
Molecular Mechanisms and Mediators of Hepatotoxicity Resulting from an Excess of Lipids and Non-Alcoholic Fatty Liver Disease


Abstract
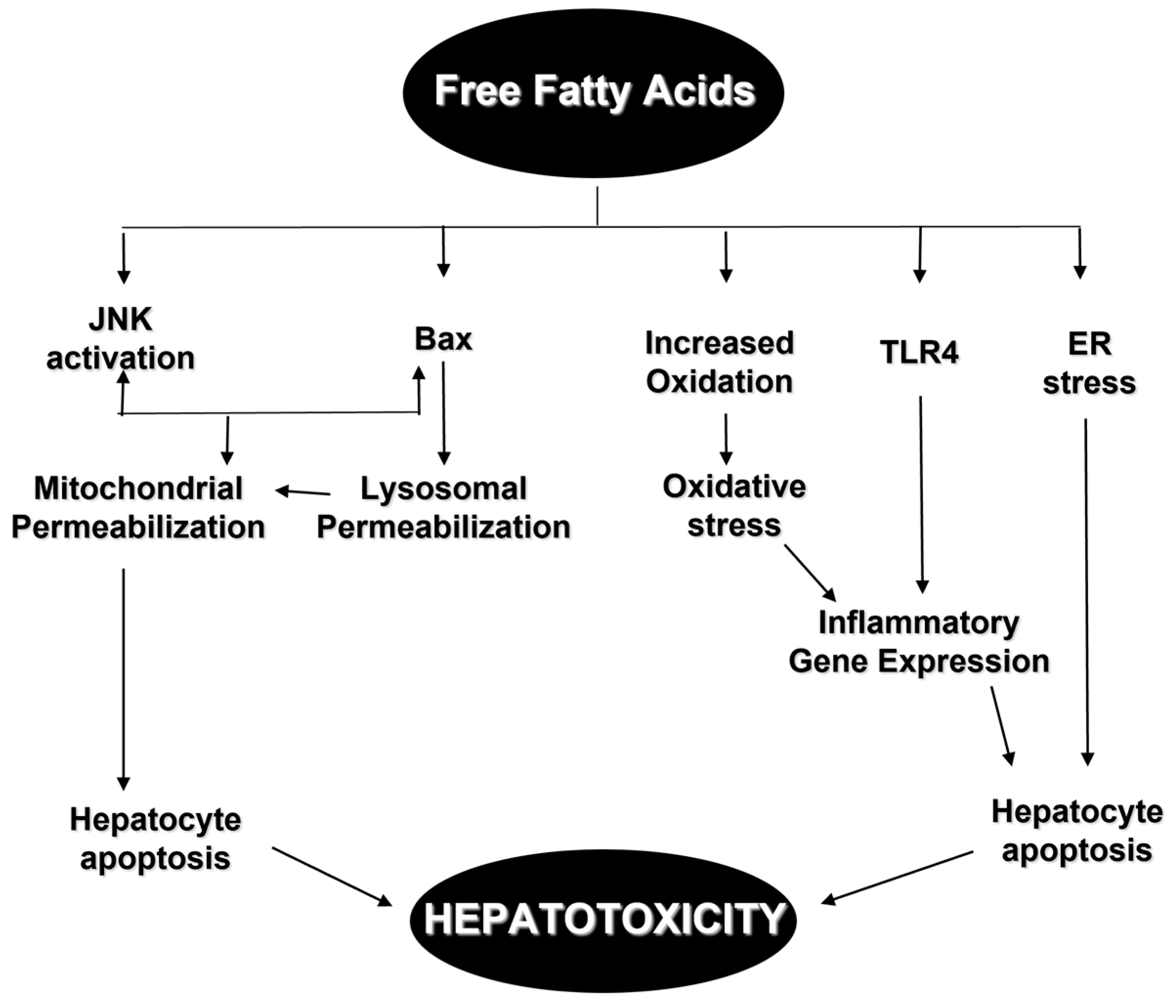
:1. Introduction
2. Mediators of Hepatotoxicity from Excess of Lipids
2.1. Toll-like Receptors
2.2. Death Receptors
2.3. Mitochondrial Dysfunction and Reactive Oxygen Species
2.4. Lysosomal Permeabilization
2.5. Endoplasmic Reticulum Stress
3. Pleiotropic Signals of Hepatotoxicity and Lipids
3.1. Prostaglandins Cyclooxygenase 2
3.2. Leukotrienes
3.2.1. Ceramides
3.2.2. Fatty Acids and Neutral Fats
Lipid Receptors
4. Effects of Lipid-Altering Drugs on Hepatotoxicity
4.1. Statins
4.2. Fibrates
4.3. Ezetimibe
4.4. Niacin
5. Discussion
6. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Tarantino, G.; Finelli, C. Pathogenesis of hepatic steatosis: The link between hypercortisolism and non-alcoholic fatty liver disease. World J. Gastroenterol. 2013, 19, 6735–6743. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; Zhang, L.; La, X.; Wu, H.; Yang, R.; Li, H.; Li, Z. Ferulic Acid and P-Coumaric Acid Synergistically Attenuate Non-Alcoholic Fatty Liver Disease through HDAC1/PPARG-Mediated Free Fatty Acid Uptake. Int. J. Mol. Sci. 2022, 23, 15297. [Google Scholar] [CrossRef]
- Gluchowski, N.L.; Gabriel, K.R.; Chitraju, C.; Bronson, R.T.; Mejhert, N.; Boland, S.; Wang, K.; Lai, Z.W. Hepatocyte Deletion of Triglyceride-Synthesis Enzyme Acyl CoA: Diacylglycerol Acyltransferase 2 Reduces Steatosis Without Increasing Inflammation or Fibrosis in Mice. Hepatology 2019, 70, 1972–1985. [Google Scholar] [CrossRef]
- Lipke, K.; Kubis-Kubiak, A.; Piwowar, A. Molecular Mechanism of Lipotoxicity as an Interesting Aspect in the Development of Pathological States—Current View of Knowledge. Cells 2022, 11, 844. [Google Scholar] [CrossRef] [PubMed]
- Cotter, T.G.; Rinella, M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology 2020, 158, 1851–1864. [Google Scholar] [CrossRef] [PubMed]
- Godoy-Matos, A.F.; Júnior, W.S.S.; Valerio, C.M. NAFLD as a continuum: From obesity to metabolic syndrome and diabetes. Diabetol. Metab. Syndr. 2020, 12, 60. [Google Scholar] [CrossRef]
- Finelli, C.; Tarantino, G. What is the role of adiponectin in obesity related non-alcoholic fatty liver disease? World J. Gastroenterol. 2013, 19, 802–812. [Google Scholar] [CrossRef]
- Paul, S.; Dhamija, E.; Kedia, S. Non-alcoholic fatty liver disease associated with hepatocellular carcinoma: An increasing concern. Indian J. Med. Res. 2019, 149, 9–17. [Google Scholar] [CrossRef]
- Ramai, D.; Facciorusso, A.; Vigandt, E.; Schaf, B.; Saadedeen, W.; Chauhan, A.; di Nunzio, S.; Shah, A.; Giacomelli, L.; Sacco, R. Progressive Liver Fibrosis in Non-Alcoholic Fatty Liver Disease. Cells 2021, 10, 3401. [Google Scholar] [CrossRef]
- Mazzolini, G.; Sowa, J.-P.; Atorrasagasti, C.; Kücükoglu, Ö.; Syn, W.-K.; Canbay, A. Significance of Simple Steatosis: An Update on the Clinical and Molecular Evidence. Cells 2020, 9, 2458. [Google Scholar] [CrossRef]
- Sato, K.; Kennedy, L.; Liangpunsakul, S.; Kusumanchi, P.; Yang, Z.; Meng, F.; Glaser, S.; Francis, H.; Alpini, G. Intercellular Communication between Hepatic Cells in Liver Diseases. Int. J. Mol. Sci. 2019, 20, 2180. [Google Scholar] [CrossRef] [PubMed]
- Ramos, M.J.; Bandiera, L.; Menolascina, F.; Fallowfield, J.A. In vitro models for non-alcoholic fatty liver disease: Emerging platforms and their applications. iScience 2021, 25, 103549. [Google Scholar] [CrossRef] [PubMed]
- Gambino, R.; Bugianesi, E.; Rosso, C.; Mezzabotta, L.; Pinach, S.; Alemanno, N.; Saba, F.; Cassader, M. Different Serum Free Fatty Acid Profiles in NAFLD Subjects and Healthy Controls after Oral Fat Load. Int. J. Mol. Sci. 2016, 17, 479. [Google Scholar] [CrossRef] [PubMed]
- Cansanção, K.; Monteiro, L.S.; Leite, N.C.; Dávalos, A.; Carmo, M.G.T.; Peres, W.A.F. Advanced liver fibrosis is independently associated with palmitic acid and insulin levels in patients with non-alcoholic fatty liver disease. Nutrients 2018, 10, 1586. [Google Scholar] [CrossRef]
- Hliwa, A.; Ramos-Molina, B.; Laski, D.; Mika, A.; Sledzinski, T. The Role of Fatty Acids in Non-Alcoholic Fatty Liver Disease Progression: An Update. Int. J. Mol. Sci. 2021, 22, 6900. [Google Scholar] [CrossRef]
- Lujan, P.V.; Esmel, E.V.; Meseguer, E.S. Overview of Non-Alcoholic Fatty Liver Disease (NAFLD) and the Role of Sugary Food Consumption and Other Dietary Components in Its Development. Nutrients 2021, 13, 1442. [Google Scholar] [CrossRef]
- Zhou, X.; Zhu, X.; Li, C.; Li, Y.; Ye, Z.; Shapiro, V.S.; Copland, J.A.; Hitosugi, T.; Bernlohr, D.A.; Sun, J.; et al. Stearoyl-CoA Desaturase-Mediated Monounsaturated Fatty Acid Availability Supports Humoral Immunity. Cell Rep. 2021, 34, 108601. [Google Scholar] [CrossRef]
- Ducheix, S.; Piccinin, E.; Peres, C.; Garcia-Irigoyen, O.; Bertrand-Michel, J.; Fouache, A.; Cariello, M.; Lobaccaro, J.; Guillou, H.; Sabbà, C.; et al. Reduction in gut-derived MUFAs via intestinal stearoyl-CoA desaturase 1 deletion drives susceptibility to NAFLD and hepatocarcinoma. Hepatol. Commun. 2022, 6, 2937–2949. [Google Scholar] [CrossRef]
- Geng, Y.; Faber, K.N.; de Meijer, V.E.; Blokzijl, H.; Moshage, H. How does hepatic lipid accumulation lead to lipotoxicity in non-alcoholic fatty liver disease? Hepatol. Int. 2021, 15, 21–35. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Tarantino, G.; Finelli, C.; Colao, A.; Capone, D.; Tarantino, M.; Grimaldi, E.; Chianese, D.; Gioia, S.; Pasanisi, F.; Contaldo, F.; et al. Are hepatic steatosis and carotid intima media thickness associated in obese patients with normal or slightly elevated gamma-glutamyl-transferase? J. Transl. Med. 2012, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Hu, Y.; Peng, J. Targeting programmed cell death in metabolic dysfunction-associated fatty liver disease (MAFLD): A promising new therapy. Cell Mol. Biol. Lett. 2021, 26, 17. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Sun, H.; Xue, T.; Gan, C.; Liu, H.; Xie, Y.; Yao, Y.; Ye, T. Liver Fibrosis: Therapeutic Targets and Advances in Drug Therapy. Front. Cell Dev. Biol. 2021, 9, 730176. [Google Scholar] [CrossRef]
- Lee, J.; Vali, Y.; Boursier, J.; Duffin, K.; Verheij, J.; Brosnan, M.J.; Zwinderman, K.; Anstee, Q.M.; Bossuyt, P.M.; Zafarmand, M.H. Accuracy of cytokeratin 18 (M30 and M65) in detecting non-alcoholic steatohepatitis and fibrosis: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0238717. [Google Scholar] [CrossRef]
- Natarajan, S.K.; Bruett, T.; Muthuraj, P.G.; Sahoo, P.K.; Power, J.; Mott, J.L.; Hanson, C.; Anderson-Berry, A. Saturated free fatty acids induce placental trophoblast lipoapoptosis. PLoS ONE 2021, 16, e0249907. [Google Scholar] [CrossRef]
- Hao, Q.; Chen, J.; Lu, H.; Zhou, X. The ARTS of p53-dependent mitochondrial apoptosis. J. Mol. Cell Biol. 2022, 14, mjac074. [Google Scholar] [CrossRef]
- Sharma, B.; John, S. Nonalcoholic Steatohepatitis (NASH). In Treasure Island; StatPearls Publishing: Tampa, FL, USA, 2022. [Google Scholar]
- Mendez-Sanchez, N.; Cruz-Ramon, V.C.; Ramirez-Perez, O.L.; Hwang, J.P.; Barranco-Fragoso, B.; Cordova-Gallardo, J. New Aspects of Lipotoxicity in Nonalcoholic Steatohepatitis. Int. J. Mol. Sci. 2018, 19, 2034. [Google Scholar] [CrossRef]
- Huang, X.; Yang, G.; Zhao, L.; Yuan, H.; Chen, H.; Shen, T.; Tang, W.; Man, Y.; Ma, J.; Ma, Y.; et al. Protein Phosphatase 4 Promotes Hepatocyte Lipoapoptosis by Regulating RAC1/MLK3/JNK Pathway. Oxidative Med. Cell Longev. 2021, 2021, 5550498. [Google Scholar] [CrossRef]
- Saponaro, C.; Sabatini, S.; Gaggini, M.; Carli, F.; Rosso, C.; Positano, V.; Armandi, A.; Caviglia, G.P.; Faletti, R.; Bugianesi, E.; et al. Adipose tissue dysfunction and visceral fat are associated with hepatic insulin resistance and severity of NASH even in lean individuals. Liver Int. 2022, 42, 2418–2427. [Google Scholar] [CrossRef]
- Patel, K.K.; Sehgal, V.S.; Kashfi, K. Molecular targets of statins and their potential side effects: Not all the glitter is gold. Eur. J. Pharmacol. 2022, 922, 174906. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Liu, B.; Tang, G.; Jin, P.; Liu, D. Two cases report of febuxostat-induced acute liver injury: Acute heart failure as a probable risk factor? Drug Chem. Toxicol. 2023, 1–5, ahead of print. [Google Scholar] [CrossRef]
- Wang, X.; Rao, J.; Tan, Z.; Xun, T.; Zhao, J.; Yang, X. Inflammatory signaling on cytochrome P450-mediated drug metabolism in hepatocytes. Front. Pharmacol. 2022, 13, 1043836. [Google Scholar] [CrossRef] [PubMed]
- Nie, L.; Cai, S.-Y.; Shao, J.-Z.; Chen, J. Toll-Like Receptors, Associated Biological Roles, and Signaling Networks in Non-Mammals. Front. Immunol. 2018, 9, 1523. [Google Scholar] [CrossRef]
- McKernan, K.; Varghese, M.; Patel, R.; Singer, K. Role of TLR4 in the induction of inflammatory changes in adipocytes and macrophages. Adipocyte 2020, 9, 212–222. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhu, C.; Wang, X.; Kim, K.; Bartolome, A.; Dongiovanni, P.; Yates, K.P.; Valenti, L.; Carrer, M.; Sadowski, T.; et al. Hepatocyte TLR4 triggers inter-hepatocyte Jagged1/Notch signaling to determine NASH-induced fibrosis. Sci. Transl. Med. 2021, 13, abe1692. [Google Scholar] [CrossRef]
- Zhou, Y.; Feng, Y.; Yang, L.; Zheng, P.; Hang, L.; Jiang, F.; Yuan, J.; Zhu, L. High-fat diet combined with dextran sulfate sodium failed to induce a more serious NASH phenotype than high-fat diet alone. Front. Pharmacol. 2022, 13, 1022172. [Google Scholar] [CrossRef]
- Carotti, S.; Guarino, M.P.; Vespasiani-Gentilucci, U.; Morini, S. Starring role of toll-like receptor-4 activation in the gut-liver axis. World J. Gastrointest. Pathophysiol. 2015, 6, 99–109. [Google Scholar] [CrossRef]
- Song, Q.; Zhang, X. The Role of Gut–Liver Axis in Gut Microbiome Dysbiosis Associated NAFLD and NAFLD-HCC. Biomedicines 2022, 10, 524. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Thorburn, A. Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand (TRAIL) Pathway Signaling. J. Thorac. Oncol. 2007, 2, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Tummers, B.; Green, D.R. Caspase-8: Regulating life and death. Immunol. Rev. 2017, 277, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Matsuoka, S.; Yamazaki, M.; Shibata, T.; Nirei, K.; Takahashi, H.; Kaneko, T.; Fujisawa, M.; Higuchi, T.; Nakamura, H.; et al. Apoptosis and non-alcoholic fatty liver diseases. World J. Gastroenterol. 2018, 24, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Pizzino, G.; Irrera, N.; Cucinotta, M.; Pallio, G.; Mannino, F.; Arcoraci, V.; Squadrito, F.; Altavilla, D.; Bitto, A. Oxidative Stress: Harms and Benefits for Human Health. Oxid. Med. Cell Longev. 2017, 2017, 8416763. [Google Scholar] [CrossRef]
- Snezhkina, A.V.; Kudryavtseva, A.V.; Kardymon, O.L.; Savvateeva, M.V.; Melnikova, N.V.; Krasnov, G.S.; Dmitriev, A.A. ROS Generation and Antioxidant Defense Systems in Normal and Malignant Cells. Oxidative Med. Cell Longev. 2019, 2019, 6175804. [Google Scholar] [CrossRef] [PubMed]
- Leyane, T.S.; Jere, S.W.; Houreld, N.N. Oxidative Stress in Ageing and Chronic Degenerative Pathologies: Molecular Mechanisms Involved in Counteracting Oxidative Stress and Chronic Inflammation. Int. J. Mol. Sci. 2022, 23, 7273. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Ren, J.; Zhou, W.; Huang, J.; Wu, G.; Yang, F.; Yuan, S.; Fang, J.; Liu, J.; Jin, Y.; et al. Lean non-alcoholic fatty liver disease (Lean-NAFLD) and the development of metabolic syndrome: A retrospective study. Sci. Rep. 2022, 12, 10977. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Tian, R.; She, Z.; Cai, J.; Li, H. Role of oxidative stress in the pathogenesis of nonalcoholic fatty liver disease. Free Radic. Biol. Med. 2020, 152, 116–141. [Google Scholar] [CrossRef]
- Kaarniranta, K.; Pawlowska, E.; Szczepanska, J.; Jablkowska, A.; Blasiak, J. Role of Mitochondrial DNA Damage in ROS-Mediated Pathogenesis of Age-Related Macular Degeneration (AMD). Int. J. Mol. Sci. 2019, 20, 2374. [Google Scholar] [CrossRef]
- Li, A.; Zheng, N.; Ding, X. Mitochondrial abnormalities: A hub in metabolic syndrome-related cardiac dysfunction caused by oxidative stress. Heart Fail. Rev. 2022, 27, 1387–1394. [Google Scholar] [CrossRef]
- Kim, H.-E.; Du, F.; Fang, M.; Wang, X. Formation of apoptosome is initiated by cytochrome c-induced dATP hydrolysis and subsequent nucleotide exchange on Apaf-1. Proc. Natl. Acad. Sci. USA 2005, 102, 17545–17550. [Google Scholar] [CrossRef] [PubMed]
- Parrish, A.B.; Freel, C.D.; Kornbluth, S. Cellular Mechanisms Controlling Caspase Activation and Function. Cold Spring Harb. Perspect. Biol. 2013, 5, a008672. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Passarella, S.; Shanmugam, H.; Noviello, M.; Bonfrate, L.; Wang, D.Q.-H.; Portincasa, P. Nonalcoholic Fatty Liver Disease (NAFLD). Mitochondria as Players and Targets of Therapies? Int. J. Mol. Sci. 2021, 22, 5375. [Google Scholar] [CrossRef] [PubMed]
- Ramanathan, R.; Ali, A.H.; Ibdah, J.A. Mitochondrial Dysfunction Plays Central Role in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 7280. [Google Scholar] [CrossRef]
- Talukdar, R.; Sareen, A.; Zhu, H.; Yuan, Z.; Dixit, A.; Cheema, H.; George, J.; Barlass, U.; Sah, R.; Garg, S.K.; et al. Release of Cathepsin B in Cytosol Causes Cell Death in Acute Pancreatitis. Gastroenterology 2016, 151, 747–758.e5. [Google Scholar] [CrossRef]
- Moles, A.; Tarrats, N.; Fernández-Checa, J.C.; Marí, M. Cathepsins B and D drive hepatic stellate cell proliferation and promote their fibrogenic potential. Hepatology 2009, 49, 1297–1307. [Google Scholar] [CrossRef]
- Phillips, M.J.; Voeltz, G.K. Structure and function of ER membrane contact sites with other organelles. Nat. Rev. Mol. Cell Biol. 2016, 17, 69–82. [Google Scholar] [CrossRef]
- Bravo, R.; Parra, V.; Gatica, D.; Rodriguez, A.E.; Torrealba, N.; Paredes, F.; Wang, Z.V.; Zorzano, A.; Hill, J.A.; Jaimovich, E.; et al. Endoplasmic Reticulum and the Unfolded Protein Response: Dynamics and Metabolic Integration. Int. Rev. Cell Mol. Biol. 2013, 301, 215–290. [Google Scholar] [CrossRef]
- Cirone, M. ER Stress, UPR Activation and the Inflammatory Response to Viral Infection. Viruses 2021, 13, 798. [Google Scholar] [CrossRef]
- Szegezdi, E.; Logue, S.E.; Gorman, A.M.; Samali, A. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Rep. 2006, 7, 880–885. [Google Scholar] [CrossRef]
- Ben-Dror, K.; Birk, R. Oleic acid ameliorates palmitic acid-induced ER stress and inflammation markers in naive and cerulein-treated exocrine pancreas cells. Biosci. Rep. 2019, 39, bsr20190054. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, N.; Carter-Kent, C.; Feldstein, A.E. Apoptosis in nonalcoholic fatty liver disease: Diagnostic and therapeutic implications. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Sunshine, H.; Iruela-Arispe, M.L. Membrane lipids and cell signaling. Curr. Opin. Infect. Dis. 2017, 28, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Pei, K.; Gui, T.; Kan, D.; Feng, H.; Jin, Y.; Yang, Y.; Zhang, Q.; Du, Z.; Gai, Z.; Wu, J.; et al. An Overview of Lipid Metabolism and Nonalcoholic Fatty Liver Disease. BioMed Res. Int. 2020, 2020, 4020249. [Google Scholar] [CrossRef]
- Zarghi, A.; Arfaei, S. Selective COX-2 Inhibitors: A Review of Their Structure-Activity Relationships. Iran. J. Pharm. Res. 2011, 10, 655–683. [Google Scholar]
- Gunaydin, C.; Bilge, S.S. Effects of Nonsteroidal Anti-Inflammatory Drugs at the Molecular Level. Eurasian J. Med. 2018, 50, 116–121. [Google Scholar] [CrossRef]
- Fuertes-Agudo, M.; Luque-Tévar, M.; Cucarella, C.; Brea, R.; Boscá, L.; Quintana-Cabrera, R.; Martín-Sanz, P.; Casado, M. COX-2 Expression in Hepatocytes Improves Mitochondrial Function after Hepatic Ischemia-Reperfusion Injury. Antioxidants 2022, 11, 1724. [Google Scholar] [CrossRef]
- Artru, F.; McPhail, M.J.W.; Triantafyllou, E.; Trovato, F.M. Lipids in Liver Failure Syndromes: A Focus on Eicosanoids, Specialized Pro-Resolving Lipid Mediators and Lysophospholipids. Front. Immunol. 2022, 13, 867261. [Google Scholar] [CrossRef]
- Tourkochristou, E.; Assimakopoulos, S.F.; Thomopoulos, K.; Marangos, M.; Triantos, C. NAFLD and HBV interplay-related mechanisms underlying liver disease progression. Front. Immunol. 2022, 13, 965548. [Google Scholar] [CrossRef]
- Gehrke, N.; Wörns, M.A.; Mann, A.; Hövelmeyer, N.; Waisman, A.; Straub, B.K.; Galle, P.R.; Schattenberg, J.M. Hepatocyte Bcl-3 protects from death-receptor mediated apoptosis and subsequent acute liver failure. Cell Death Dis. 2022, 13, 510. [Google Scholar] [CrossRef]
- Cheng, H.; Huang, H.; Guo, Z.; Chang, Y.; Li, Z. Role of prostaglandin E2 in tissue repair and regeneration. Theranostics 2021, 11, 8836–8854. [Google Scholar] [CrossRef] [PubMed]
- Tallima, H. Clarification of Arachidonic Acid Metabolic Pathway Intricacies. ACS Omega 2021, 6, 15559–15563. [Google Scholar] [CrossRef]
- Hassoun, D.; Rose, L.; Blanc, F.-X.; Magnan, A.; Loirand, G.; Sauzeau, V. Bronchial smooth muscle cell in asthma: Where does it fit? BMJ Open Respir. Res. 2022, 9, e001351. [Google Scholar] [CrossRef]
- Marbach-Breitrück, E.; Rohwer, N.; Infante-Duarte, C.; Romero-Suarez, S.; Labuz, D.; Machelska, H.; Kutzner, L.; Schebb, N.H.; Rothe, M.; Reddanna, P.; et al. Knock-In Mice Expressing a 15-Lipoxygenating Alox5 Mutant Respond Differently to Experimental Inflammation Than Reported Alox5−/− Mice. Metabolites 2021, 11, 698. [Google Scholar] [CrossRef]
- Chang, T.-T.; Yang, H.-Y.; Chen, C.; Chen, J.-W. CCL4 Inhibition in Atherosclerosis: Effects on Plaque Stability, Endothelial Cell Adhesiveness, and Macrophages Activation. Int. J. Mol. Sci. 2020, 21, 6567. [Google Scholar] [CrossRef]
- Sun, Q.-Y.; Zhou, H.-H.; Mao, X.-Y. Emerging Roles of 5-Lipoxygenase Phosphorylation in Inflammation and Cell Death. Oxidative Med. Cell Longev. 2019, 2019, 2749173. [Google Scholar] [CrossRef]
- Di Pasqua, L.G.; Cagna, M.; Berardo, C.; Vairetti, M.; Ferrigno, A. Detailed Molecular Mechanisms Involved in Drug-Induced Non-Alcoholic Fatty Liver Disease and Non-Alcoholic Steatohepatitis: An Update. Biomedicines 2022, 10, 194. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.; Nadler, J.L.; Mirmira, R.G.; Casimiro, I. Regulation of Tissue Inflammation by 12-Lipoxygenases. Biomolecules 2021, 11, 717. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kho, A.R.; Lee, S.H.; Hong, D.K.; Kang, B.S.; Park, M.K.; Lee, C.J.; Yang, H.W.; Woo, S.Y.; Park, S.W.; et al. Acid Sphingomyelinase Inhibitor, Imipramine, Reduces Hippocampal Neuronal Death after Traumatic Brain Injury. Int. J. Mol. Sci. 2022, 23, 14749. [Google Scholar] [CrossRef]
- Taniguchi, M.; Nagaya, S.; Yuyama, K.; Kotani, A.; Igarashi, Y.; Okazaki, T. Ceramide Metabolism Regulated by Sphingomyelin Synthase 2 Is Associated with Acquisition of Chemoresistance via Exosomes in Human Leukemia Cells. Int. J. Mol. Sci. 2022, 23, 10648. [Google Scholar] [CrossRef]
- Pal, P.; Atilla-Gokcumen, G.E.; Frasor, J. Emerging Roles of Ceramides in Breast Cancer Biology and Therapy. Int. J. Mol. Sci. 2022, 23, 11178. [Google Scholar] [CrossRef] [PubMed]
- Fisher-Wellman, K.H.; Hagen, J.T.; Neufer, P.D.; Kassai, M.; Cabot, M.C. On the nature of ceramide-mitochondria interactions—Dissection using comprehensive mitochondrial phenotyping. Cell Signal. 2020, 78, 109838. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-S.; Park, H.Y.; Kwon, S.P.; Kim, B.; Lee, Y.; Kim, S.; Shin, K.-O.; Park, K. NADPH Oxidase-Mediated Activation of Neutral Sphingomyelinase Is Responsible for Diesel Particulate Extract-Induced Keratinocyte Apoptosis. Int. J. Mol. Sci. 2020, 21, 1001. [Google Scholar] [CrossRef]
- Sztolsztener, K.; Konstantynowicz-Nowicka, K.; Harasim-Symbor, E.; Chabowski, A. Time-Dependent Changes in Hepatic Sphingolipid Accumulation and PI3K/Akt/mTOR Signaling Pathway in a Rat Model of NAFLD. Int. J. Mol. Sci. 2021, 22, 12478. [Google Scholar] [CrossRef]
- Mao, J.; Yi, M.; Wang, R.; Huang, Y.; Chen, M. Protective Effects of Costunolide Against D-Galactosamine and Lipopolysaccharide-Induced Acute Liver Injury in Mice. Front. Pharmacol. 2018, 9, 1469. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Talbot, C.L.; Chandravanshi, B.; Ksiazek, A.; Sood, A.; Chowdhury, K.H.; Maschek, J.A.; Cox, J.; Babu, A.K.S.; Paz, H.A.; et al. Cordyceps inhibits ceramide biosynthesis and improves insulin resistance and hepatic steatosis. Sci. Rep. 2022, 12, 7273. [Google Scholar] [CrossRef]
- Beckmann, N.; Becker, K.A. Ceramide and Related Molecules in Viral Infections. Int. J. Mol. Sci. 2021, 22, 5676. [Google Scholar] [CrossRef]
- De Carvalho, C.C.C.R.; Caramujo, M.J. The Various Roles of Fatty Acids. Molecules 2018, 23, 2583. [Google Scholar] [CrossRef]
- Heeren, J.; Scheja, L. Metabolic-associated fatty liver disease and lipoprotein metabolism. Mol. Metab. 2021, 50, 101238. [Google Scholar] [CrossRef]
- Santos-Laso, A.; Gutiérrez-Larrañaga, M.; Alonso-Peña, M.; Medina, J.M.; Iruzubieta, P.; Arias-Loste, M.T.; López-Hoyos, M.; Crespo, J. Pathophysiological Mechanisms in Non-Alcoholic Fatty Liver Disease: From Drivers to Targets. Biomedicines 2021, 10, 46. [Google Scholar] [CrossRef]
- Suriano, F.; Vieira-Silva, S.; Falony, G.; Roumain, M.; Paquot, A.; Pelicaen, R.; Régnier, M.; Delzenne, N.M.; Raes, J.; Muccioli, G.G.; et al. Novel insights into the genetically obese (ob/ob) and diabetic (db/db) mice: Two sides of the same coin. Microbiome 2021, 9, 147. [Google Scholar] [CrossRef]
- Paeschke, S.; Winter, K.; Bechmann, I.; Klöting, N.; Blüher, M.; Baum, P.; Kosacka, J.; Nowicki, M. Leptin Receptor-Deficient db/db Mice Show Significant Heterogeneity in Response to High Non-heme Iron Diet. Front. Nutr. 2021, 8, 741249. [Google Scholar] [CrossRef] [PubMed]
- Di Benedetto, G.; Burgaletto, C.; Serapide, M.F.; Caltabiano, R.; Munafò, A.; Bellanca, C.M.; Di Mauro, R.; Bernardini, R.; Cantarella, G. TRAIL-R Deficient Mice Are Protected from Neurotoxic Effects of Amyloid-β. Int. J. Mol. Sci. 2022, 23, 11625. [Google Scholar] [CrossRef]
- Samsonov, M.V.; Podkuychenko, N.V.; Khapchaev, A.Y.; Efremov, E.E.; Yanushevskaya, E.V.; Vlasik, T.N.; Lankin, V.Z.; Stafeev, I.S.; Skulachev, M.V.; Shestakova, M.V.; et al. AICAR Protects Vascular Endothelial Cells from Oxidative Injury Induced by the Long-Term Palmitate Excess. Int. J. Mol. Sci. 2021, 23, 211. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Zhong, W.; Zhang, W.; Hao, L.; Guo, W.; Yue, R.; Sun, X.; Sun, Z.; Bataller, R.; Zhou, Z. Loss of long-chain acyl-CoA synthetase 1 promotes hepatocyte death in alcohol-induced steatohepatitis. Metabolism 2022, 138, 155334. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, S.; Han, K.; Wang, L.; Liu, X. Induction of Apoptosis by Matrine Derivative ZS17 in Human Hepatocellular Carcinoma BEL-7402 and HepG2 Cells through ROS-JNK-P53 Signalling Pathway Activation. Int. J. Mol. Sci. 2022, 23, 15991. [Google Scholar] [CrossRef]
- Liu, N.; Zhu, Y.; Song, W.; Ren, W.; Tian, Z. Cardioprotection Attributed to Aerobic Exercise-Mediated Inhibition of ALCAT1 and Oxidative Stress-Induced Apoptosis in MI Rats. Biomedicines 2022, 10, 2250. [Google Scholar] [CrossRef]
- Ho, Q.W.C.; Zheng, X.; Ali, Y. Ceramide Acyl Chain Length and Its Relevance to Intracellular Lipid Regulation. Int. J. Mol. Sci. 2022, 23, 9697. [Google Scholar] [CrossRef]
- Zeng, X.; Zhu, M.; Liu, X.; Chen, X.; Yuan, Y.; Li, L.; Liu, J.; Lu, Y.; Cheng, J.; Chen, Y. Oleic acid ameliorates palmitic acid induced hepatocellular lipotoxicity by inhibition of ER stress and pyroptosis. Nutr. Metab. 2020, 17, 11. [Google Scholar] [CrossRef]
- Paramitha, P.N.; Zakaria, R.; Maryani, A.; Kusaka, Y.; Andriana, B.B.; Hashimoto, K.; Nakazawa, H.; Kato, S.; Sato, H. Raman Study on Lipid Droplets in Hepatic Cells Co-Cultured with Fatty Acids. Int. J. Mol. Sci. 2021, 22, 7378. [Google Scholar] [CrossRef]
- Yadav, A.K.; Sata, T.N.; Verma, D.; Mishra, A.K.; Sah, A.K.; Hossain, M.; Pant, K.; Venugopal, S.K. Free fatty acid-induced miR-181a-5p stimulates apoptosis by targeting XIAP and Bcl2 in hepatic cells. Life Sci. 2022, 301, 120625. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhang, J.-D.; Xiao, H.; He, S.; He, T.-T.; Ren, X.-M.; Yan, B.-H.; Luo, L.; Yin, Y.-L.; Cao, L.-Y. Triclocarban and triclosan exacerbate high-fat diet-induced hepatic lipid accumulation at environmental related levels: The potential roles of estrogen-related receptors pathways. Sci. Total. Environ. 2023, 858, 160079. [Google Scholar] [CrossRef]
- Römer, A.; Linn, T.; Petry, S.F. Lipotoxic Impairment of Mitochondrial Function in β-Cells: A Review. Antioxidants 2021, 10, 293. [Google Scholar] [CrossRef]
- Alves-Bezerra, M.; Cohen, D.E. Triglyceride Metabolism in the Liver. Compr. Physiol. 2017, 8, c170012. [Google Scholar] [CrossRef]
- Bjune, K.; Wierød, L.; Naderi, S. Triciribine increases LDLR expression and LDL uptake through stabilization of LDLR mRNA. Sci. Rep. 2018, 8, 16174. [Google Scholar] [CrossRef] [PubMed]
- Grewal, T.; Buechler, C. Emerging Insights on the Diverse Roles of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) in Chronic Liver Diseases: Cholesterol Metabolism and beyond. Int. J. Mol. Sci. 2022, 23, 1070. [Google Scholar] [CrossRef]
- Pirahanchi, Y.; Sinawe, H.; Dimri, M. Biochemistry, LDL Cholesterol. In Treasure Island; StatPearls Publishing: Tampa, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK519561/ (accessed on 18 March 2023).
- Rutkowska, L.; Sałacińska, K.; Salachna, D.; Matusik, P.; Pinkier, I.; Kępczyński, Ł.; Piotrowicz, M.; Starostecka, E.; Lewiński, A.; Gach, A. Identification of New Genetic Determinants in Pediatric Patients with Familial Hypercholesterolemia Using a Custom NGS Panel. Genes 2022, 13, 999. [Google Scholar] [CrossRef]
- Dato, V.A.; Chiabrando, G.A. The Role of Low-Density Lipoprotein Receptor-Related Protein 1 in Lipid Metabolism, Glucose Homeostasis and Inflammation. Int. J. Mol. Sci. 2018, 19, 1780. [Google Scholar] [CrossRef]
- Calvier, L.; Herz, J.; Hansmann, G. Interplay of Low-Density Lipoprotein Receptors, LRPs, and Lipoproteins in Pulmonary Hypertension. JACC Basic Transl. Sci. 2022, 7, 164–180. [Google Scholar] [CrossRef] [PubMed]
- Zarkasi, K.A.; Abdullah, N.; Murad, N.A.A.; Ahmad, N.; Jamal, R. Genetic Factors for Coronary Heart Disease and Their Mechanisms: A Meta-Analysis and Comprehensive Review of Common Variants from Genome-Wide Association Studies. Diagnostics 2022, 12, 2561. [Google Scholar] [CrossRef]
- Schäfer, I.; Kaisler, J.; Scheller, A.; Kirchhoff, F.; Haghikia, A.; Faissner, A. Conditional Deletion of LRP1 Leads to Progressive Loss of Recombined NG2-Expressing Oligodendrocyte Precursor Cells in a Novel Mouse Model. Cells 2019, 8, 1550. [Google Scholar] [CrossRef]
- Wijers, M.; Zanoni, P.; Liv, N.; Vos, D.Y.; Jäckstein, M.Y.; Smit, M.; Wilbrink, S.; Wolters, J.C.; van der Veen, Y.T.; Huijkman, N.; et al. The hepatic WASH complex is required for efficient plasma LDL and HDL cholesterol clearance. J. Clin. Investig. 2019, 4, 126462. [Google Scholar] [CrossRef] [PubMed]
- Lutsiv, T.; McGinley, J.N.; Neil, E.S.; Foster, M.T.; Thompson, H.J. Thwarting Metabolic Dysfunction-Associated Fatty Liver Disease (MAFLD) with Common Bean: Dose- and Sex-Dependent Protection against Hepatic Steatosis. Nutrients 2023, 15, 526. [Google Scholar] [CrossRef]
- Németh, K.; Tóth, B.; Sarnyai, F.; Koncz, A.; Lenzinger, D.; Kereszturi, É.; Visnovitz, T.; Kestecher, B.M.; Osteikoetxea, X.; Csala, M.; et al. High fat diet and PCSK9 knockout modulates lipid profile of the liver and changes the expression of lipid homeostasis related genes. Nutr. Metab. 2023, 20, 19. [Google Scholar] [CrossRef]
- Chen, J.; Su, Y.; Pi, S.; Hu, B.; Mao, L. The Dual Role of Low-Density Lipoprotein Receptor-Related Protein 1 in Atherosclerosis. Front. Cardiovasc. Med. 2021, 8, 682389. [Google Scholar] [CrossRef]
- Olivieri, C.; Ruzza, M.; Tolaj, F.; DaDalt, L.; Magni, P. Molecular and Functional Characterization of Human SW 872 Adipocytes as a Model System for Testing Nutraceutical Products. Biol. Life Sci. Forum 2022, 12, 19. [Google Scholar] [CrossRef]
- Konaniah, E.S.; Kuhel, D.G.; Basford, J.E.; Weintraub, N.L.; Hui, D.Y. Deficiency of LRP1 in Mature Adipocytes Promotes Diet-Induced Inflammation and Atherosclerosis—Brief Report. Arter. Thromb. Vasc. Biol. 2017, 37, 1046–1049. [Google Scholar] [CrossRef] [PubMed]
- Clark, L.E.; Clark, S.A.; Lin, C.; Liu, J.; Coscia, A.; Nabel, K.G.; Yang, P.; Neel, D.V.; Lee, H.; Brusic, V.; et al. VLDLR and ApoER2 are receptors for multiple alphaviruses. Nature 2022, 602, 475–480. [Google Scholar] [CrossRef]
- Passarella, D.; Ciampi, S.; Di Liberto, V.; Zuccarini, M.; Ronci, M.; Medoro, A.; Foderà, E.; Frinchi, M.; Mignogna, D.; Russo, C.; et al. Low-Density Lipoprotein Receptor-Related Protein 8 at the Crossroad between Cancer and Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 8921. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Ishii, K.; Hirota, Y.; Honda, T.; Makino, M.; Kawasaki, T.; Nakajima, K.; Hattori, M. Reelin-Nrp1 Interaction Regulates Neocortical Dendrite Development in a Context-Specific Manner. J. Neurosci. 2020, 40, 8248–8261. [Google Scholar] [CrossRef]
- Oshio, Y.; Hattori, Y.; Kamata, H. Very low-density lipoprotein receptor increases in a liver-specific manner due to protein deficiency but does not affect fatty liver in mice. Sci. Rep. 2021, 11, 8003. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Cai, X.; Ren, F.; Ye, Y.; Wang, F.; Zheng, C.; Qian, Y.; Zhang, M. The Macrophage-Osteoclast Axis in Osteoimmunity and Osteo-Related Diseases. Front. Immunol. 2021, 12, 664871. [Google Scholar] [CrossRef]
- Shin, K.C.; Hwang, I.; Choe, S.S.; Park, J.; Ji, Y.; Kim, J.I.; Lee, G.Y.; Choi, S.H.; Ching, J.; Kovalik, J.-P.; et al. Macrophage VLDLR mediates obesity-induced insulin resistance with adipose tissue inflammation. Nat. Commun. 2017, 8, 1087. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.; Zhan, Y.; Hou, Z.; Wang, C.; Fan, W.; Guo, T.; Li, Z.; Fang, L.; Lv, S.; Li, S.; et al. VLDLR disturbs quiescence of breast cancer stem cells in a ligand-independent function. Front. Oncol. 2022, 12, 887035. [Google Scholar] [CrossRef]
- Clinical Course and Diagnosis of Drug Induced Liver Disease. In LiverTox: Clinical and Research Information on Drug-Induced Liver Injury; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2019.
- Malakouti, M.; Kataria, A.; Ali, S.K.; Schenker, S. Elevated Liver Enzymes in Asymptomatic Patients—What Should I Do? J. Clin. Transl. Hepatol. 2017, 5, 394–403. [Google Scholar] [CrossRef]
- Chhetry, M.; Jialal, I. Lipid Lowering Drug Therapy. In Treasure Island; StatPearls Publishing: Tampa, FL, USA, 2022. [Google Scholar]
- Spigoni, V.; Aldigeri, R.; Antonini, M.; Micheli, M.M.; Fantuzzi, F.; Fratter, A.; Pellizzato, M.; Derlindati, E.; Zavaroni, I.; Bonadonna, R.C.; et al. Effects of a New Nutraceutical Formulation (Berberine, Red Yeast Rice and Chitosan) on Non-HDL Cholesterol Levels in Individuals with Dyslipidemia: Results from a Randomized, Double Blind, Placebo-Controlled Study. Int. J. Mol. Sci. 2017, 18, 1498. [Google Scholar] [CrossRef]
- Banach, M.; Catapano, A.L.; Cicero, A.F.; Escobar, C.; Foger, B.; Katsiki, N.; Latkovskis, G.; Rakowski, M.; Reiner, Z.; Sahebkar, A.; et al. Red yeast rice for dyslipidaemias and cardiovascular risk reduction: A position paper of the International Lipid Expert Panel. Pharmacol. Res. 2022, 183, 106370. [Google Scholar] [CrossRef]
- Feingold, K.R. Cholesterol Lowering Drugs. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2021. [Google Scholar]
- Linnenberger, R.; Hoppstädter, J.; Wrublewsky, S.; Ampofo, E.; Kiemer, A.K. Statins and Bempedoic Acid: Different Actions of Cholesterol Inhibitors on Macrophage Activation. Int. J. Mol. Sci. 2021, 22, 12480. [Google Scholar] [CrossRef]
- Chou, R.; Cantor, A.; Dana, T.; Wagner, J.; Ahmed, A.Y.; Fu, R.; Ferencik, M. Statin Use for the Primary Prevention of Cardiovascular Disease in Adults. JAMA 2022, 328, 754–771. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.H.; Zhang, B.L.; Cheng, Y.; Fu, S.K.; Jin, H.M. Statin use and the risk of CVD events, stroke, and all-cause mortality in patients with diabetes: A systematic review and meta-analysis. Nutr. Metab. Cardiovasc. Dis. 2022, 32, 2470–2482. [Google Scholar] [CrossRef]
- Selvaraj, S.; Oh, J.-H.; Yoon, S.; Borlak, J. Diclofenac Disrupts the Circadian Clock and through Complex Cross-Talks Aggravates Immune-Mediated Liver Injury—A Repeated Dose Study in Minipigs for 28 Days. Int. J. Mol. Sci. 2023, 24, 1445. [Google Scholar] [CrossRef] [PubMed]
- Merćep, I.; Strikić, D.; Slišković, A.M.; Reiner, Ž. New Therapeutic Approaches in Treatment of Dyslipidaemia—A Narrative Review. Pharmaceuticals 2022, 15, 839. [Google Scholar] [CrossRef]
- Lipsy, R.J. Introduction. J. Manag. Care Pharm. 2003, 9, 2–5. [Google Scholar] [CrossRef] [PubMed]
- Averbukh, L.D.; Turshudzhyan, A.; Wu, D.C.; Wu, G.Y. Statin-induced Liver Injury Patterns: A Clinical Review. J. Clin. Transl. Hepatol. 2022, 10, 543–552. [Google Scholar] [CrossRef]
- Lala, V.; Zubair, M.; Minter, D.A. Liver Function Tests. In Treasure Island; StatPearls Publishing: Tampa, FL, USA, 2022. [Google Scholar]
- Toth, P.P.; Patti, A.M.; Giglio, R.V.; Nikolic, D.; Castellino, G.; Rizzo, M.; Banach, M. Management of Statin Intolerance in 2018: Still More Questions Than Answers. Am. J. Cardiovasc. Drugs 2018, 18, 157–173. [Google Scholar] [CrossRef] [PubMed]
- Ah, Y.-M.; Jeong, M.; Choi, H.D. Comparative safety and efficacy of low- or moderate-intensity statin plus ezetimibe combination therapy and high-intensity statin monotherapy: A meta-analysis of randomized controlled studies. PLoS ONE 2022, 17, e0264437. [Google Scholar] [CrossRef]
- Qin, L.; Wang, Y.; Liang, Y.; Li, Q.; Xie, X.; Zhang, H. Astragaloside IV Alleviates Atorvastatin-Induced Hepatotoxicity via AMPK/SIRT1 Pathway. Pharmacology 2023, 108, 74–82. [Google Scholar] [CrossRef]
- Malone, M.; Lahmar, A.; Siddique, A.; Rozboril, M.; Kresak, J.L. A Case of Statin-Associated Autoimmune Myopathy: Management and Treatment. J. Prim. Care Community Health 2023, 14, 21501319221148635. [Google Scholar] [CrossRef]
- Banach, M.; Rizzo, M.; Toth, P.P.; Farnier, M.; Davidson, M.H.; Al-Rasadi, K.; Aronow, W.S.; Athyros, V.; Djuric, D.M.; Ezhov, M.V.; et al. Position paper Statin intolerance—An attempt at a unified definition. Position paper from an International Lipid Expert Panel. Arch. Med. Sci. 2015, 1, 49807. [Google Scholar] [CrossRef]
- Fu, S.; Wu, D.; Jiang, W.; Li, J.; Long, J.; Jia, C.; Zhou, T. Molecular Biomarkers in Drug-Induced Liver Injury: Challenges and Future Perspectives. Front. Pharmacol. 2020, 10, 1667. [Google Scholar] [CrossRef]
- Ward, N.C.; Watts, G.F.; Eckel, R.H. Statin Toxicity. Mechanistic Insights and Clinical Implications. Circ. Res. 2019, 124, 328–350. [Google Scholar] [CrossRef]
- McKenney, J.M.; Davidson, M.H.; Jacobson, T.A.; Guyton, J.R. Final Conclusions and Recommendations of the National Lipid Association Statin Safety Assessment Task Force. Am. J. Cardiol. 2006, 97, S89–S94. [Google Scholar] [CrossRef]
- Azemawah, V.; Movahed, M.R.; Centuori, P.; Penaflor, R.; Riel, P.L.; Situ, S.; Shadmehr, M.; Hashemzadeh, M. State of the Art Comprehensive Review of Individual Statins, Their Differences, Pharmacology, and Clinical Implications. Cardiovasc. Drugs Ther. 2019, 33, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Feingold, K.R. Triglyceride Lowering Drugs. In Endotext; Feingold, K.R., Anawalt, B., Blackman, M.R., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2021. [Google Scholar]
- Rikhi, R.; Shapiro, M.D. Newer and Emerging LDL-C Lowering Agents and Implications for ASCVD Residual Risk. J. Clin. Med. 2022, 11, 4611. [Google Scholar] [CrossRef] [PubMed]
- Lim, G.B. Limited benefit of triglyceride lowering with fibrates in statin-treated patients. Nat. Rev. Cardiol. 2023, 20, 4. [Google Scholar] [CrossRef]
- Quintanilla Rodriguez, B.S.; Correa, R. Gemfibrozil. In Treasure Island; StatPearls Publishing: Tampa, FL, USA, 2022. [Google Scholar]
- Shin, K.-H.; Choi, H.D. Comparison of Efficacy and Safety of Statin–Ezetimibe Combination Therapy with Statin Monotherapy in Patients with Diabetes: A Meta-Analysis of Randomized Controlled Studies. Am. J. Cardiovasc. Drugs 2022, 22, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Soulele, K.; Karalis, V. On the population pharmacokinetics and the enterohepatic recirculation of total ezetimibe. Xenobiotica 2019, 49, 446–456. [Google Scholar] [CrossRef]
- Kanagalingam, T.; Lazarte, J.; Wong, D.K.; Hegele, R.A. Liver Injury Associated With Ezetimibe Monotherapy. CJC Open 2020, 3, 195–197. [Google Scholar] [CrossRef]
- Bach, R.G.; Cannon, C.P.; Giugliano, R.P.; White, J.A.; Lokhnygina, Y.; Bohula, E.A.; Califf, R.M.; Braunwald, E.; Blazing, M.A. Effect of Simvastatin-Ezetimibe Compared With Simvastatin Monotherapy After Acute Coronary Syndrome Among Patients 75 Years or Older. JAMA Cardiol. 2019, 4, 846–854. [Google Scholar] [CrossRef]
- Qian, J.; Li, Z.; Zhang, X.; Chen, J.; Ding, C.; Yang, P.; Liu, Y.; Shi, M.; Ren, X.; Ge, J. Efficacy and Tolerability of Ezetimibe/Atorvastatin Fixed-dose Combination Versus Atorvastatin Monotherapy in Hypercholesterolemia: A Phase III, Randomized, Active-controlled Study in Chinese Patients. Clin. Ther. 2022, 44, 1282–1296. [Google Scholar] [CrossRef]
- Yu, M.; Liang, C.; Kong, Q.; Wang, Y.; Li, M. Efficacy of combination therapy with ezetimibe and statins versus a double dose of statin monotherapy in participants with hypercholesterolemia: A meta-analysis of literature. Lipids Health Dis. 2020, 19, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Djadjo, S.; Bajaj, T. Niacin. In Treasure Island; StatPearls Publishing: Tampa, FL, USA, 2022. [Google Scholar]
- Li, H.; Xu, X.; Lu, L.; Sun, R.; Guo, Q.; Chen, Q.; Wang, J.; He, Z.; Zhang, Y. The comparative impact among different intensive statins and combination therapies with niacin/ezetimibe on carotid intima-media thickness: A systematic review, traditional meta-analysis, and network meta-analysis of randomized controlled trials. Eur. J. Clin. Pharmacol. 2021, 77, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Ronsein, G.E.; Vaisar, T.; Davidson, W.S.; Bornfeldt, K.E.; Probstfield, J.L.; O’brien, K.D.; Zhao, X.-Q.; Heinecke, J.W. Niacin Increases Atherogenic Proteins in High-Density Lipoprotein of Statin-Treated Subjects. Arter. Thromb. Vasc. Biol. 2021, 41, 2330–2341. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Drug(s) | Key Contraindication | Recommended Liver Function Monitoring |
|---|---|---|
| Statins | Active or chronic liver disease | Obtain AST/ALT initially, 12 weeks after starting, then annually or sooner if clinically indicated. Baseline, with follow-up, only as clinically indicated |
| Fibrates | Gall bladder disease, hepatic disease (biliary cirrhosis), or severe renal impairment including dialysis | Liver tests should be monitored periodically |
| Ezetimibe | Active liver disease or unexplained AST/ALT elevations (when co-administered with a statin) | When co-administered with statin therapy, monitor according to recommendations for individual statins |
| Niacin | Active liver disease, active peptic ulcer disease, or severe gout | Obtain AST/ALT initially, 6–8 weeks after reaching 1500 mg daily, 6–8 weeks after reaching max daily dose, then annually or sooner if clinically indicated |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Finelli, C. Molecular Mechanisms and Mediators of Hepatotoxicity Resulting from an Excess of Lipids and Non-Alcoholic Fatty Liver Disease. Gastrointest. Disord. 2023, 5, 243-260. https://doi.org/10.3390/gidisord5020020
Finelli C. Molecular Mechanisms and Mediators of Hepatotoxicity Resulting from an Excess of Lipids and Non-Alcoholic Fatty Liver Disease. Gastrointestinal Disorders. 2023; 5(2):243-260. https://doi.org/10.3390/gidisord5020020
Chicago/Turabian StyleFinelli, Carmine. 2023. "Molecular Mechanisms and Mediators of Hepatotoxicity Resulting from an Excess of Lipids and Non-Alcoholic Fatty Liver Disease" Gastrointestinal Disorders 5, no. 2: 243-260. https://doi.org/10.3390/gidisord5020020