
UV/VIS-Spectroscopic Inline Measurement for the Detection of Fouling Processes during the Polymerization of N-Vinylpyrrolidone




Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
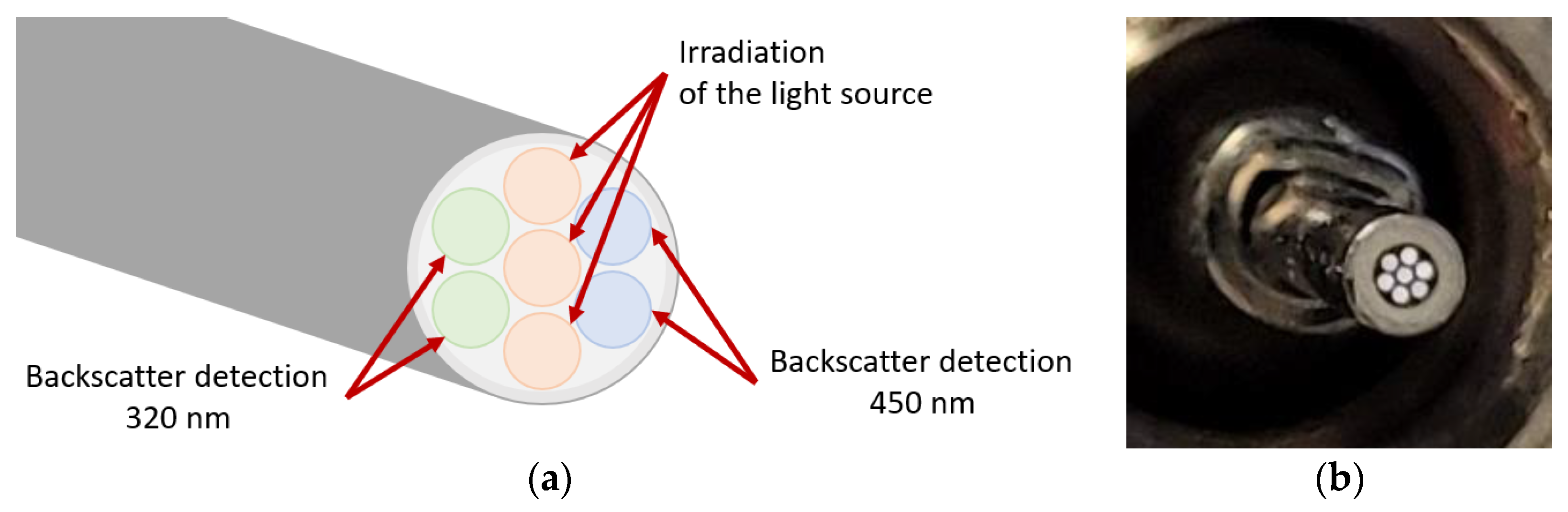
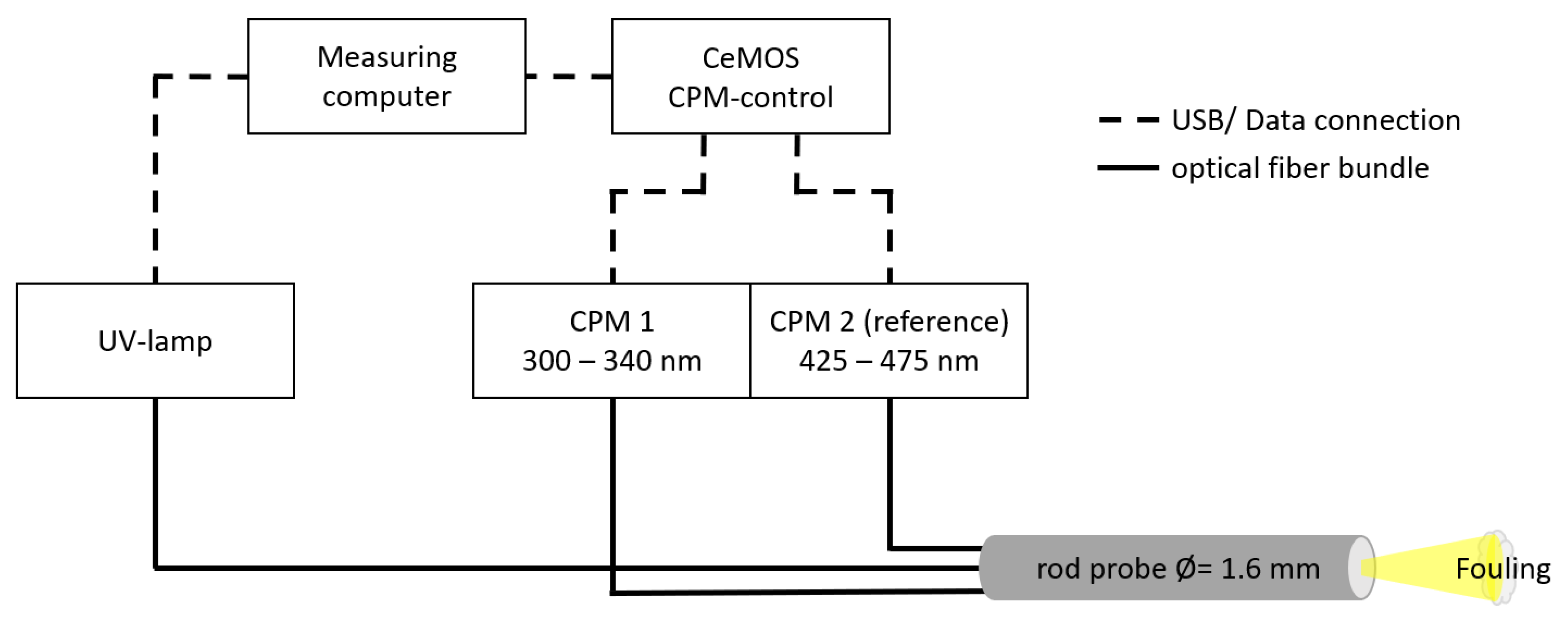
2.2. Optical Measurement Setup
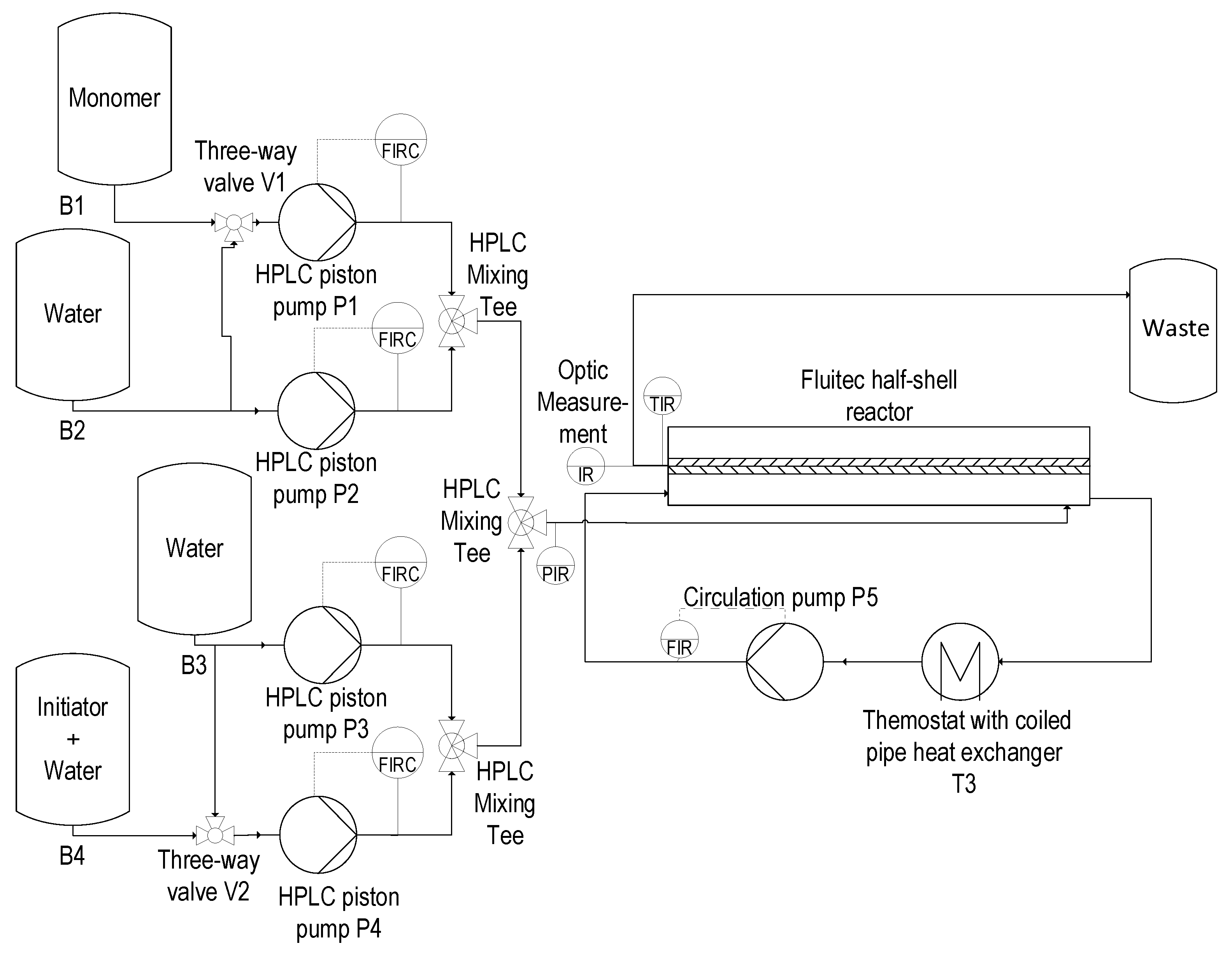
2.3. Reactor Setup
3. Results
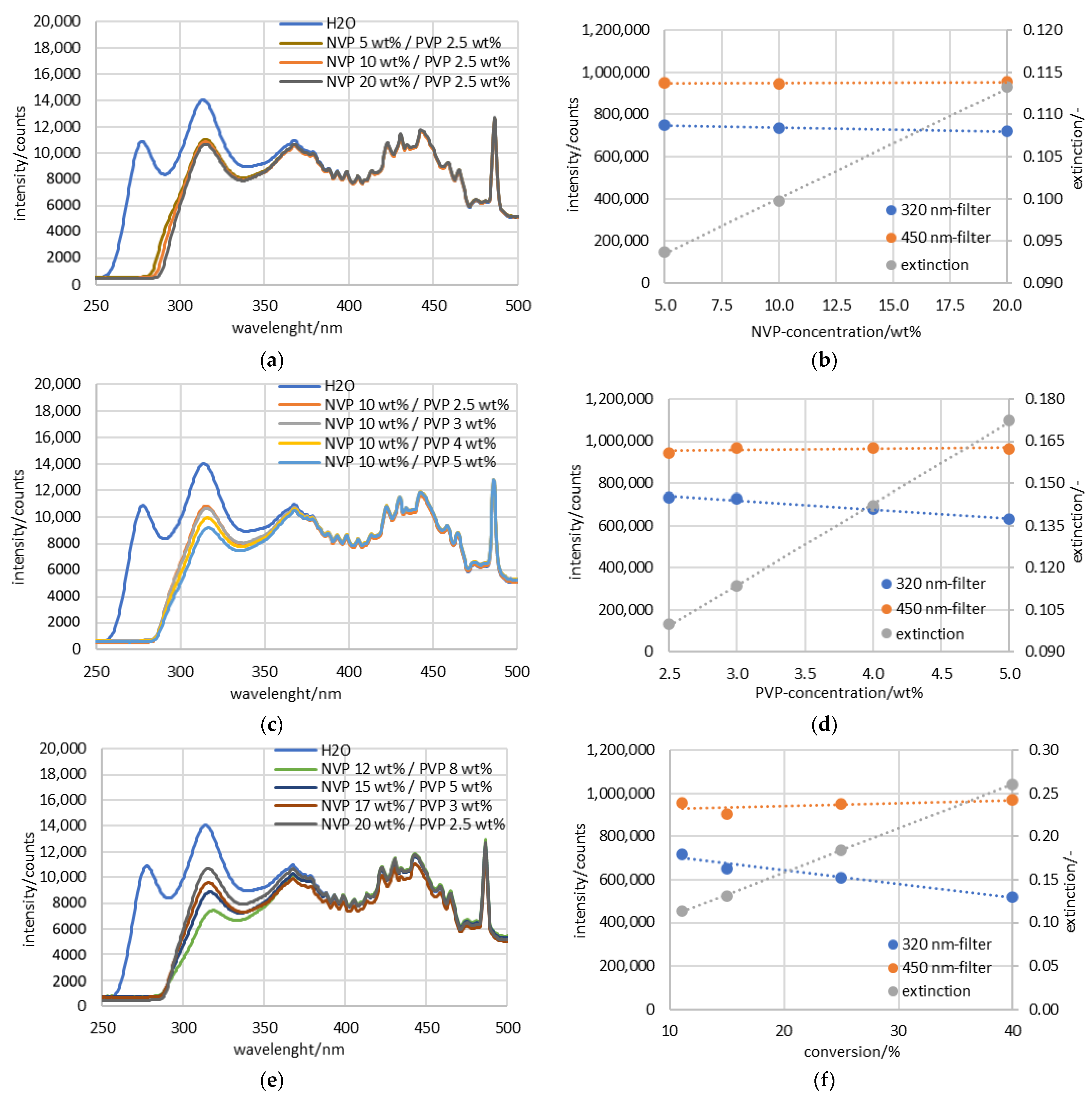
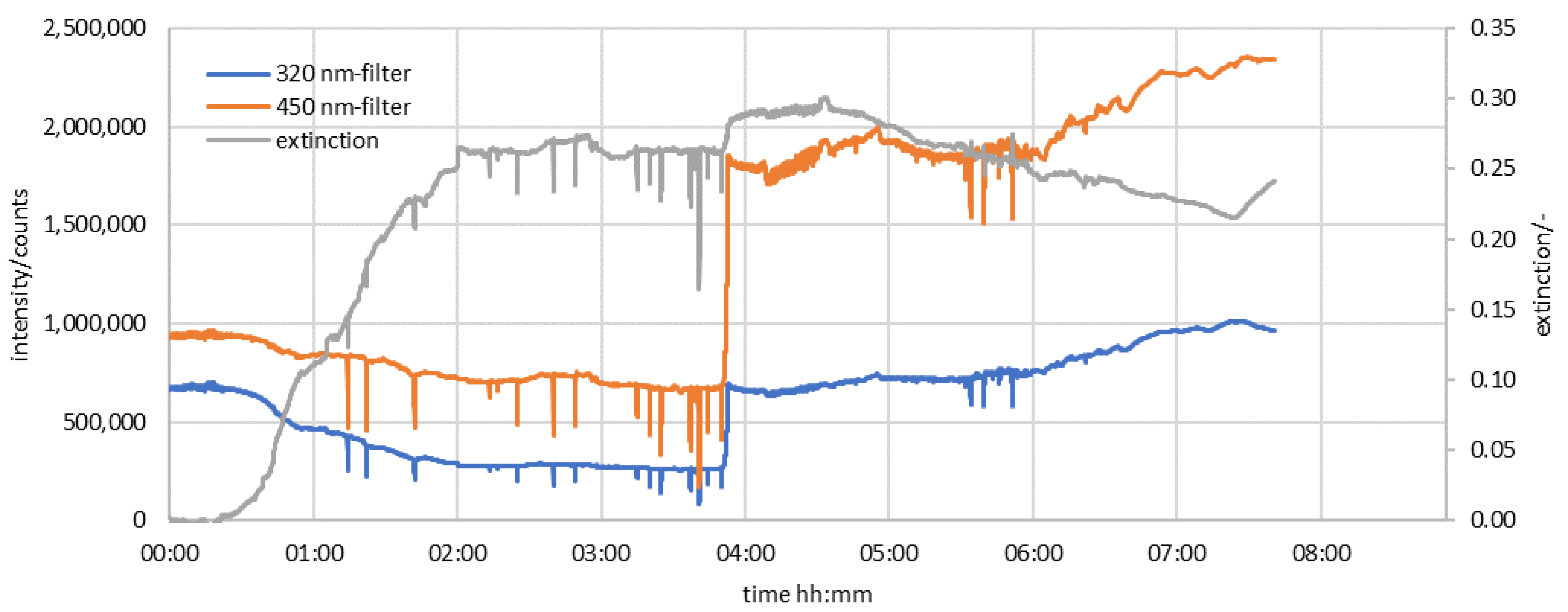
3.1. Preliminary Examination of the UV-Spectra
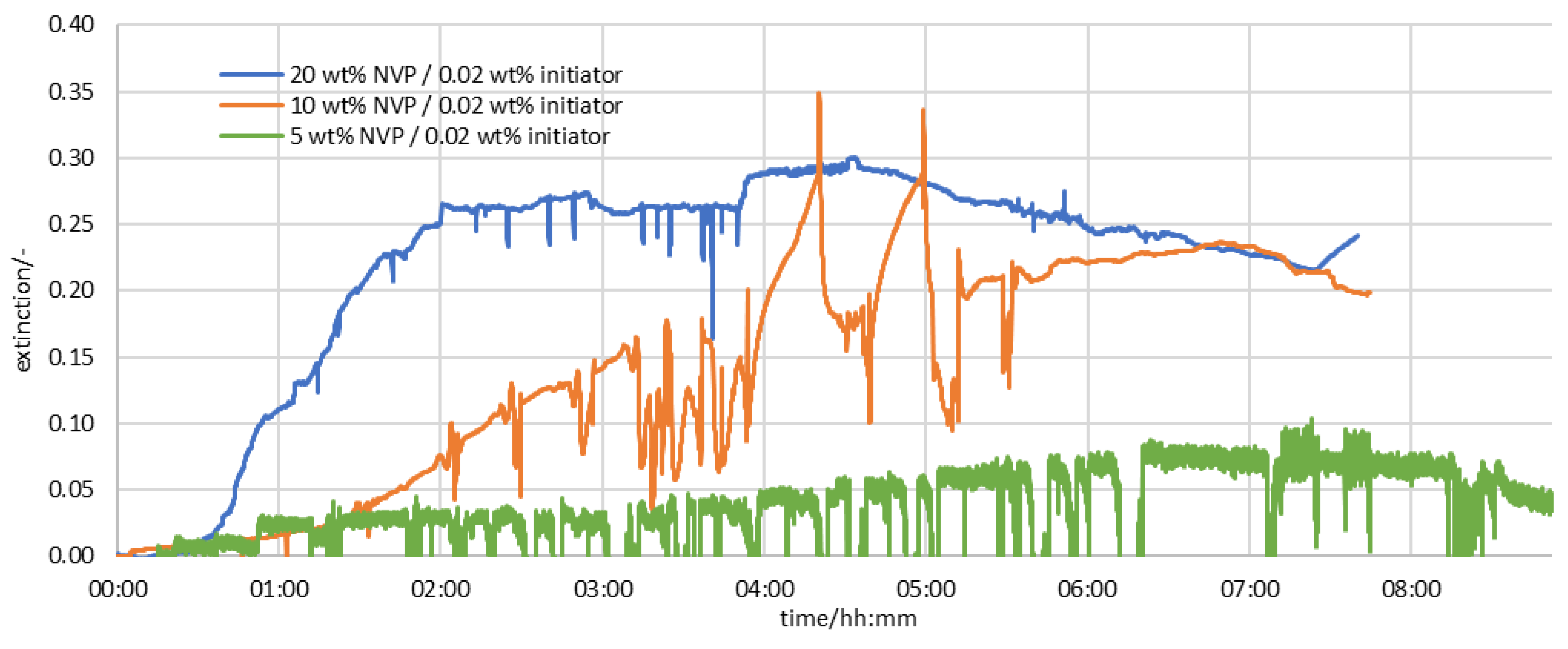
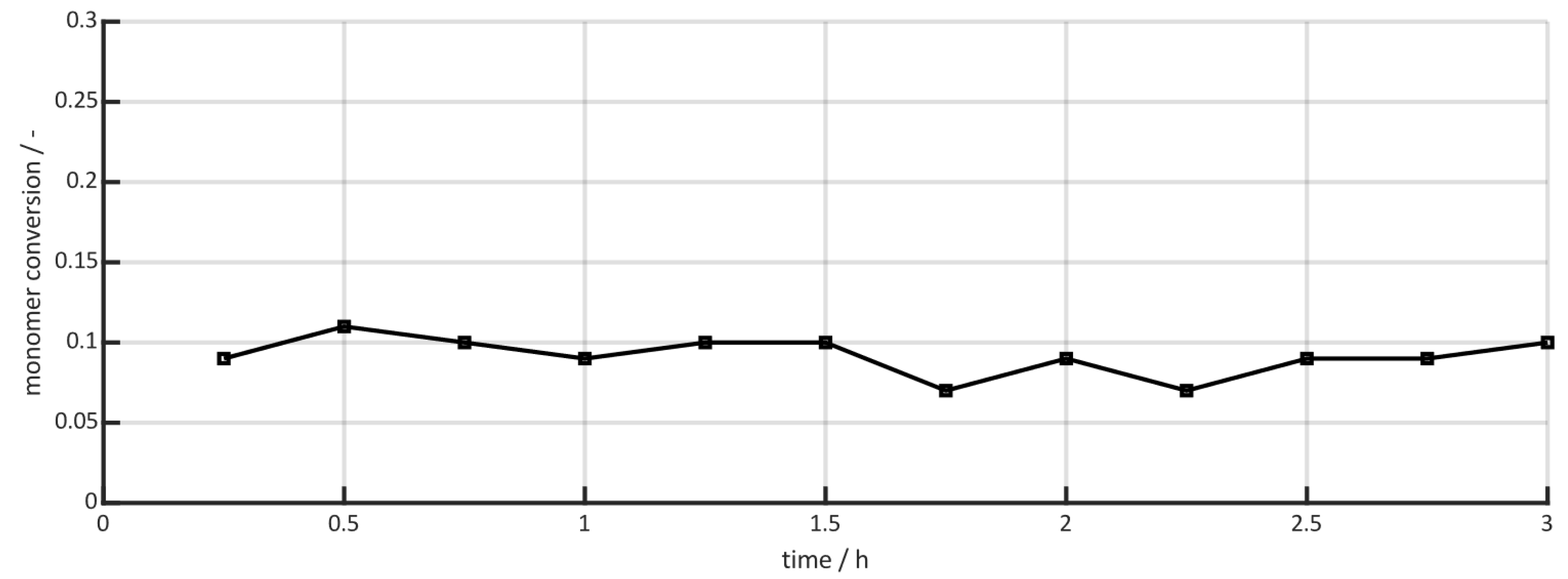
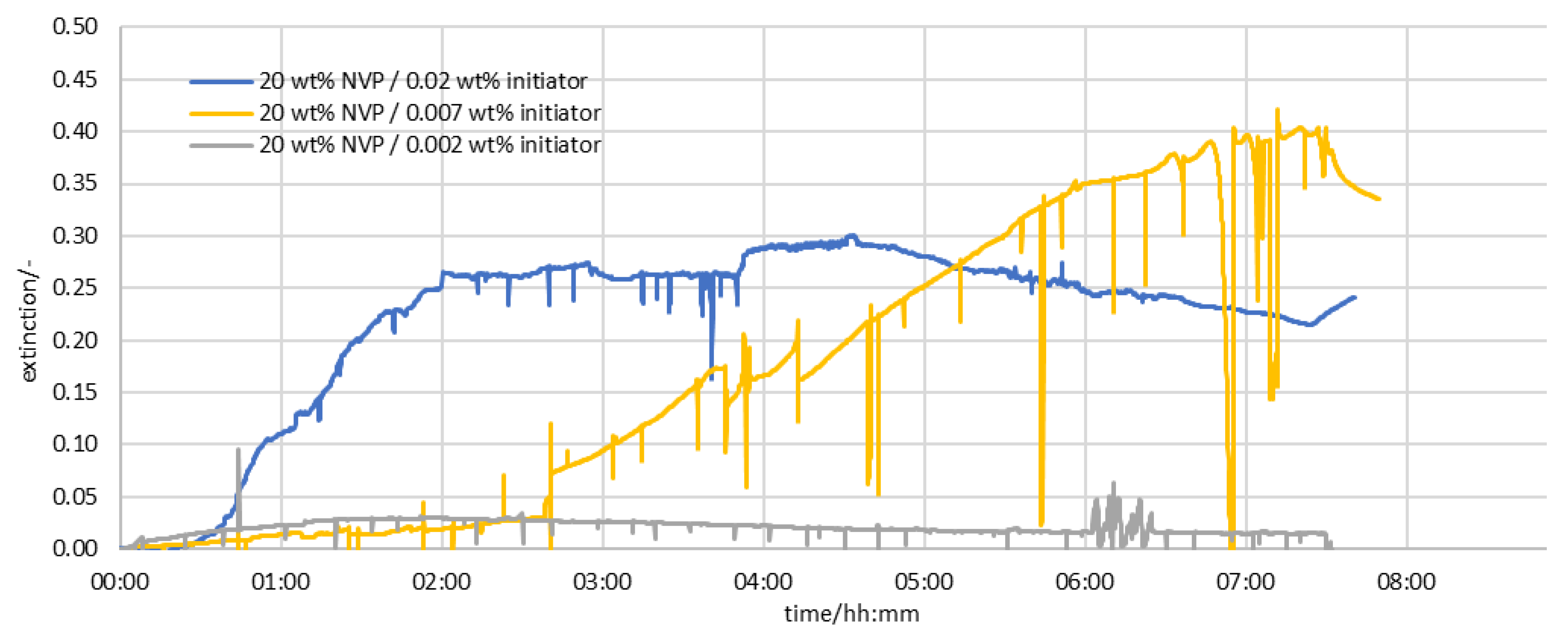
3.2. Results of the Measurement Series in the Half-Shell Reactor
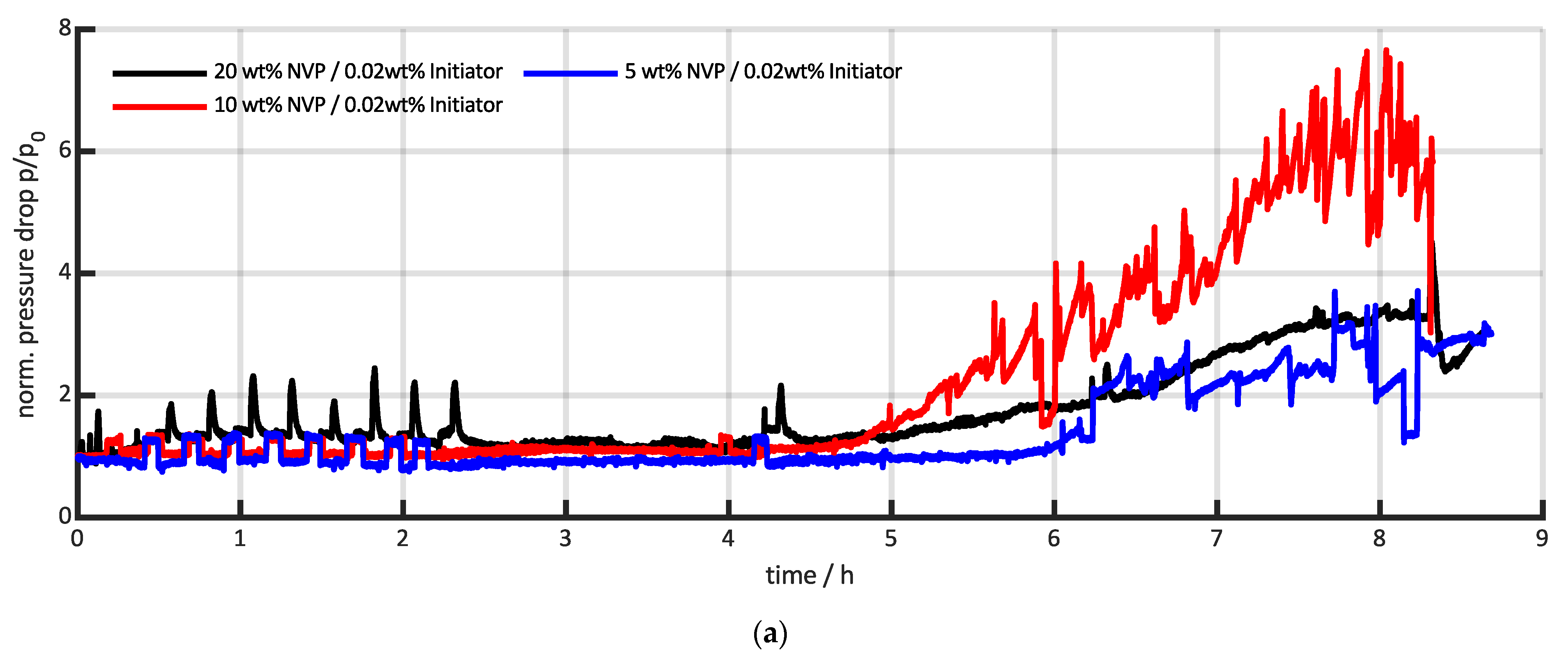
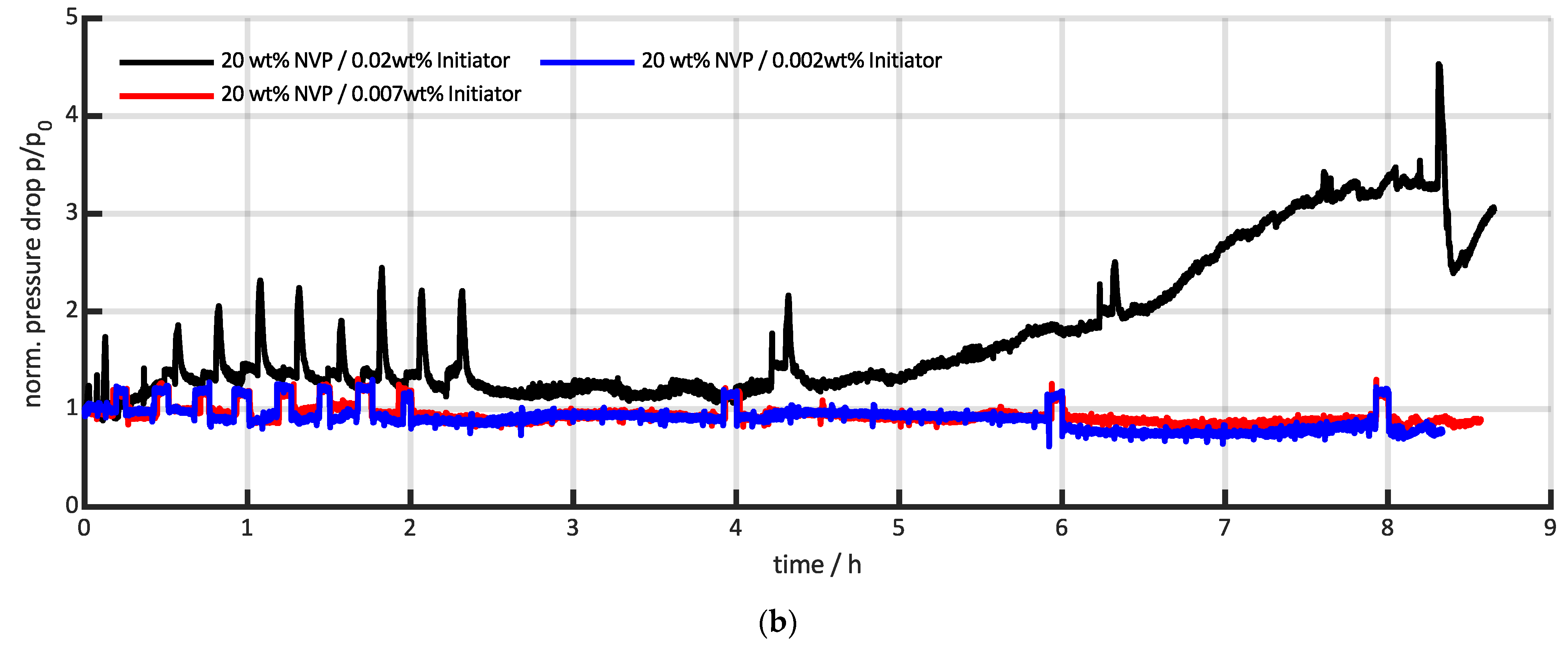
3.3. Comparison with Conventional Pressure Measurements
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bakeev, K.A. Process Analytical Technology: Spectroscopic Tools and Implementation Strategies for the Chemical and Pharmaceutical Industries, 2nd ed.; Wiley: Chichester, UK, 2010; ISBN 9780470722077. [Google Scholar]
- Simon, L.L.; Pataki, H.; Marosi, G.; Meemken, F.; Hungerbühler, K.; Baiker, A.; Tummala, S.; Glennon, B.; Kuentz, M.; Steele, G.; et al. Assessment of Recent Process Analytical Technology (PAT) Trends: A Multiauthor Review. Org. Process Res. Dev. 2015, 19, 3–62. [Google Scholar] [CrossRef]
- Kessler, R.W.; Kessler, W.; Zikulnig-Rusch, E. A Critical Summary of Spectroscopic Techniques and their Robust-ness in Industrial PAT Applications. Chem. Ing. Tech. 2016, 88, 710–721. [Google Scholar] [CrossRef]
- Parson, W.W. Modern Optical Spectroscopy; Springer: Berlin/Heidelberg, Germany, 2015; ISBN 978-3-662-46776-3. [Google Scholar]
- Günzler, H.; Gremlich, H.-U. IR-Spektroskopie; Wiley: Hoboken, NJ, USA, 2003; ISBN 9783527308019. [Google Scholar]
- Ritgen, U. Analytische Chemie I, 1. Aufl; Springer: Berlin/Heidelberg, Germany, 2020; pp. 63–83. ISBN 3662604949. [Google Scholar]
- Perkampus, H.-H. UV-VIS Spectroscopy and Its Applications; Springer: Berlin/Heidelberg, Germany; New York, NY, USA, 1992; ISBN 978-3-642-77479-9. [Google Scholar]
- Shinde, G.; Godage, R.K.; Jadhav, R.S.; Manoj, B.; Aniket, B. A Review on Advances in UV Spectroscopy. Res. J. Scie. Technol. 2020, 12, 47. [Google Scholar] [CrossRef]
- Zander, C. Fouling during Solution Polymerization in Continuously Operated Reactors; University of Stuttgart: Stuttgart, Germany, 2021. [Google Scholar]
- Lim, S.Y.; Ghazali, N.F. Product Removal Strategy and Fouling Mechanism for Cellulose Hydrolysis in Enzymatic Membrane Reactor. Waste Biomass Valor. 2020, 11, 5575–5590. [Google Scholar] [CrossRef]
- Peinemann, K.-V. Membranes for Water Treatment, 1st ed.; John Wiley & Sons Incorporated: Weinheim, Germany, 2010; ISBN 978-3-527-31483-6. [Google Scholar]
- Neßlinger, V.; Welzel, S.; Rieker, F.; Meinderink, D.; Nieken, U.; Grundmeier, G. Thin Organic-Inorganic Anti-Fouling Hybrid-Films for Microreactor Components. Macro React. Eng. 2022, 10, 2200043. [Google Scholar] [CrossRef]
- Welzel, S.; Zander, C.; Hungenberg, K.-D.; Nieken, U. Modeling of the Branching Point Distribution During the Polymerization of N -Vinylpyrrolidone. Macro React. Eng. 2022, 16, 2200005. [Google Scholar] [CrossRef]
- Osenberg, M.; Forster, J.; Rust, S.; Fritsch, T.; Tebrugge, J.; Pauer, W.; Musch, T. Ultrasound Sensor for Process and Fouling Monitoring in Emulsion Polymerization Processes. In Proceedings of the 2022 IEEE Sensors, Dallas, TX, USA, 30 October–2 November 2022; IEEE: Piscataway, NJ, USA, 2022; pp. 1–4, ISBN 978-1-6654-8464-0. [Google Scholar]
- Böttcher, A.; Petri, J.; Langhoff, A.; Scholl, S.; Augustin, W.; Hohlen, A.; Johannsmann, D. Fouling Pathways in Emulsion Polymerization Differentiated with a Quartz Crystal Microbalance (QCM) Integrated into the Reactor Wall. Macro React. Eng. 2022, 16, 2100045. [Google Scholar] [CrossRef]
- Hohlen, A.; Augustin, W.; Scholl, S. Quantification of Polymer Fouling on Heat Transfer Surfaces During Synthesis. Macromol. React. Eng. 2020, 14, 1900035. [Google Scholar] [CrossRef] [Green Version]
- Schmitt, L.; Meyer, C.; Schorz, S.; Manser, S.; Scholl, S.; Rädle, M. Use of a Scattered Light Sensor for Monitoring the Dispersed Surface in Crystallization. Chem. Ing. Tech. 2022, 94, 1177–1184. [Google Scholar] [CrossRef]
- Guffart, J.; Bus, Y.; Nachtmann, M.; Lettau, M.; Schorz, S.; Nieder, H.; Repke, J.-U.; Rädle, M. Photometric Inline Monitoring of Pigment Concentration in Highly Filled Lacquers. Chem. Ing. Tech. 2020, 92, 729–735. [Google Scholar] [CrossRef]
- Manser, S.; Kommert, S.; Keck, S.; Spoor, E.; Rädle, M. New Conceptional Study of a Portable Highly Sensitive Pho-tometric Raman Sensor. Sensors 2022, 22, 6098. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feed Pump | Mass Flux/g/min | Weight Fractions (NVP/Water/Initiator) |
|---|---|---|
| P1 | 6.2 | 1/0/0 |
| P2 | 9.3 | 0/1/0 |
| P3 | 9.3 | 0/1/0 |
| P4 | 6.2 | 0/0.999/0.001 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spoor, E.; Welzel, S.; Nieken, U.; Rädle, M. UV/VIS-Spectroscopic Inline Measurement for the Detection of Fouling Processes during the Polymerization of N-Vinylpyrrolidone. Reactions 2023, 4, 176-188. https://doi.org/10.3390/reactions4010011
Spoor E, Welzel S, Nieken U, Rädle M. UV/VIS-Spectroscopic Inline Measurement for the Detection of Fouling Processes during the Polymerization of N-Vinylpyrrolidone. Reactions. 2023; 4(1):176-188. https://doi.org/10.3390/reactions4010011
Chicago/Turabian StyleSpoor, Erik, Stefan Welzel, Ulrich Nieken, and Matthias Rädle. 2023. "UV/VIS-Spectroscopic Inline Measurement for the Detection of Fouling Processes during the Polymerization of N-Vinylpyrrolidone" Reactions 4, no. 1: 176-188. https://doi.org/10.3390/reactions4010011