

How Topological Differences between Two Oxide Surfaces Determine Selectivity—The Case of the Dehydra-Decyclization of Tetrahydrofuran

Abstract
:1. Introduction
2. Materials and Methods
2.1. DFT Simulations
2.2. Microkinetic Simulations
3. Results
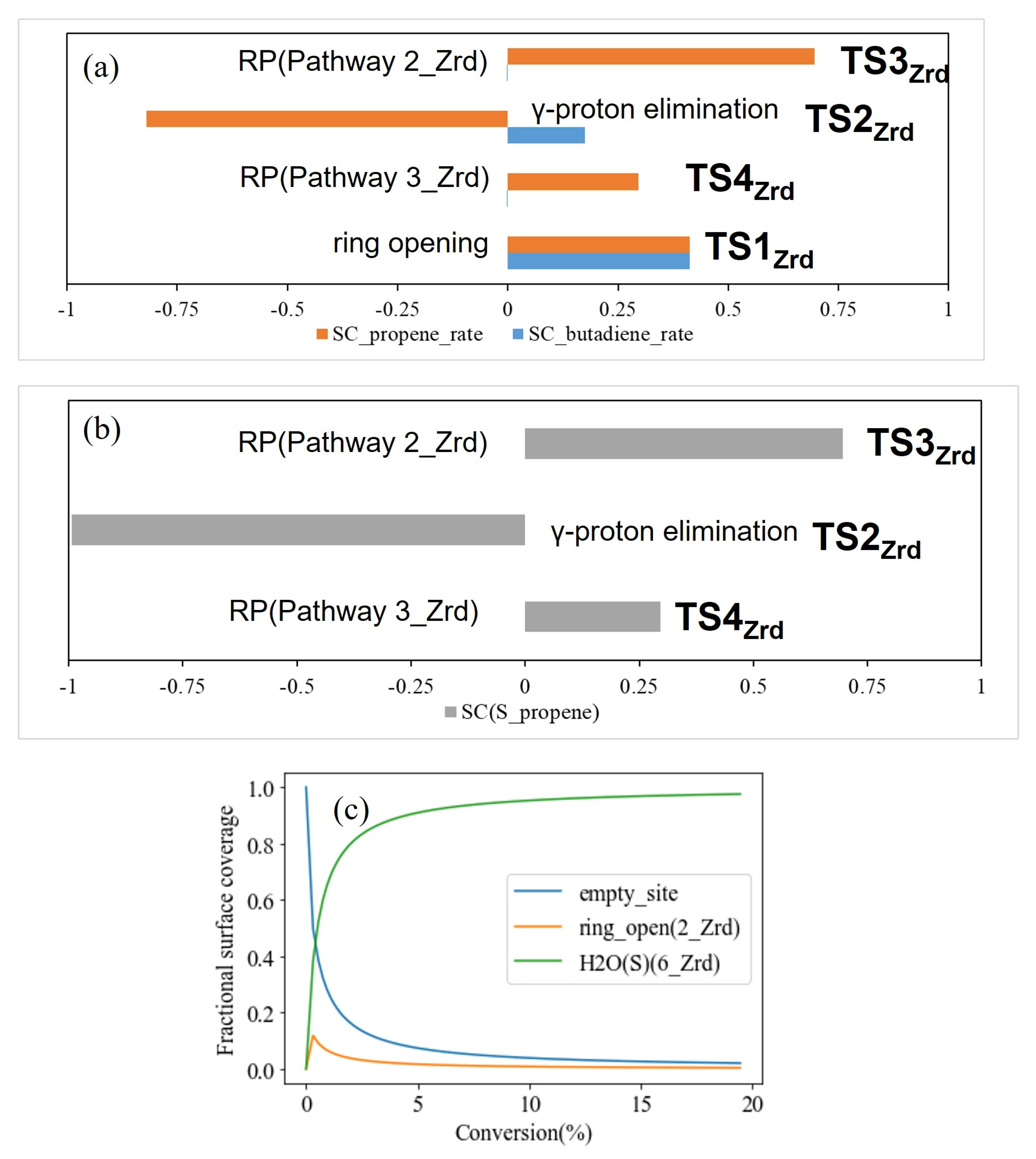
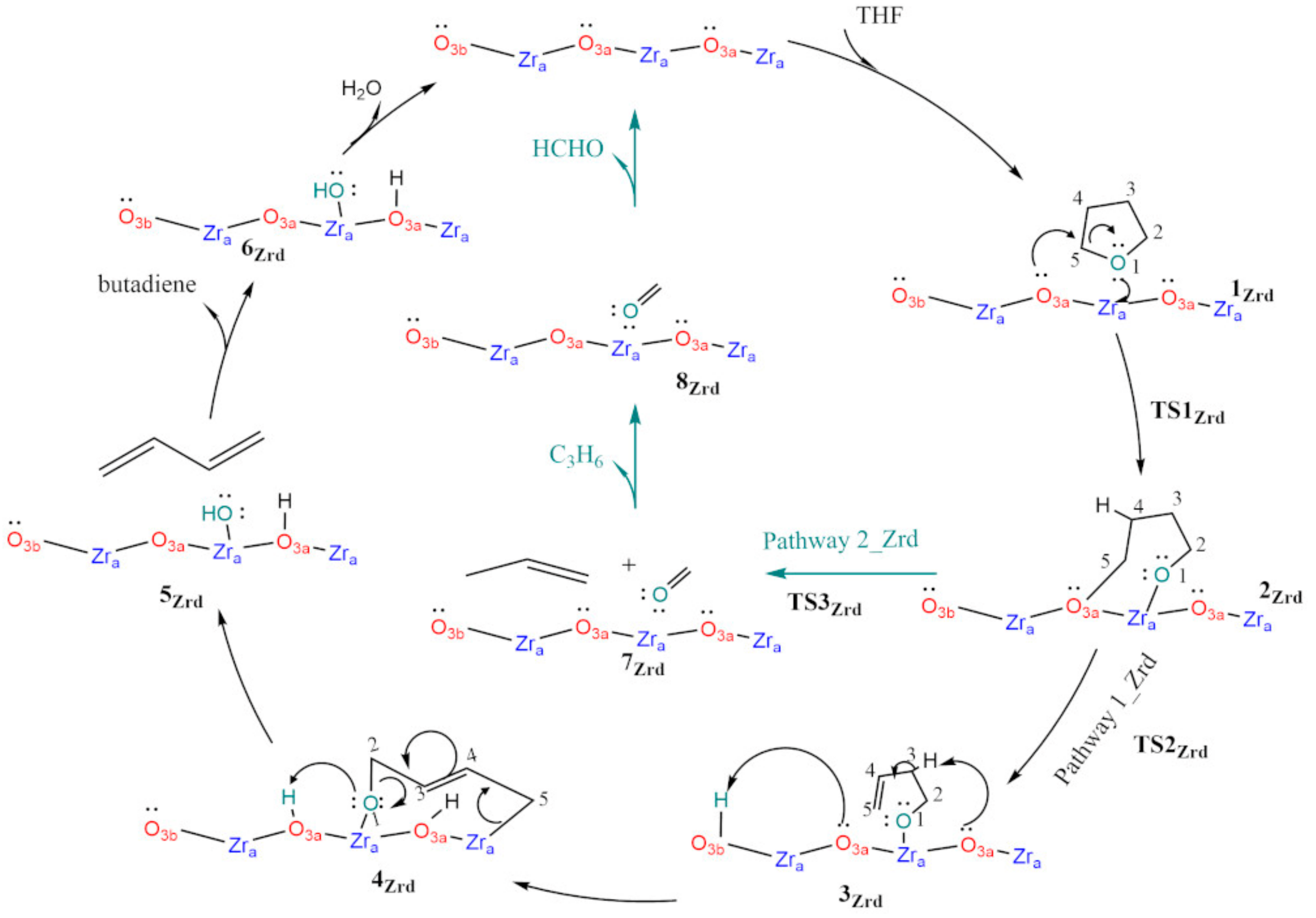
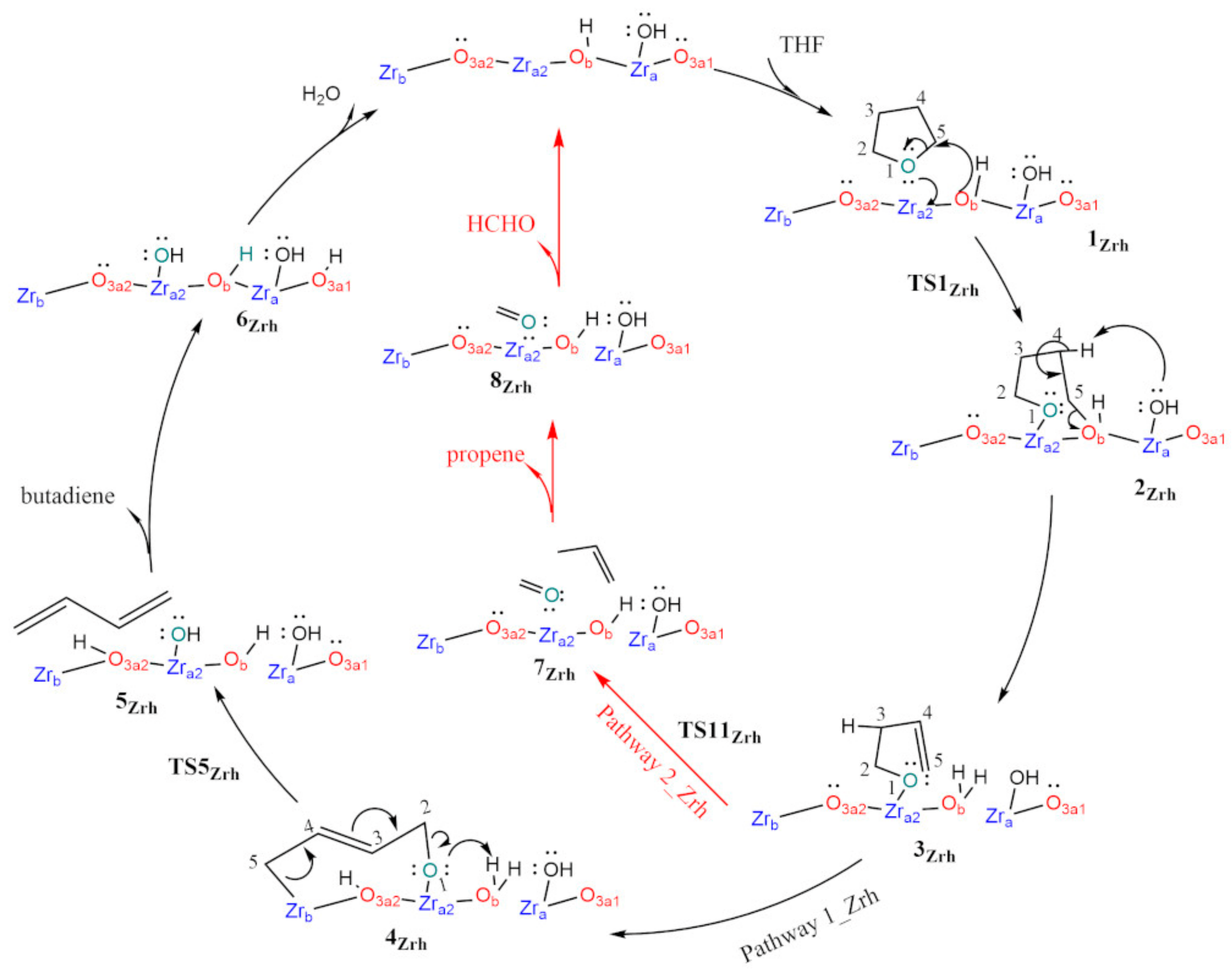
3.1. Dry t-ZrO2 (101)
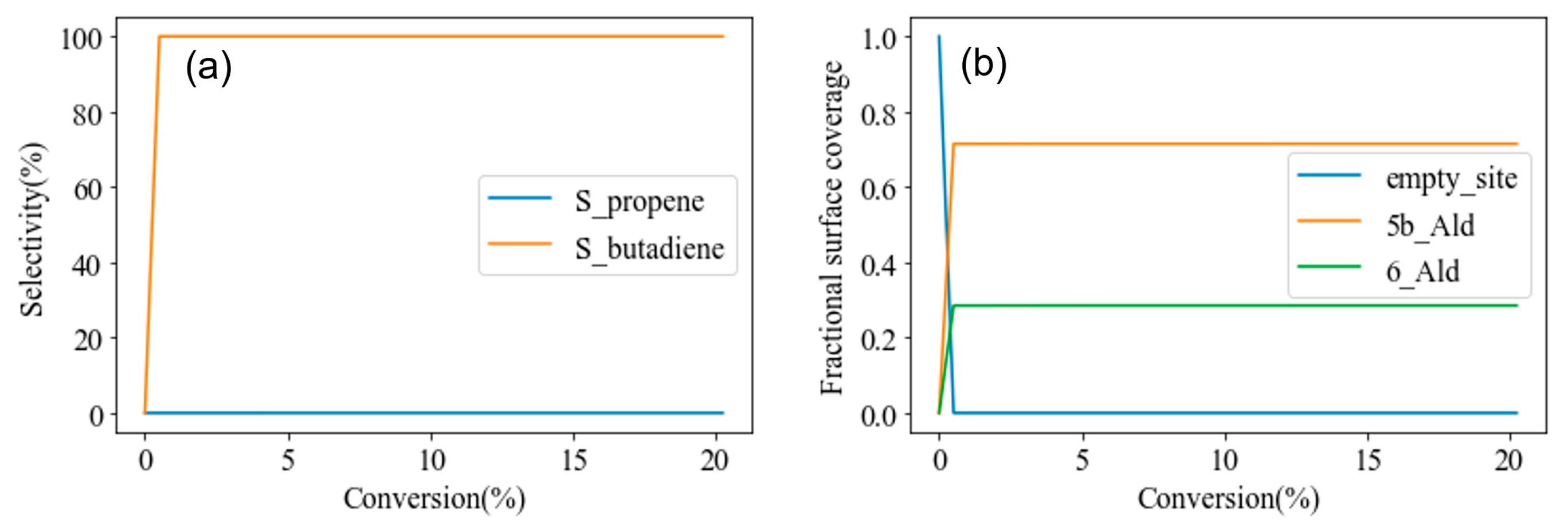
3.2. Hydroxylated t-ZrO2 (101)
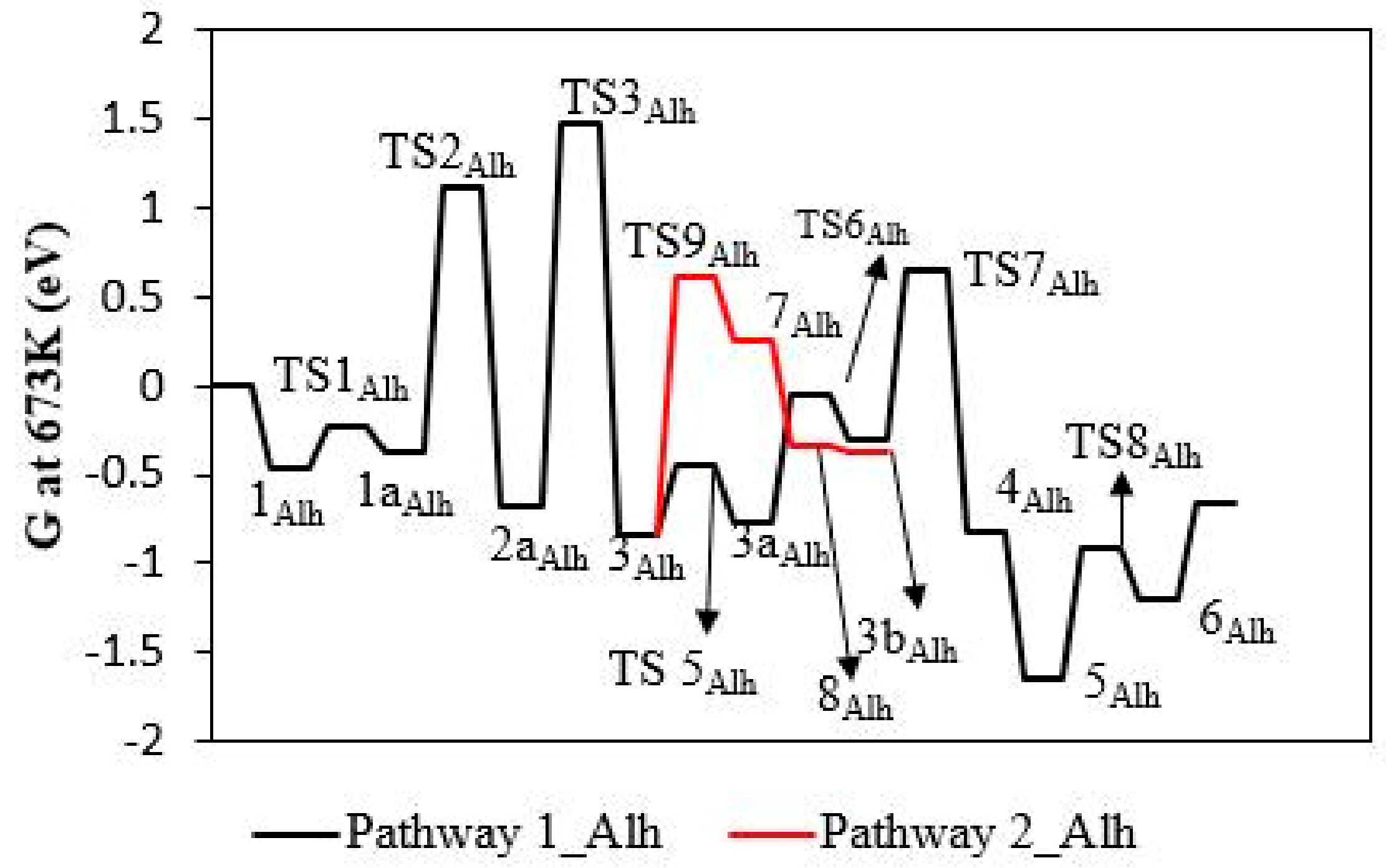
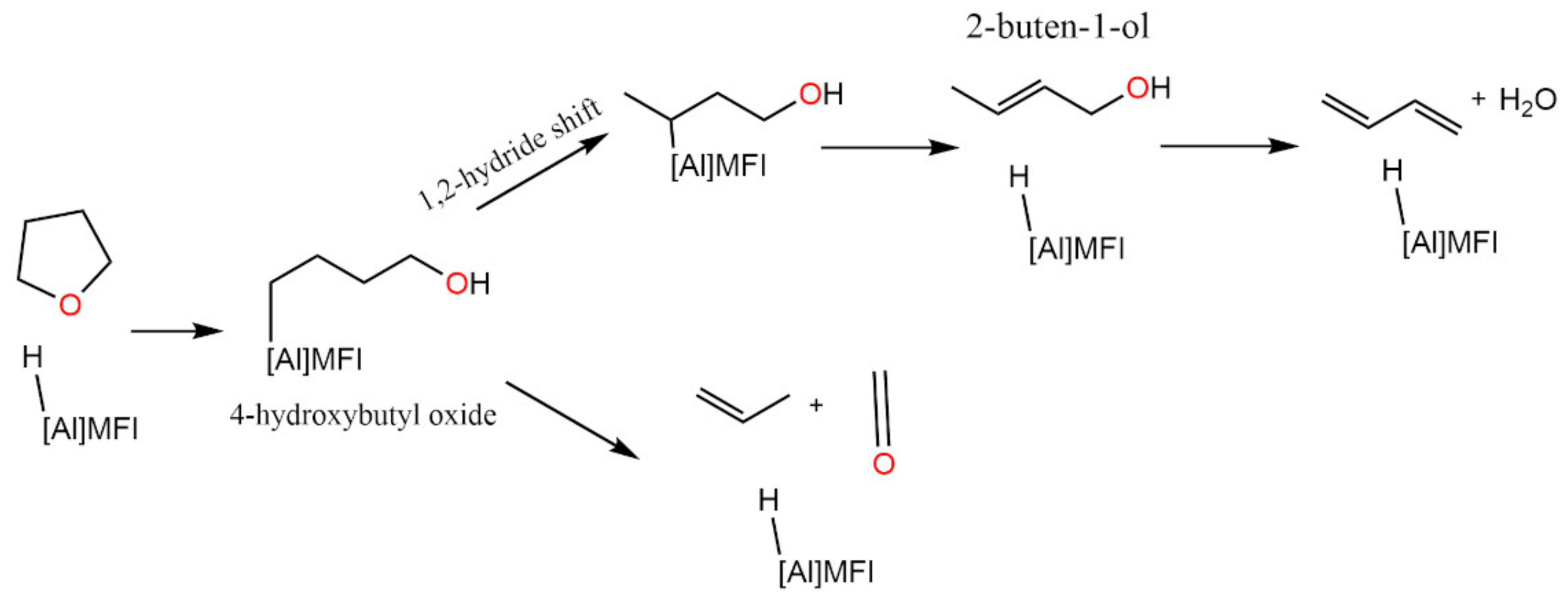
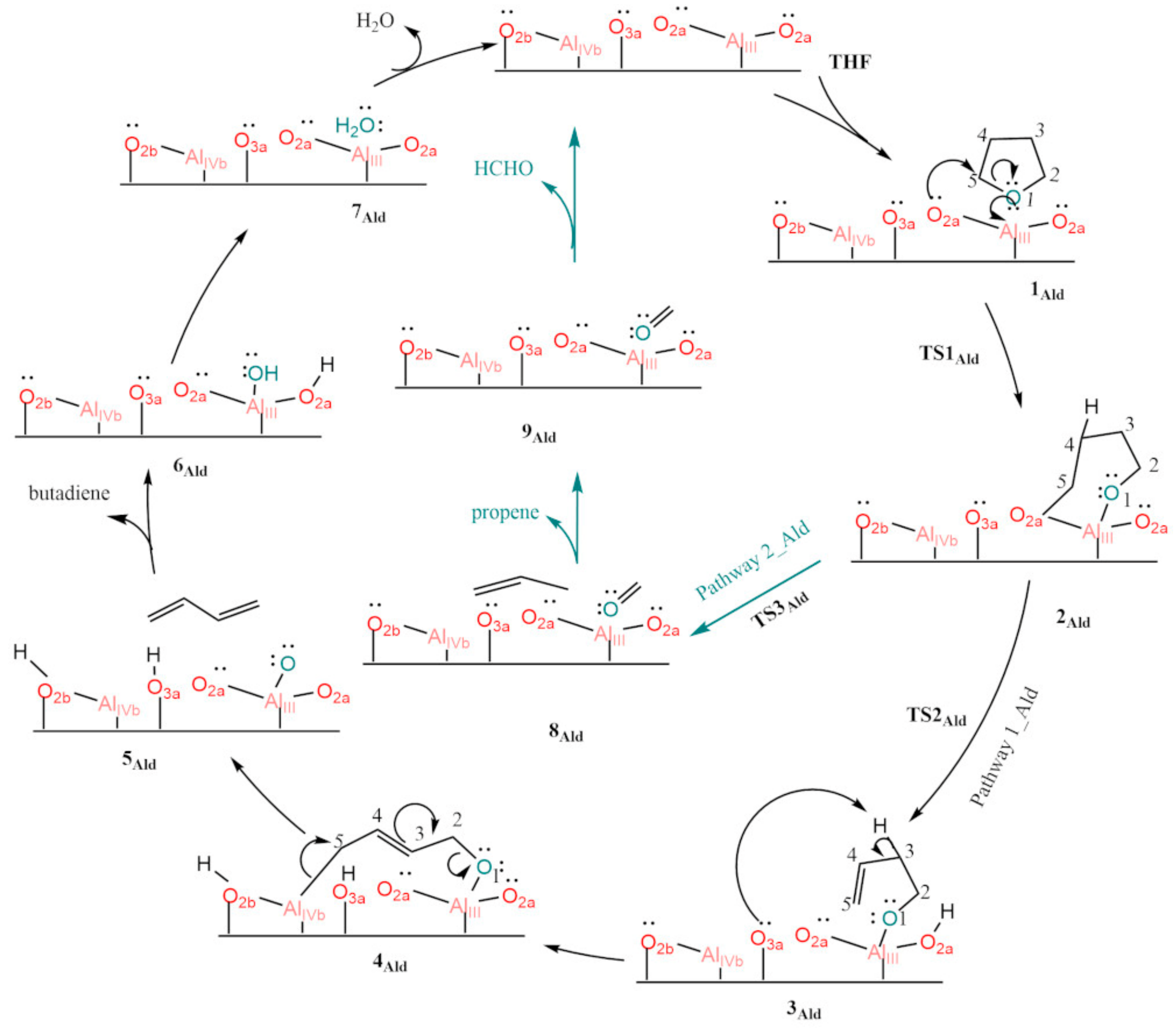
3.3. Dry γ-Al2O3 (110)
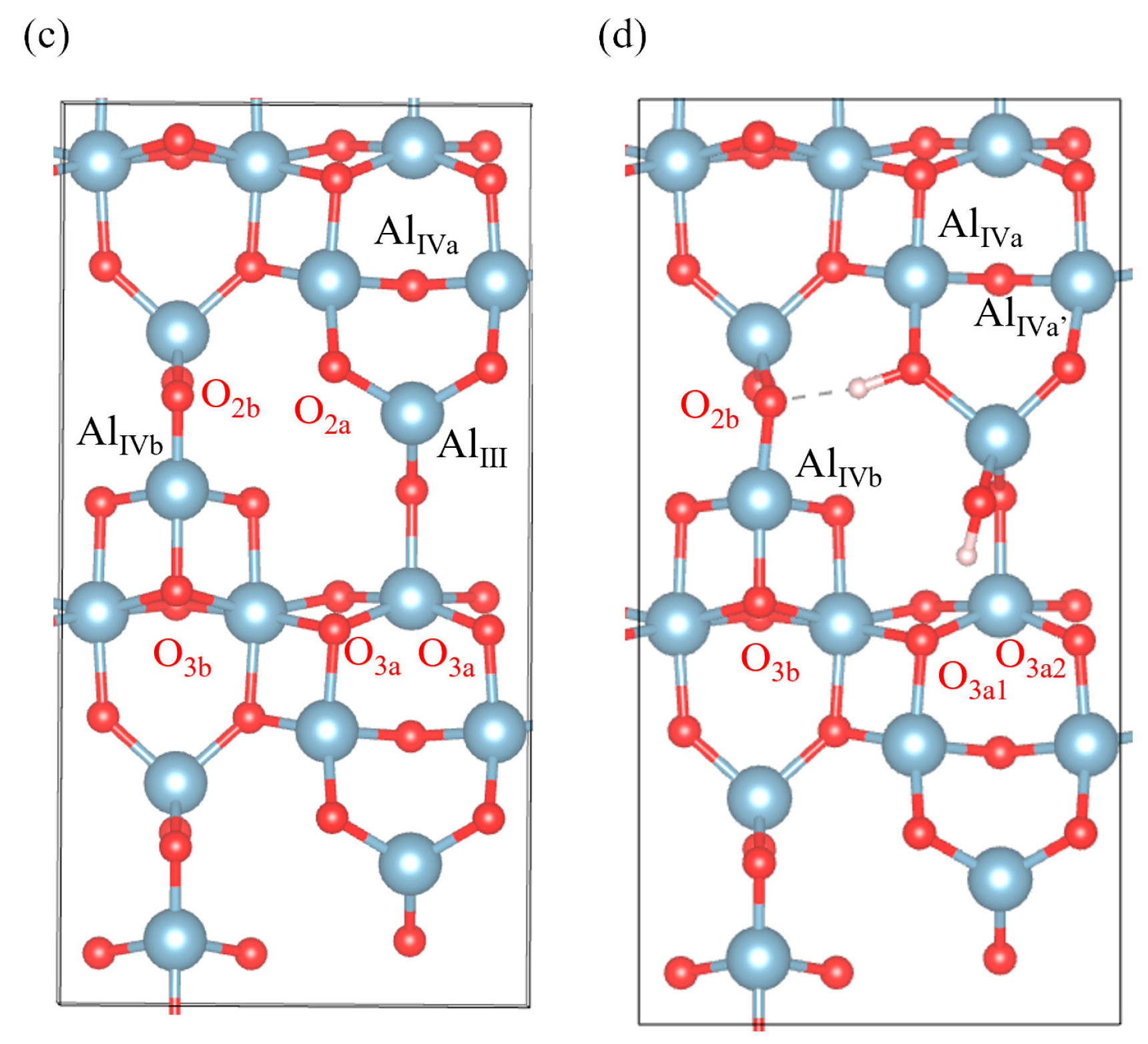
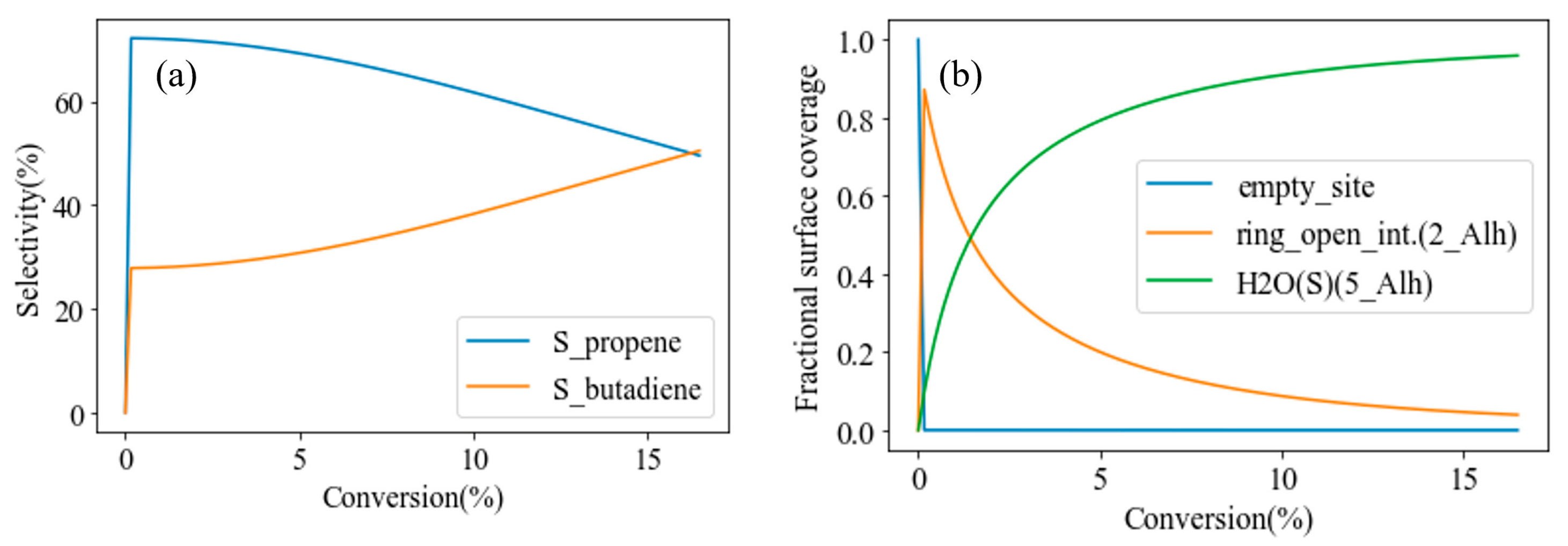
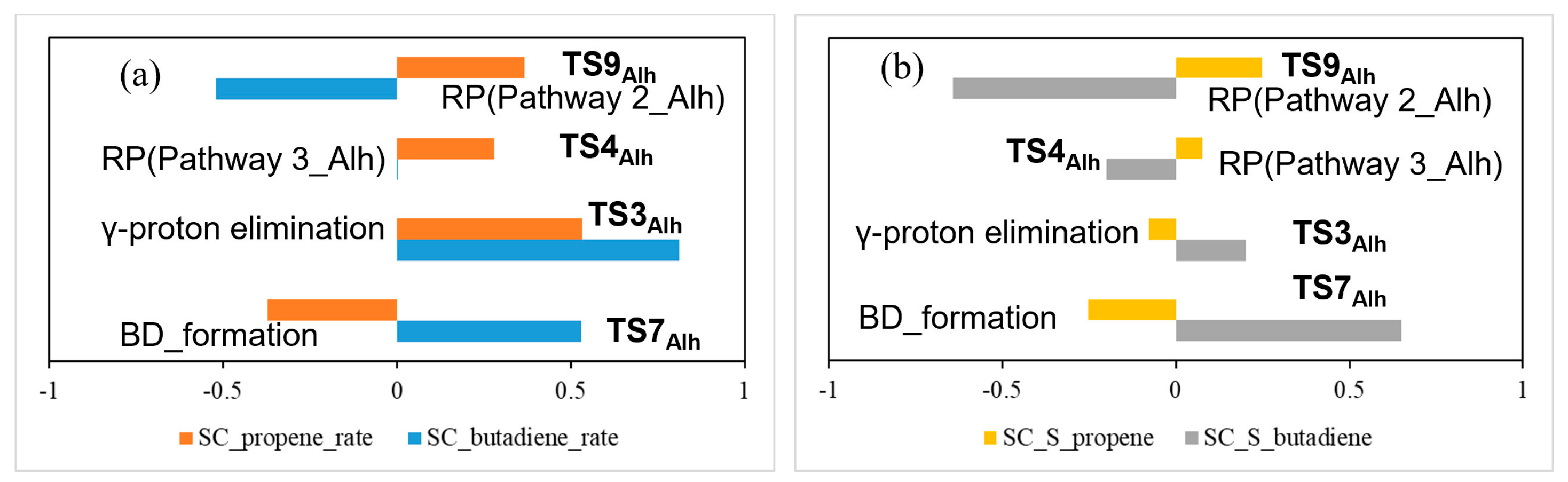
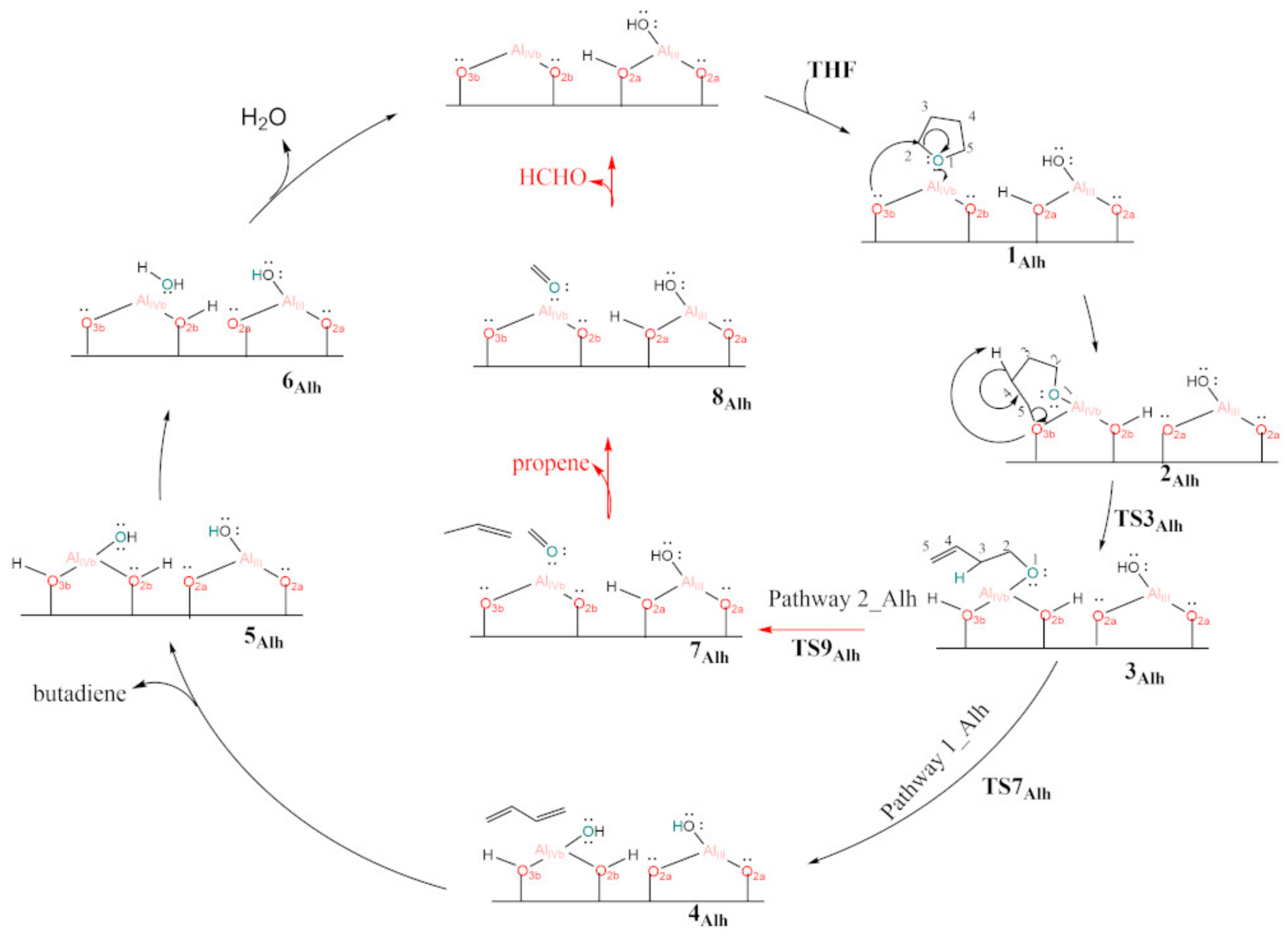
3.4. Hydroxylated γ-Al2O3 (110)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Choudhary, V.; Pinar, A.B.; Sandler, S.I.; Vlachos, D.G.; Lobo, R.F. Xylose Isomerization to Xylulose and Its Dehydration to Furfural in Aqueous Media. ACS Catal. 2011, 1, 1724–1728. [Google Scholar] [CrossRef]
- Choudhary, V.; Sandler, S.I.; Vlachos, D.G. Conversion of Xylose to Furfural Using Lewis and Brønsted Acid Catalysts in Aqueous Media. ACS Catal. 2012, 2, 2022–2028. [Google Scholar] [CrossRef]
- Wang, S.; Vorotnikov, V.; Vlachos, D.G. Coverage-Induced Conformational Effects on Activity and Selectivity: Hydrogenation and Decarbonylation of Furfural on Pd(111). ACS Catal. 2015, 5, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Jia, P.; Wang, T. Furfural: A Promising Platform Compound for Sustainable Production of C4 and C5 Chemicals. ACS Catal. 2016, 6, 7621–7640. [Google Scholar] [CrossRef]
- Abdelrahman, O.A.; Park, D.S.; Vinter, K.P.; Spanjers, C.S.; Ren, L.; Cho, H.J.; Vlachos, D.G.; Fan, W.; Tsapatsis, M.; Dauenhauer, P.J. Biomass-Derived Butadiene by Dehydra-Decyclization of Tetrahydrofuran. ACS Sustain. Chem. Eng. 2017, 5, 3732–3736. [Google Scholar] [CrossRef]
- Kumbhalkar, M.D.; Buchanan, J.S.; Huber, G.W.; Dumesic, J.A. Ring Opening of Biomass-Derived Cyclic Ethers to Dienes over Silica/Alumina. ACS Catal. 2017, 7, 5248–5256. [Google Scholar] [CrossRef]
- Kumar, G.; Liu, D.; Xu, D.; Ren, L.; Tsapatsis, M.; Dauenhauer, P.J. Dehydra-Decyclization of 2-Methyltetrahydrofuran to Pentadienes on Boron-Containing Zeolites. Green Chem. 2020, 22, 4147–4160. [Google Scholar] [CrossRef]
- Li, S.; Abdelrahman, O.A.; Kumar, G.; Tsapatsis, M.; Vlachos, D.G.; Caratzoulas, S.; Dauenhauer, P.J. Dehydra-Decyclization of Tetrahydrofuran on H-ZSM5: Mechanisms, Pathways, and Transition State Entropy. ACS Catal. 2019, 9, 10279–10293. [Google Scholar] [CrossRef]
- Wang, C.; Li, S.; Mao, X.; Caratzoulas, S.; Gorte, R.J. H-D Exchange of Simple Aromatics as a Measure of Brønsted-Acid Site Strengths in Solids. Catal. Lett. 2018, 148, 3548–3556. [Google Scholar] [CrossRef]
- Ji, Y.; Lawal, A.; Nyholm, A.; Gorte, R.J.; Abdelrahman, O.A. Dehydra-Decyclization of Tetrahydrofurans to Diene Monomers over Metal Oxides. Catal. Sci. Technol. 2020, 10, 5903–5912. [Google Scholar] [CrossRef]
- Ji, Y.; Batchu, S.P.; Lawal, A.; Vlachos, D.G.; Gorte, R.J.; Caratzoulas, S.; Abdelrahman, O.A. Selective Dehydra-Decyclization of Cyclic Ethers to Conjugated Dienes over Zirconia. J. Catal. 2022, 410, 10–21. [Google Scholar] [CrossRef]
- Digne, M.; Sautet, P.; Raybaud, P.; Euzen, P.; Toulhoat, H. Hydroxyl Groups on γ-Alumina Surfaces: A DFT Study. J. Catal. 2002, 211, 1–5. [Google Scholar] [CrossRef]
- Christensen, A.; Carter, E.A. First-principles study of the surfaces of zirconia. Phys. Rev. B 1998, 58, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Hofmann, A.; Clark, S.J.; Oppel, M.; Hahndorf, I. Hydrogen Adsorption on the Tetragonal ZrO2(101) Surface: A Theoretical Study of an Important Catalytic Reactanty. Phys. Chem. Chem. Phys. 2002, 4, 3500–3508. [Google Scholar] [CrossRef]
- Yang, Y.; Lin, F.; Tran, H.; Chin, Y.H.C. Butanal Condensation Chemistry Catalyzed by Brønsted Acid Sites on Polyoxometalate Clusters. ChemCatChem 2017, 9, 287–299. [Google Scholar] [CrossRef]
- Lin, F.; Yang, Y.; Chin, Y.H. Kinetic Requirements of Aldehyde Transfer Hydrogenation Catalyzed by Microporous Solid Brønsted Acid Catalysts. ACS Catal. 2017, 7, 6909–6914. [Google Scholar] [CrossRef]
- Blochl, P.E.; Forst, C.J.; Schimpl, J. Projector Augmented Wave Method: Ab Initio Molecular Dynamics with Full Wave Functions. Bull. Mater. Sci. 2003, 26, 33–41. [Google Scholar] [CrossRef] [Green Version]
- Ernzerhof, M.; Scuseria, G.E. Assessment of the Perdew–Burke–Ernzerhof Exchange-Correlation Functional. J. Chem. Phys. 1999, 110, 5029–5036. [Google Scholar] [CrossRef] [Green Version]
- Becke, A.D.; Johnson, E.R. A Density-Functional Model of the Dispersion Interaction. J. Chem. Phys. 2005, 123, 154101. [Google Scholar] [CrossRef]
- Henkelman, G.; Jónsson, H. A Dimer Method for Finding Saddle Points on High Dimensional Potential Surfaces Using Only First Derivatives. J. Chem. Phys. 1999, 111, 7010–7022. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 2000, 113, 9901–9904. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Supramolecular Binding Thermodynamics by Dispersion-Corrected Density Functional Theory. Chem. A Eur. J. 2012, 18, 9955–9964. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Gomes, J.; Sharada, S.M.; Bell, A.T.; Head-Gordon, M. Improved Force-Field Parameters for QM/MM Simulations of the Energies of Adsorption for Molecules in Zeolites and a Free Rotor Correction to the Rigid Rotor Harmonic Oscillator Model for Adsorption Enthalpies. J. Phys. Chem. C 2015, 119, 1840–1850. [Google Scholar] [CrossRef]
- Schoonheydt, R.A.; Weckhuysen, B.M. Editorial Highlight: Molecules in Confined Spaces. Phys. Chem. Chem. Phys. 2009, 11, 2794–2798. [Google Scholar] [CrossRef] [PubMed]
- Lym, J.; Wittreich, G.R.; Vlachos, D.G. A Python Multiscale Thermochemistry Toolbox (PMuTT) for Thermochemical and Kinetic Parameter Estimation. Comput. Phys. Commun. 2020, 247, 106864. [Google Scholar] [CrossRef]
- Chen, H.Y.T.; Tosoni, S.; Pacchioni, G. Adsorption of Ruthenium Atoms and Clusters on Anatase TiO2 and Tetragonal ZrO2(101) Surfaces: A Comparative DFT Study. J. Phys. Chem. C 2015, 119, 10856–10868. [Google Scholar] [CrossRef]
- Digne, M.; Sautet, P.; Raybaud, P.; Euzen, P.; Toulhoat, H. Use of DFT to Achieve a Rational Understanding of Acid-Basic Properties of γ-Alumina Surfaces. J. Catal. 2004, 226, 54–68. [Google Scholar] [CrossRef]
- Coltrin, M.E.; Kee, R.J.; Rupley, F.M. Surface Chemkin: A General Formalism and Software for Analyzing Heterogeneous Chemical Kinetics at a Gas-surface Interface. Int. J. Chem. Kinet. 1991, 23, 1111–1128. [Google Scholar] [CrossRef]
- Valero, M.C.; Digne, M.; Sautet, P.; Raybaud, P. DFT Study of the Interaction of a Single Palladium Atom with γ -Alumina Surfaces: The Role of Hydroxylation. Oil Gas Sci. Technol.-Rev. l’IFP 2006, 61, 535–545. [Google Scholar] [CrossRef]
- Wischert, R.; Copéret, C.; Delbecq, F.; Sautet, P. Optimal Water Coverage on Alumina: A Key to Generate Lewis Acid-Base Pairs That Are Reactive towards the C-H Bond Activation of Methane. Angew. Chem. Int. Ed. 2011, 50, 3202–3205. [Google Scholar] [CrossRef]
- Wischert, R.; Laurent, P.; Copéret, C.; Delbecq, F.; Sautet, P. γ-Alumina: The Essential and Unexpected Role of Water for the Structure, Stability, and Reactivity of “Defect” Sites. J. Am. Chem. Soc. 2012, 134, 14430–14449. [Google Scholar] [CrossRef] [PubMed]
- Batchu, S.P.; Wang, H.-L.; Chen, W.; Zheng, W.; Caratzoulas, S.; Lobo, R.F.; Vlachos, D.G. Ethane Dehydrogenation on Single and Dual Centers of Ga-Modified γ-Al2O3. ACS Catal. 2021, 11, 1380–1391. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Experiment/Model | Ea (kJ/mol) |
|---|---|
| Experiment | 65.2 ± 12.1 |
| Dry-ZrO2 | 94.2 ± 0.3 |
| Hydroxylated-ZrO2 | 73.3 ± 0.2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Batchu, S.P.; Caratzoulas, S.; Vlachos, D.G. How Topological Differences between Two Oxide Surfaces Determine Selectivity—The Case of the Dehydra-Decyclization of Tetrahydrofuran. Chemistry 2023, 5, 422-437. https://doi.org/10.3390/chemistry5010031
Batchu SP, Caratzoulas S, Vlachos DG. How Topological Differences between Two Oxide Surfaces Determine Selectivity—The Case of the Dehydra-Decyclization of Tetrahydrofuran. Chemistry. 2023; 5(1):422-437. https://doi.org/10.3390/chemistry5010031
Chicago/Turabian StyleBatchu, Sai Praneet, Stavros Caratzoulas, and Dionisios G. Vlachos. 2023. "How Topological Differences between Two Oxide Surfaces Determine Selectivity—The Case of the Dehydra-Decyclization of Tetrahydrofuran" Chemistry 5, no. 1: 422-437. https://doi.org/10.3390/chemistry5010031