Synthesis of a-substituted indolylacetamide using acetonitriles as acetamide enolate equivalents through O-transfer reactions. Chem. Commun. 2021, 57, 7493–7496. [Google Scholar] [CrossRef]
- Yamashiro, T.; Abe, T.; Tanioka, M.; Kamino, S.; Sawada, D. cis-3-Azido-2-methoxyindolines as safe and stable precursors to overcome he instability of fleeting 3-azidoindoles. Chem. Commun. 2021, 57, 13381–13384. [Google Scholar] [CrossRef] [PubMed]
- Yamashiro, T.; Abe, T.; Sawada, D. Synthesis of 2-monosubstituted indolin-3-ones by cine-substitution of 3-azido-2-methoxyindolines. Org. Chem. Front. 2022, 9, 1897–1903. [Google Scholar] [CrossRef]
- Abe, T.; Yamashiro, T.; Shimizu, K.; Sawada, D. Indole editing enabled by HFIP-mediated ring-switch reactions of 3-amino-2-hydroxyindolines. Chem. Eur. J. 2022, 28, e202201113. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Nakajima, R.; Yamashiro, T.; Sawada, D. First total synthesis of reassigned echinosulfonic acid D. J. Nat. Prod. 2022, 85, 2122–2125. [Google Scholar] [CrossRef] [PubMed]
- Hirao, S.; Yamashiro, T.; Kohira, K.; Mishima, N.; Abe, T. 2,3-Dimethoxyindolines: A latent electrophile for SNAr reactions triggered by indium catalysts. Chem. Commun. 2020, 56, 5139–5142. [Google Scholar] [CrossRef]
- Abe, T.; Hirao, S. Rapid access to indole-fused bicyclo[2.2.2]octanones by merging the umpolung strategy and molecular iodine as a green catalyst. Chem. Commun. 2020, 56, 5139–5142. [Google Scholar] [CrossRef]
- Sestelo, J.P.; Sarandeses, L.A.; Martínez, M.M.; Alonso-Marañón, L. Indium(III) as p-acid catalyst for the electrophilic activation of carbon-carbon unsaturated systems. Org. Biomol. Chem. 2018, 16, 5733–5747. [Google Scholar] [CrossRef]
- Shen, Z.-L.; Wang, S.-Y.; Chok, Y.-K.; Xu, Y.-H.; Loh, T.-P. Organoindium reagents: The preparation and application in organic synthesis. Chem. Rev. 2013, 113, 271–401. [Google Scholar] [CrossRef]
- Zhao, K.; Shen, L.; Shen, Z.-L.; Loh, T.-P. Transition metal-catalyzed cross-coupling reactions using organoindium reagents. Chem. Rev. 2017, 46, 586–602. [Google Scholar] [CrossRef]
- Gohain, M.; Marais, C.; Bezuidenhoudt, C.B. An Al(OTf)3-catalyzed environmentally benign process for the propargylation of indoles. Tetrahedron Lett. 2012, 53, 4704–4707. [Google Scholar] [CrossRef]
- Ajvazi, N.; Stavber, S. Alcohols in direct carbon-carbon and carbon-heteroatom bond-forming reactions: Recent advances. Arkivoc 2018, 2018, 288–329. [Google Scholar] [CrossRef] [Green Version]
- Bandini, M. Electrophilicity: The “dark-side” of indole chemistry. Org. Biomol. Chem. 2013, 11, 5206–5212. [Google Scholar] [CrossRef] [PubMed]
- Cerveri, A.; Bandini, M. Recent advances in the catalytic functionalization of “electrophilic” indoles. Chin. J. Chem. 2020, 38, 287–294. [Google Scholar] [CrossRef]
- Kawasaki, T.; Kodama, A.; Nishida, T.; Shimizu, K.; Somei, M. Preparation of 1-hydroxyindole derivatives and a new route to 2-substituted indoles. Heterocycles 1991, 32, 221–227. [Google Scholar]
- Vo, Q.V.; Trenerry, C.; Rochfort, S.; Wadespm, J.; Leyton, C.; Hughes, A.B. Synthesis and anti-inflammatory activity of indole glucosinolates. Bioorg. Med. Chem. 2014, 22, 856–864. [Google Scholar] [CrossRef]
- Pezzella, A.; Panzella, L.; Natangelo, A.; Arzillo, M.; Napolitano, A.; d’Ischia, M. 5,6-Dihydroxyindole tetramers with “anomalous” interunit bonding patterns by oxidative coupling of 5,5′,6,6′-tetrahydroxy-2,7′-biindolyl: Emerging complexities on the way toward an improved model of eumelanin build up. J. Org. Chem. 2007, 72, 9225–9230. [Google Scholar] [CrossRef] [PubMed]
- Arzillo, M.; Pezzella, A.; Crescezi, O.; Napolitano, A.; Land, E.J.; Barone, V.; d’Ischia, M. Cyclic structural motifs in 5,6-dihydroxyindole polymerization uncovered: Biomimetic modular buildup of a unique five-membered macrocycle. Org. Lett. 2010, 12, 3250–3253. [Google Scholar] [CrossRef]
- Chen, C.-T.; Chuang, C.; Cao, J.; Ball, V.; Ruch, D.; Buehler, M.J. Excitonic effects from geometric order and disorder explain broadband optical absorption in eumelanin. Nat. Commun. 2014, 5, 3859. [Google Scholar] [CrossRef] [Green Version]
- Jamison, C.R.; Badillo, J.J.; Lipshultz, J.M.; Comito, R.J.; MacMillan, D.W.C. Catalyst-controlled oligomerization for the collective synthesis of polypyrroloindoline natural products. Nat. Chem. 2017, 9, 1165–1169. [Google Scholar] [CrossRef]
- Chang, K.-J.; Kang, B.-N.; Lee, M.-H.; Jeong, K.-S. Oligoindole-based foldamers with a helical conformation induced by chloride. J. Am. Chem. Soc. 2005, 127, 12214–12215. [Google Scholar] [CrossRef]
- Wu, J.; Zhou, W.; Jiang, F.; Chang, Y.; Zhou, Q.; Li, D.; Ye, G.; Li, C.; Nie, G.; Xu, J.; et al. Three-dimensional porous carbon derived from polyindole hollow nanospheres for high-performance supercapacitor electrode. ACS Appl. Energy Mater. 2018, 1, 4572–4579. [Google Scholar] [CrossRef]
- Bürger, M.; Ehrhardt, N.; Barber, T.; Ball, L.T.; Namyslo, J.C.; Jones, P.G.; Werz, D.B. Phosphine-catalyzed aryne oligomerization: Direct access to a,w-bisfunctionalized oligo(ortho-arylenes). J. Am. Chem. Soc. 2021, 143, 16796–16803. [Google Scholar] [CrossRef] [PubMed]
- Boknevitz, K.; Darrigan, C.; Chrostowska, A.; Liu, S.-Y. Cation–p binding ability of BN indole. Chem. Commun. 2020, 56, 3749–3752. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.; Wang, Y.; Wang, C.; Li, Y.; Xu, Y.; Yang, L. A recyclable hydroxyl functionalized polyindole hydrogel for sodium hydroxide extraction via the synergistic effect of cation-p interactions and hydrogen boning. Chem. Commun. 2018, 54, 9785–9788. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Structures of C3–N1′ bisindole alkaloids.

Scheme 1.
State-of-the-art N-electrophilic indoles. (a) Previous works by Somei; (b) Remaining challenge.
Scheme 1.
State-of-the-art N-electrophilic indoles. (a) Previous works by Somei; (b) Remaining challenge.

Scheme 2.
Our working hypothesis inspired by our previous SNAr reaction under indium catalyst.

Scheme 3.
Substrate scope.

Scheme 4.
Oligomerization.

Scheme 5.
Gram-scale synthesis of bisindole 3ab.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Optimization of reaction conditions.
 | ||||
|---|---|---|---|---|
| Run 1 | Catalyst | Solvent | Time (h) | Yield (%) of 3aa 2 |
| 1 | In(OTf)3 | MeCN | 1.5 | 72 |
| 2 | InF3·3H2O | MeCN | 1.5 | 14 |
| 3 | InBr3 | MeCN | 1.5 | 18 |
| 4 | InCl3·4H2O | MeCN | 1.5 | 8 |
| 5 | Ga(OTf)3 | MeCN | 1.5 | 79 |
| 6 | La(OTf)3 | MeCN | 1.5 | 31 |
| 7 | Bi(OTf)3 | MeCN | 1.5 | 60 |
| 8 | AgOTf | MeCN | 1.5 | 15 |
| 9 | Yb(OTf)3 | MeCN | 1.5 | 0 |
| 10 | Cu(OTf)2 | MeCN | 1.5 | 72 |
| 11 | Zn(OTf)2 | MeCN | 1.5 | 7 |
| 12 | Al(OTf)3 | MeCN | 1.5 | 83 (87) 3 |
| 13 | AlCl3 | MeCN | 1.5 | 23 |
| 14 | Al(O-iPr)3 | MeCN | 1.5 | 9 |
| 15 | Al(OTf)3 | PhCl | 1.5 | 69 |
| 16 | Al(OTf)3 | 1,4-dioxane | 1.5 | 43 |
| 17 | Al(OTf)3 | CHCl3 | 1.5 | 71 |
| 18 | TfOH | MeCN | 1.5 | 54 |
| 19 | --- | MeCN | 24 | nr |
| 20 | Al(OTf)3 | --- | 24 | 0 |
1 1a (0.1 mmol), 2a (0.1 mmol), and catalyst (0.001 × X mmol) in solvent (5 mL). 2 NMR yields. 3 Isolated yields.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Tokushige, K.; Yamashiro, T.; Hirao, S.; Abe, T. Aluminum-Catalyzed Cross Selective C3–N1′ Coupling Reactions of N-Methoxyindoles with Indoles. Chemistry 2023, 5, 452-462. https://doi.org/10.3390/chemistry5010033
AMA Style
Tokushige K, Yamashiro T, Hirao S, Abe T. Aluminum-Catalyzed Cross Selective C3–N1′ Coupling Reactions of N-Methoxyindoles with Indoles. Chemistry. 2023; 5(1):452-462. https://doi.org/10.3390/chemistry5010033
Chicago/Turabian StyleTokushige, Keisuke, Toshiki Yamashiro, Seiya Hirao, and Takumi Abe. 2023. "Aluminum-Catalyzed Cross Selective C3–N1′ Coupling Reactions of N-Methoxyindoles with Indoles" Chemistry 5, no. 1: 452-462. https://doi.org/10.3390/chemistry5010033