
Comprehensive Review of Acute Pancreatitis Pain Syndrome
 , ,
, ,
Abstract
:1. Introduction
1.1. Epidemiology
1.2. Morphology
1.3. Endocrine and Excocrine Function
1.4. Vascularization and Lymphatics
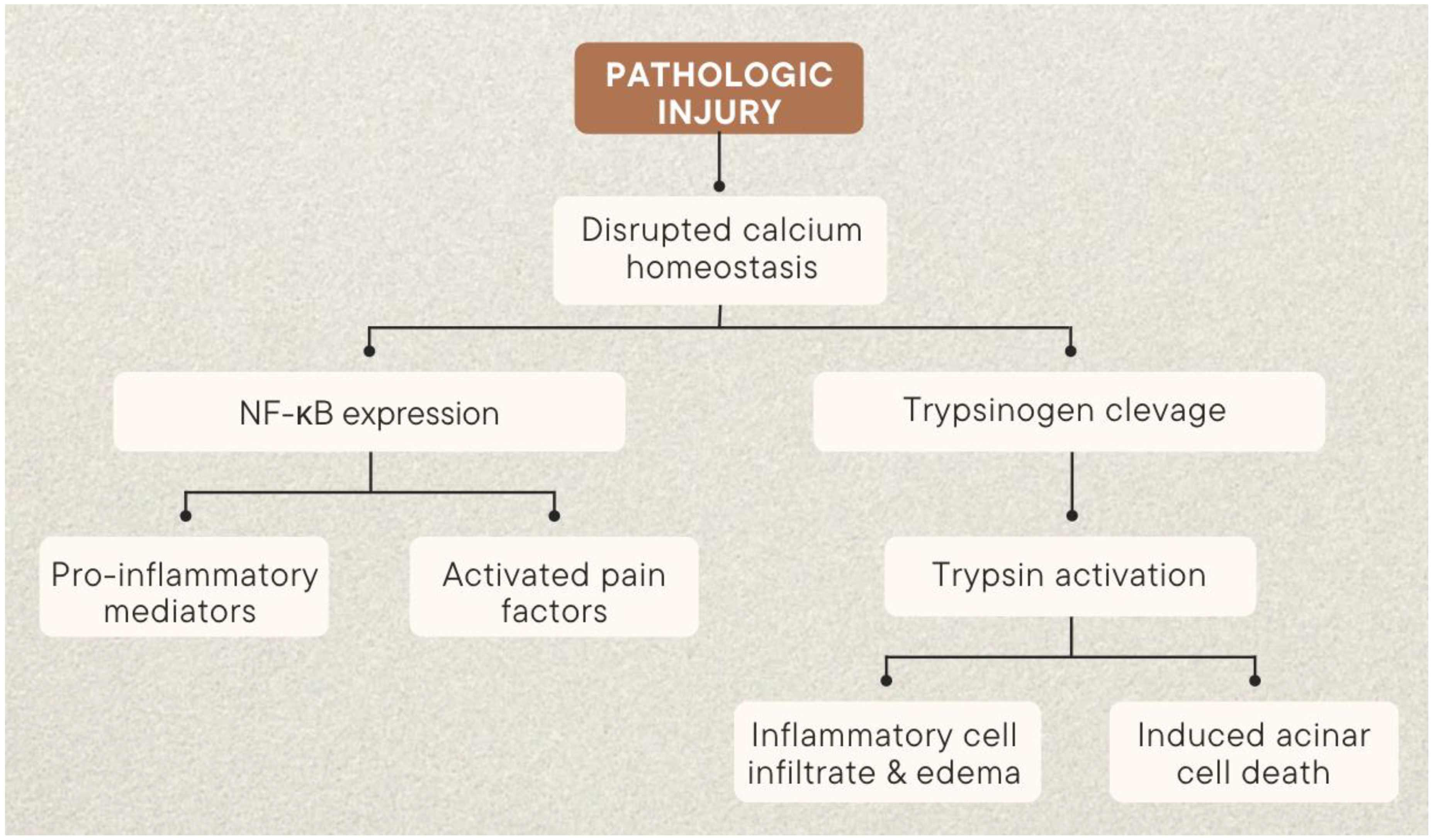
2. Pathophysiology of Acute Pancreatitis Pain
2.1. Trypsin
2.2. Calcium Signaling
2.3. Calcineurin
2.4. Co-Localization and Autophagy
2.5. Inflammatory Pathway
2.6. Neuropathic Pain
2.7. Inflammatory Pain
2.8. Visceral Pain
2.9. Gut Flora Dysbiosis
3. Initial Treatment
3.1. Nutrition
3.2. Intravenous Fluids
3.3. Pain Management
- Tailor treatment to the individual as one size will not fit all;
- Assess pain intensity and pancreatitis severity to select analgesics;
- Step-down approach required for prompt pain relief;
- Use opioid-sparing strategies wherever possible.
4. Pharmacological Treatment
4.1. Lidocaine

{kind=link}
| Trials Evaluating Pancreatitis Pain Pharmalogical Treatment | |
|---|---|
| Lidocaine | Animal studies indicate intra-arterial significantly decreases amylase and lipase serum concentrations. RCT show topical administration was comparative to controls [128] |
| Hydromorphone/Opioid Agonists | RCT’s demonstrate no difference in pancreatitis complications or adverse events between opioid and non-opioid analgesics [114,145,146] |
| Ketamine | Case studies demonstrate success in the treatment of pancreatic pain in both quick and long-lasting analgesia. RCT’s are ongoing [155] |
| Magnesium | There is currently one clinical trial investigating magnesium sulfate’s effects on preventing post-ERCP pancreatitis [173] |
4.2. Hydromorphone/Opioid Agonists
4.3. Ketamine
4.4. Magnesium
5. Opioid Use in Patients with Pancreatitis
6. Novel Treatments
6.1. Endoscopic Therapy
6.2. Pancreatic Enzyme Replacement Therapy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Banks, P.A.; Bollen, T.L.; Dervenis, C.; Gooszen, H.G.; Johnson, C.D.; Sarr, M.G.; Tsiotos, G.G.; Vege, S.S. Acute Pancreatitis Classification Working Group. Classification of acute pancreatitis—2012: Revision of the Atlanta classification and definitions by international consensus. Gut 2013, 62, 102–111. [Google Scholar] [CrossRef]
- Roberts, S.E.; Morrison-Rees, S.; John, A.; Williams, J.G.; Brown, T.H.; Samuel, D.G. The incidence and aetiology of acute pancreatitis across Europe. Pancreatology 2017, 17, 155–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannuzzi, J.P.; King, J.A.; Leong, J.H.; Quan, J.; Windsor, J.W.; Tanyingoh, D.; Coward, S.; Forbes, N.; Heitman, S.J.; Shaheen, A.-A.; et al. Global Incidence of Acute Pancreatitis Is Increasing Over Time: A Systematic Review and Meta-Analysis. Gastroenterology 2022, 162, 122–134. [Google Scholar] [CrossRef]
- Yadav, D.; Lowenfels, A.B. The Epidemiology of Pancreatitis and Pancreatic Cancer. Gastroenterology 2013, 144, 1252–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, C.-Y.; Yeh, C.-N.; Hsu, J.-T.; Jan, Y.-Y.; Hwang, T.-L. Timing of mortality in severe acute pancreatitis: Experience from 643 patients. World J. Gastroenterol. 2007, 13, 1966–1969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khatua, B.; El-Kurdi, B.; Singh, V. Obesity and pancreatitis. Curr. Opin. Gastroenterol. 2017, 33, 374–382. [Google Scholar] [CrossRef] [PubMed]
- Talathi, S.S.; Zimmerman, R.; Young, M. Anatomy, Abdomen and Pelvis, Pancreas; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Paoli, C.; Carrer, A. Organotypic Culture of Acinar Cells for the Study of Pancreatic Cancer Initiation. Cancers 2020, 12, 2606. [Google Scholar] [CrossRef]
- Strobel, O.; Rosow, D.E.; Rakhlin, E.Y.; Lauwers, G.Y.; Trainor, A.G.; Alsina, J.; Castillo, C.F.; Warshaw, A.L.; Thayer, S.P. Pancreatic Duct Glands Are Distinct Ductal Compartments That React to Chronic Injury and Mediate Shh-Induced Metaplasia. Gastroenterology 2010, 138, 1166–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.; Ahmed, S. Functional, Diagnostic and Therapeutic Aspects of Gastrointestinal Hormones. Gastroenterol. Res. 2019, 12, 233–244. [Google Scholar] [CrossRef] [PubMed]
- Rorsman, P.; Huising, M.O. The somatostatin-secreting pancreatic delta-cell in health and disease. Nat. Rev. Endocrinol. 2018, 14, 404–414. [Google Scholar] [CrossRef] [PubMed]
- El Sayed, S.A.; Mukherjee, S. Physiology, Pancreas; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Hundt, M.; Wu, C.; Young, M. Anatomy, Abdomen and Pelvis, Biliary Ducts; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Jansson, L.; Barbu, A.; Bodin, B.; Drott, C.J.; Espes, D.; Gao, X.; Grapensparr, L.; Källskog, Ö.; Lau, J.; Liljebäck, H.; et al. Pancreatic islet blood flow and its measurement. Ups. J. Med. Sci. 2016, 121, 81–95. [Google Scholar] [CrossRef] [Green Version]
- Jansson, L.; Carlsson, P. Pancreatic Blood Flow with Special Emphasis on Blood Perfusion of the Islets of Langerhans. Compr. Physiol. 2019, 9, 799–837. [Google Scholar]
- Szmola, R.; Sahin-Toth, M. Chymotrypsin C (caldecrin) promotes degradation of human cationic trypsin: Identity with Rinderknecht’s enzyme Y. Proc. Natl. Acad. Sci. USA 2007, 104, 11227–11232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wartmann, T.; Mayerle, J.; Kähne, T.; Sahin–Tóth, M.; Ruthenbürger, M.; Matthias, R.; Kruse, A.; Reinheckel, T.; Peters, C.; Kruse, A.; et al. Cathepsin L Inactivates Human Trypsinogen, Whereas Cathepsin L-Deletion Reduces the Severity of Pancreatitis in Mice. Gastroenterology 2010, 138, 726–737. [Google Scholar] [CrossRef] [Green Version]
- Saluja, A.; Dudeja, V.; Dawra, R.; Sah, R.P. Early Intra-Acinar Events in Pathogenesis of Pancreatitis. Gastroenterology 2019, 156, 1979–1993. [Google Scholar] [CrossRef] [PubMed]
- Baumann, B.; Wagner, M.; Aleksic, T.; Von Wichert, G.; Weber, C.K.; Adler, G.; Wirth, T. Constitutive IKK2 activation in acinar cells is sufficient to induce pancreatitis in vivo. J. Clin. Investig. 2007, 117, 1502–1513. [Google Scholar] [CrossRef]
- Marrache, F.; Tu, S.P.; Penclyala, S.; Bhagat, G.; Oesterreicher, C.; Penz-Oesterreicher, M.; Betz, K.; Stoffers, D.; Ai, W.; Takaishi, S.; et al. Overexpression of interleukin-1beta in the murine pancreas results in chronic pancreatitis. Gastroenterology 2008, 135, 1277–1287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Criddle, D.N.; Gerasimenko, J.V.; Baumgartner, H.K.; Jaffar, M.; Voronina, S.; Sutton, R.; Petersen, O.H.; Gerasimenko, O.V. Calcium signalling and pancreatic cell death: Apoptosis or necrosis? Cell Death Differ. 2007, 14, 1285–1294. [Google Scholar] [CrossRef] [Green Version]
- Straub, S.V.; Giovannucci, D.; Yule, D. Calcium wave propagation in pancreatic acinar cells: Functional interaction of inositol 1,4,5-trisphosphate receptors, ryanodine receptors, and mitochondria. J. Gen. Physiol. 2000, 116, 547–560. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.A.; Criddle, D.N.; Sherwood, M.; Chvanov, M.; Mukherjee, R.; McLaughlin, E.; Booth, D.; Gerasimenko, J.V.; Raraty, M.G.T.; Ghaneh, P.; et al. Direct Activation of Cytosolic Ca2+ Signaling and Enzyme Secretion by Cholecystokinin in Human Pancreatic Acinar Cells. Gastroenterology 2008, 135, 632–641. [Google Scholar] [CrossRef]
- Sah, R.P.; Garg, P.; Saluja, A.K. Pathogenic mechanisms of acute pancreatitis. Curr. Opin. Gastroenterol. 2012, 28, 507–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.S.; Hong, J.H.; Li, Q.; Shin, D.M.; Abramowitz, J.; Birnbaumer, L.; Muallem, S. Deletion of TRPC3 in Mice Reduces Store-Operated Ca2+ Influx and the Severity of Acute Pancreatitis. Gastroenterology 2009, 137, 1509–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewarchik, C.M.; Orabi, A.I.; Jin, S.; Wang, D.; Muili, K.A.; Shah, A.U.; Eisses, J.F.; Malik, A.; Bottino, R.; Jayaraman, T.; et al. The ryanodine receptor is expressed in human pancreatic acinar cells and contributes to acinar cell injury. Am. J. Physiol. Liver Physiol. 2014, 307, G574–G581. [Google Scholar]
- Husain, S.Z.; Grant, W.M.; Gorelick, F.S.; Nathanson, M.H.; Shah, A.U. Caerulein-induced intracellular pancreatic zymogen activation is dependent on calcineurin. Am. J. Physiol. Liver Physiol. 2007, 292, G1594–G1599. [Google Scholar] [CrossRef]
- Susini, C.; Estival, A.; Scemama, J.L.; Ruellan, C.; Vaysse, N.; Clemente, F.; Esteve, J.P.; Fourmy, D.; Ribet, A. Studies on human pancreatic acini: Action of secretagogues on amylase release and cellular cyclic AMP accumulation. Pancreas 1986, 1, 124–129. [Google Scholar]
- Shah, A.U.; Sarwar, A.; Orabi, A.I.; Gautam, S.; Grant, W.M.; Park, A.J.; Shah, A.U.; Liu, J.; Mistry, P.K.; Jain, D.; et al. Protease activation during in vivo pancreatitis is dependent on calcineurin activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G967–G973. [Google Scholar] [CrossRef] [Green Version]
- Muili, K.A.; Ahmad, M.; Orabi, A.I.; Mahmood, S.M.; Shah, A.U.; Molkentin, J.D.; Husain, S.Z. Pharmacological and genetic inhibition of calcineurin protects against carbachol-induced pathological zymogen activation and acinar cell injury. Am. J. Physiol. Liver Physiol. 2012, 302, G898–G905. [Google Scholar] [CrossRef] [Green Version]
- Muili, K.A.; Wang, D.; Orabi, O.I.; Sarwar, S.; Luo, Y.; Javed, T.A.; Eisses, J.F.; Mahmood, S.M.; Jin, S.; Singh, V.P.; et al. Bile Acids Induce Pancreatic Acinar Cell Injury and Pancreatitis by Activating Calcineurin*. J. Biol. Chem. 2013, 288, 570–580. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, O.; Baccino, F.M.; Steer, M.L.; Meldolesi, J. Supramaximal caerulein stimulation and ultrastructure of rat pancreatic acinar cell: Early morphological changes during development of experimental pancreatitis. Am. J. Physiol. Liver Physiol. 1984, 246, G457–G467. [Google Scholar] [CrossRef]
- Miehle, W. Serology of rheumatism. Fortschr. Med. 1978, 96, 2024–2028. [Google Scholar] [PubMed]
- Willemer, S.; Klöppel, G.; Kern, H.F.; Adler, G. Immunocytochemical and morphometric analysis of acinar zymogen granules in human acute pancreatitis. Virchows Arch. A Pathol. Anat. Histopathol. 1989, 415, 115–123. [Google Scholar] [CrossRef]
- Halangk, W.; Lerch, M.M.; Brandt-Nedelev, B.; Roth, W.; Ruthenbuerger, M.; Reinheckel, T.; Domschke, W.; Lippert, H.; Peters, C.; Deussing, J. Role of cathepsin B in intracellular trypsinogen activation and the onset of acute pancreatitis. J. Clin. Investig. 2000, 106, 773–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saluja, A.K.; Donovan, E.A.; Yamanaka, K.; Yamaguchi, Y.; Hofbauer, B.; Steer, M.L. Cerulein-induced in vitro activation of trypsinogen in rat pancreatic acini is mediated by cathepsin B. Gastroenterology 1997, 113, 304–310. [Google Scholar] [CrossRef]
- Hietaranta, A.J.; Saluja, A.K.; Bhagat, L.; Singh, V.P.; Song, A.M.; Steer, M.L. Relationship between NF-kB and trypsinogen activation in rat pancreas after supramaximal caerulein stimulation. Biochem. Biophys. Res. Commun. 2001, 280, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Gukovsky, I.; Gukovskaya, A.S.; Blinman, T.A.; Zaninovic, V.; Pandol, S.J. Early NF-kB activation is associated with hormone-induced pancreatitis. Am. J. Physiol. 1998, 275, G1402–G1414. [Google Scholar] [PubMed]
- Rakonczay, Z., Jr.; Hegyi, P.; Takács, T.; McCarroll, J.; Saluja, A.K. The role of NF-kB activation in the pathogenesis of acute pancreatitis. Gut 2008, 57, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Tando, Y.; Algül, H.; Schneider, G.; Weber, C.K.; Weidenbach, H.; Adler, G.; Schmid, R.M. Induction of IkB-kinase by cholecystokinin is mediated by trypsinogen activation in rat pancreatic lobules. Digestion 2002, 66, 237–245. [Google Scholar] [CrossRef]
- Gukovskaya, A.S.; Hosseini, S.; Satoh, A.; Cheng, J.H.; Nam, K.J.; Gukovsky, I.; Pandol, S.J. Ethanol differentially regulates NF-kB activation in pancreatic acinar cells through calcium and protein kinase C pathways. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G204–G213. [Google Scholar] [CrossRef] [Green Version]
- Sah, R.P.; Dudeja, V.; Dawra, R.K.; Saluja, A.K. Cerulein-Induced Chronic Pancreatitis Does Not Require Intra-Acinar Activation of Trypsinogen in Mice. Gastroenterology 2013, 144, 1076–1085.e2. [Google Scholar] [CrossRef] [Green Version]
- Ji, B.; Gaiser, S.; Gaiser, X.; Ernst, S.A.; Logsdon, C.D. Intracellular trypsin induces pancreatic acinar cell death but not NF-kB activation. J. Biol. Chem. 2009, 284, 17488–17498. [Google Scholar] [CrossRef] [Green Version]
- Dawra, R.; Sah, R.P.; Dudeja, V.; Rishi, L.; Talukdar, R.; Garg, P.; Saluja, A.K. Intra-acinar trypsinogen activation mediates early stages of pancreatic injury but not inflammation in mice with acute pancreatitis. Gastroenterology 2011, 141, 2210–2217.e2. [Google Scholar] [CrossRef] [Green Version]
- Földi, M.; Gede, N.; Kiss, S.; Vincze, Á.; Bajor, J.; Szabó, I.; Szepes, Z.; Izbéki, F.; Gervain, J.; Hamvas, J.; et al. The characteristics and prognostic role of acute abdominal on-admission pain in acute pancreatitis: A prospective cohort analysis of 1432 cases. Eur. J. Pain 2021, 26, 610–623. [Google Scholar] [CrossRef] [PubMed]
- Mackey, S.; Carroll, I.; Emir, B.; Murphy, T.K.; Whalen, E.; Dumenci, L. Sensory Pain Qualities in Neuropathic Pain. J. Pain 2012, 13, 58–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dworkin, R.H.; Jensen, M.P.; Gammaitoni, A.R.; Olaleye, D.O.; Galer, B.S. Symptom Profiles Differ in Patients With Neuropathic Versus Non-neuropathic Pain. J. Pain 2007, 8, 118–126. [Google Scholar] [CrossRef] [PubMed]
- VanDenKerkhof, E.G.; Stitt, L.; Clark, A.J.; Gordon, A.; Lynch, M.; Morley-Forster, P.K.; Nathan, H.J.; Smyth, C.; Toth, C.; Ware, M.A.; et al. Sensitivity of the DN4 in Screening for Neuropathic Pain Syndromes. Clin. J. Pain 2018, 34, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Bockman, D.; Buchler, M.; Malfertheiner, P.; Beger, H. Analysis of nerves in chronic pancreatitis. Gastroenterology 1988, 94, 1459–1469. [Google Scholar] [CrossRef]
- Di Sebastiano, P.; Fink, T.; Weihe, E.; Friess, H.; Innocenti, P.; Beger, H.G.; Büchler, M.W. Immune cell infiltration and growth-associated protein 43 expression correlate with pain in chronic pancreatitis. Gastroenterology 1997, 112, 1648–1655. [Google Scholar] [CrossRef]
- Gougol, A.; Machicado, J.D.; Matta, B.; Paragomi, P.; Pothoulakis, I.; Slivka, A.; Whitcomb, D.C.; Yadav, D.; Papachristou, G.I. Prevalence and Associated Factors of Abdominal Pain and Disability at 1-Year Follow-up After an Attack of Acute Pancreatitis. Pancreas 2019, 48, 1348–1353. [Google Scholar] [CrossRef] [PubMed]
- Ceyhan, G.O.; Bergmann, F.; Kadihasanoglu, M.; Altintas, B.; Demir, I.E.; Hinz, U.; Müller, M.W.; Giese, T.; Büchler, M.W.; Giese, N.A.; et al. Pancreatic Neuropathy and Neuropathic Pain—A Comprehensive Pathomorphological Study of 546 Cases. Gastroenterology 2009, 136, 177–186.e1. [Google Scholar] [CrossRef]
- Atsawarungruangkit, A.; Pongprasobchai, S. Current understanding of the neuropathophysiology of pain in chronic pancreatitis. World J. Gastrointest. Pathophysiol. 2015, 6, 193–202. [Google Scholar] [CrossRef]
- Li, Q.; Peng, J. Sensory nerves and pancreatitis. Gland. Surg. 2014, 3, 284–292. [Google Scholar]
- Bennett, D.L.; Clark, A.J.; Huang, J.; Waxman, S.G.; Dib-Hajj, S.D. The Role of Voltage-Gated Sodium Channels in Pain Signaling. Physiol. Rev. 2019, 99, 1079–1151. [Google Scholar] [CrossRef]
- Olesen, S.S.; Krauss, T.; Demir, I.E.; Wilder-Smith, O.H.; Ceyhan, G.O.; Pasricha, P.J.; Drewes, A.M. Towards a neurobiological understanding of pain in chronic pancreatitis: Mechanisms and implications for treatment. PAIN Rep. 2017, 2, e625. [Google Scholar] [CrossRef]
- Watkins, L.R.; Maier, S.F. Immune regulation of central nervous system functions: From sickness responses to pathological pain. J. Intern. Med. 2005, 257, 139–155. [Google Scholar] [CrossRef] [PubMed]
- Esper, R.M.; Loeb, J.A. Rapid Axoglial Signaling Mediated by Neuregulin and Neurotrophic Factors. J. Neurosci. 2004, 24, 6218–6227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Igolnikov, I.; Gallagher, R.M.; Hainline, B. Sport-related injury and pain classification. Handb. Clin. Neurol. 2018, 158, 423–430. [Google Scholar]
- Barreto, S.G.; Saccone, G.T. Pancreatic nociception—Revisiting the physiology and pathophysiology. Pancreatology 2012, 12, 104–112. [Google Scholar] [CrossRef]
- Nechutova, H.; Petr, D.; Markéta, H.; Ivo, N.; Arnost, M.; Pavel, K.; Bohuslav, K.; Miroslav, S. Pancreatic pain. Wien. Med. Wochenschr. 2014, 164, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Barreto, S.G.; Habtezion, A.; Gukovskaya, A.; Lugea, A.; Jeon, C.; Yadav, D.; Hegyi, P.; Venglovecz, V.; Sutton, R.; Pandol, S.J. Critical thresholds: Key to unlocking the door to the prevention and specific treatments for acute pancreatitis. Gut 2020, 70, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Mai, L.; Liu, Q.; Huang, F.; He, H.; Fan, W. Involvement of Mast Cells in the Pathophysiology of Pain. Front. Cell. Neurosci. 2021, 15, 665066. [Google Scholar] [CrossRef]
- Lieb, J.G., 2nd; Forsmark, C.E. Review article: Pain and chronic pancreatitis. Aliment. Pharmacol. Ther. 2009, 29, 706–719. [Google Scholar] [CrossRef]
- Moran, R.A.; James, T.; Pasricha, P. Pancreatic pain. Curr. Opin. Gastroenterol. 2015, 31, 407–415. [Google Scholar] [CrossRef]
- Hutter, M.M.; Wick, E.C.; Day, A.L.; Amy, L.; Maa, J.; Zerega, E.C.; Richmond, A.C.; Jordan, T.H.; Grady, E.F.; Mulvihill, S.J.; et al. Transient Receptor Potential Vanilloid (TRPV-1) Promotes Neurogenic Inflammation in the Pancreas Via Activation of the Neurokinin-1 Receptor (NK-1R). Pancreas 2005, 30, 260–265. [Google Scholar] [CrossRef]
- Xue, M.; Han, L.; Qian, W.; Li, J.; Qin, Y.; Xiao, Y.; Ma, Q.; Ma, J.; Shen, X. Nitric Oxide Stimulates Acute Pancreatitis Pain via Activating the NF-kB Signaling Pathway and Inhibiting the Kappa Opioid Receptor. Oxid. Med. Cell. Longev. 2020, 2020, 9230958. [Google Scholar] [CrossRef]
- Golias, C.; Charalabopoulos, A.; Stagikas, D.; Charalabopoulos, K.; Batistatou, A. The kinin system--bradykinin: Biological effects and clinical implications. Multiple role of the kinin system-bradykinin. Hippokratia 2007, 11, 124–128. [Google Scholar] [PubMed]
- McIlwrath, S.L.; Starr, M.E.; High, A.E.; Saito, H.; Westlund, K.N. Effect of acetyl-L-carnitine on hypersensitivity in acute recurrent caerulein-induced pancreatitis and microglial activation along the brain’s pain circuitry. World J. Gastroenterol. 2021, 27, 794–814. [Google Scholar] [CrossRef] [PubMed]
- Moran, R.A.; Elmunzer, B.J. Endoscopic treatment of pain in chronic pancreatitis. Curr. Opin. Gastroenterol. 2018, 34, 469–476. [Google Scholar] [CrossRef]
- Xu, G.-Y.; Winston, J.H.; Shenoy, M.; Yin, H.; Pasricha, P.J. Enhanced excitability and suppression of A-type K+ current of pancreas-specific afferent neurons in a rat model of chronic pancreatitis. Am. J. Physiol. Liver Physiol. 2006, 291, G424–G431. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Winston, J.H.; Shenoy, M.; Yin, H.; Pendyala, S.; Pasricha, P.J. Transient Receptor Potential Vanilloid 1 Mediates Hyperalgesia and Is Up-Regulated in Rats With Chronic Pancreatitis. Gastroenterology 2007, 133, 1282–1292. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Kline, R.; Deevska, G.; Ma, F.; Nikolova-Karakashian, M.; Westlund, K. Alcohol and high fat induced chronic pancreatitis: TRPV4 antagonist reduces hypersensitivity. Neuroscience 2015, 311, 166–179. [Google Scholar] [CrossRef] [Green Version]
- Liu, Q.; Ko, C.-Y.; Zheng, C.; Ye, L.; Liu, B.; Gao, H.; Huang, D.; Chou, D. Decreased Glutamatergic Synaptic Strength in the Periaqueductal Gray Contributes to Maintenance of Visceral Pain in Male Rats with Experimental Pancreatitis. Neuroscience 2019, 428, 60–69. [Google Scholar] [CrossRef]
- Black, D.; Trevethick, M. The kappa opioid receptor is associated with the perception of visceral pain. Gut 1998, 43, 312–313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogerwerf, W.A.; Shenoy, M.; Winston, J.H.; Xiao, S.-Y.; He, Z.; Pasricha, P.J. Trypsin mediates nociception via the proteinase-activated receptor 2: A potentially novel role in pancreatic pain. Gastroenterology 2004, 127, 883–891. [Google Scholar] [CrossRef]
- Gill, S.R.; Pop, M.; DeBoy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic Analysis of the Human Distal Gut Microbiome. Science 2006, 312, 1355–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinane, C.M.; Cotter, P.D. Role of the gut microbiota in health and chronic gastrointestinal disease: Understanding a hidden metabolic organ. Ther. Adv. Gastroenterol. 2013, 6, 295–308. [Google Scholar] [CrossRef] [Green Version]
- Glassner, K.L.; Abraham, B.P.; Quigley, E.M. The microbiome and inflammatory bowel disease. J. Allergy Clin. Immunol. 2020, 145, 16–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menees, S.; Chey, W. The gut microbiome and irritable bowel syndrome. F1000Research 2018, 7, 1029. [Google Scholar] [CrossRef] [Green Version]
- Sánchez-Alcoholado, L.; Ramos-Molina, B.; Otero, A.; Laborda-Illanes, A.; Ordóñez, R.; Medina, J.A.; Gómez-Millán, J.; Queipo-Ortuño, M.I. The Role of the Gut Microbiome in Colorectal Cancer Development and Therapy Response. Cancers 2020, 12, 1406. [Google Scholar] [CrossRef]
- Jiang, C.; Li, G.; Huang, P.; Liu, Z.; Zhao, B. The Gut Microbiota and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Frati, F.; Salvatori, C.; Incorvaia, C.; Bellucci, A.; Di Cara, G.; Marcucci, F.; Esposito, S. The Role of the Microbiome in Asthma: The Gut–Lung Axis. Int. J. Mol. Sci. 2018, 20, 123. [Google Scholar] [CrossRef] [Green Version]
- Aoun, A.; Darwish, F.; Hamod, N. The Influence of the Gut Microbiome on Obesity in Adults and the Role of Probiotics, Prebiotics, and Synbiotics for Weight Loss. Prev. Nutr. Food Sci. 2020, 25, 113–123. [Google Scholar] [CrossRef]
- Gupta, V.K.; Cunningham, K.Y.; Hur, B.; Bakshi, U.; Huang, H.; Warrington, K.J.; Taneja, V.; Myasoedova, E.; Davis, J.M.; Sung, J. Gut microbial determinants of clinically important improvement in patients with rheumatoid arthritis. Genome Med. 2021, 13, 149. [Google Scholar] [CrossRef]
- Boziki, M.K.; Kesidou, E.; Theotokis, P.; Mentis, A.-F.A.; Karafoulidou, E.; Melnikov, M.; Sviridova, A.; Rogovski, V.; Boyko, A.; Grigoriadis, N. Microbiome in Multiple Sclerosis; Where Are We, What We Know and Do Not Know. Brain Sci. 2020, 10, 234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, C.D. Antibiotic prophylaxis in severe acute pancreatitis. Br. J. Surg. 1996, 83, 883–884. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Huang, L.; Luo, M.; Xia, X. Bacterial translocation in acute pancreatitis. Crit. Rev. Microbiol. 2019, 45, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Roy, C.C.; Kien, C.L.; Bouthillier, L.; Levy, E. Short-chain fatty acids: Ready for prime time? Nutr. Clin. Pract. 2006, 21, 351–366. [Google Scholar] [CrossRef]
- Cook, S.; Sellin, J.H. Review article: Short chain fatty acids in health and disease. Aliment. Pharmacol. Ther. 1998, 12, 499–507. [Google Scholar] [CrossRef]
- Knudsen, K.E.B.; Lærke, H.N.; Hedemann, M.S.; Nielsen, T.S.; Ingerslev, A.K.; Nielsen, D.S.G.; Theil, P.K.; Purup, S.; Hald, S.; Schioldan, A.G.; et al. Impact of Diet-Modulated Butyrate Production on Intestinal Barrier Function and Inflammation. Nutrients 2018, 10, 1499. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, S.; Toh, H.; Hase, K.; Oshima, K.; Nakanishi, Y.; Yoshimura, K.; Tobe, T.; Clarke, J.M.; Topping, D.L.; Suzuki, T.; et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature 2011, 469, 543–547. [Google Scholar] [CrossRef]
- Levy, M.; Thaiss, C.A.; Elinav, E. Metabolites: Messengers between the microbiota and the immune system. Genes Dev. 2016, 30, 1589–1597. [Google Scholar] [CrossRef] [Green Version]
- Berg, F.F.V.D.; van Dalen, D.; Hyoju, S.K.; van Santvoort, H.C.; Besselink, M.G.; Wiersinga, W.J.; Zaborina, O.; Boermeester, M.A.; Alverdy, J. Western-type diet influences mortality from necrotising pancreatitis and demonstrates a central role for butyrate. Gut 2020, 70, 915–927. [Google Scholar] [CrossRef]
- Tan, C.; Ling, Z.; Huang, Y.; Cao, Y.; Liu, Q.; Cai, T.; Yuan, H.; Liu, C.; Li, Y.; Xu, K. Dysbiosis of Intestinal Microbiota Associated With Inflammation Involved in the Progression of Acute Pancreatitis. Pancreas 2015, 44, 868–875. [Google Scholar] [CrossRef]
- Zhu, Y.; He, C.; Li, X.; Cai, Y.; Hu, J.; Liao, Y.; Zhao, J.; Xia, L.; He, W.; Liu, L.; et al. Gut microbiota dysbiosis worsens the severity of acute pancreatitis in patients and mice. J. Gastroenterol. 2018, 54, 347–358. [Google Scholar] [CrossRef]
- van Minnen, L.P.; Timmerman, H.M.; Lutgendorff, F.; Verheem, A.; Harmsen, W.; Konstantinov, S.R.; Smidt, H.; Visser, M.R.; Rijkers, G.T.; Gooszen, H.G.; et al. Modification of intestinal flora with multispecies probiotics reduces bacterial translocation and improves clinical course in a rat model of acute pancreatitis. Surgery 2007, 141, 470–480. [Google Scholar] [CrossRef]
- Muftuoglu, M.A.T.; Isikgor, S.; Tosun, S.; Saglam, A. Effects of probiotics on the severity of experimental acute pancreatitis. Eur. J. Clin. Nutr. 2005, 60, 464–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besselink, M.G.; van Santvoort, H.C.; Buskens, E.; Boermeester, M.A.; van Goor, H.; Timmerman, H.M.; Nieuwenhuijs, V.B.; Bollen, T.L.; van Ramshorst, B.; Witteman, B.J.; et al. Probiotic prophylaxis in predicted severe acute pancreatitis: A randomised, double-blind, placebo-controlled trial. Lancet 2008, 371, 651–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaudis, H.; Pupelis, G.; Zeiza, K.; Boka, V. Early low volume oral synbiotic/prebiotic supplemented enteral stimulation of the gut in patients with severe acute pancreatitis: A prospective feasibility study. Acta Chir. Belg. 2012, 112, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.-H.; Wang, X.-H.; Peng, L.-H.; Yu, L.; Yang, Y.-S. The effects of early enteral nutrition with addition of probiotics on the prognosis of patients suffering from severe acute pancreatitis. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue 2013, 25, 224–228. [Google Scholar]
- Poropat, G.; Giljaca, V.; Hauser, G.; Štimac, D. Enteral nutrition formulations for acute pancreatitis. Cochrane Database Syst. Rev. 2015, CD010605. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.M.; Sankaran, S.J.; Plank, L.; Windsor, J.A.; Petrov, M.S. Meta-analysis of gut barrier dysfunction in patients with acute pancreatitis. Br. J. Surg. 2014, 101, 1644–1656. [Google Scholar] [CrossRef]
- Yao, Q.; Liu, P.; Peng, S.; Xu, X.; Wu, Y. Effects of immediate or early oral feeding on acute pancreatitis: A systematic review and meta-analysis. Pancreatology 2021, 22, 175–184. [Google Scholar] [CrossRef]
- Lakananurak, N.; Gramlich, L. Nutrition management in acute pancreatitis: Clinical practice consideration. World J. Clin. Cases 2020, 8, 1561–1573. [Google Scholar] [CrossRef] [PubMed]
- Blaser, A.R.; Preiser, J.-C.; Fruhwald, S.; Wilmer, A.; Wernerman, J.; Benstoem, C.; Casaer, M.P.; Starkopf, J.; van Zanten, A.; Rooyackers, O.; et al. Gastrointestinal dysfunction in the critically ill: A systematic scoping review and research agenda proposed by the Section of Metabolism, Endocrinology and Nutrition of the European Society of Intensive Care Medicine. Crit. Care 2020, 24, 224. [Google Scholar] [CrossRef] [PubMed]
- Reignier, J.; Boisramé-Helms, J.; Brisard, L.; Lascarrou, J.-B.; Hssain, A.A.; Anguel, N.; Argaud, L.; Asehnoune, K.; Asfar, P.; Bellec, F.; et al. Enteral versus parenteral early nutrition in ventilated adults with shock: A randomised, controlled, multicentre, open-label, parallel-group study (NUTRIREA-2). Lancet 2018, 391, 133–143. [Google Scholar] [CrossRef]
- Machicado, J.D.; Papachristou, G.I. Intravenous fluid resuscitation in the management of acute pancreatitis. Curr. Opin. Gastroenterol. 2020, 36, 409–416. [Google Scholar] [CrossRef]
- Working Group IAP/APA Acute Pancreatitis Guidelines. IAP/APA evidence-based guidelines for the management of acute pancreatitis. Pancreatology 2013, 13 (Suppl. 2), e1–e15. [Google Scholar] [CrossRef]
- Malbrain, M.L.N.G.; Langer, T.; Annane, D.; Gattinoni, L.; Elbers, P.; Hahn, R.G.; De Laet, I.; Minini, A.; Wong, A.; Ince, C.; et al. Intravenous fluid therapy in the perioperative and critical care setting: Executive summary of the International Fluid Academy (IFA). Ann. Intensiv. Care 2020, 10, 64. [Google Scholar] [CrossRef]
- Bentzer, P.; Griesdale, D.E.; Boyd, J.; MacLean, K.; Sirounis, D.; Ayas, N.T. Will This Hemodynamically Unstable Patient Respond to a Bolus of Intravenous Fluids? JAMA 2016, 316, 1298–1309. [Google Scholar] [CrossRef] [PubMed]
- Pandanaboyana, S.; Huang, W.; Windsor, J.A.; Drewes, A.M. Update on pain management in acute pancreatitis. Curr. Opin. Gastroenterol. 2022, 38, 487–494. [Google Scholar] [CrossRef]
- Cai, W.; Liu, F.; Wen, Y.; Han, C.; Prasad, M.; Xia, Q.; Singh, V.K.; Sutton, R.; Huang, W. Pain Management in Acute Pancreatitis: A Systematic Review and Meta-Analysis of Randomised Controlled Trials. Front. Med. 2021, 8, 782151. [Google Scholar] [CrossRef] [PubMed]
- Ona, X.B.; Comas, D.R.; Urrútia, G. Opioids for acute pancreatitis pain. Cochrane Database Syst. Rev. 2013, CD009179. [Google Scholar] [CrossRef]
- Thavanesan, N.; White, S.; Lee, S.; Ratnayake, B.; Oppong, K.W.; Nayar, M.K.; Sharp, L.; Drewes, A.M.; Capurso, G.; De-Madaria, E.; et al. Analgesia in the Initial Management of Acute Pancreatitis: A Systematic Review and Meta-Analysis of Randomised Controlled Trials. World J. Surg. 2022, 46, 878–890. [Google Scholar] [CrossRef]
- Petrov, M.S.; McIlroy, K.; Grayson, L.; Phillips, A.R.; Windsor, J.A. Early nasogastric tube feeding versus nil per os in mild to moderate acute pancreatitis: A randomized controlled trial. Clin. Nutr. 2013, 32, 697–703. [Google Scholar] [CrossRef]
- Bainbridge, D.; Martin, J.E.; Cheng, D.C. Patient-controlled versus nurse-controlled analgesia after cardiac surgery—A meta-analysis. Can. J. Anaesth. 2006, 53, 492–499. [Google Scholar] [CrossRef] [Green Version]
- Jabaudon, M.; Belhadj-Tahar, N.; Rimmelé, T.; Joannes-Boyau, O.; Bulyez, S.; Lefrant, J.-Y.; Malledant, Y.; Leone, M.; Abback, P.-S.; Tamion, F.; et al. Thoracic Epidural Analgesia and Mortality in Acute Pancreatitis: A Multicenter Propensity Analysis. Crit. Care Med. 2018, 46, e198–e205. [Google Scholar] [CrossRef]
- Vigneault, L.; Turgeon, A.F.; Côté, D.; Lauzier, F.; Zarychanski, R.; Moore, L.; McIntyre, L.A.; Nicole, P.C.; Fergusson, D. Perioperative intravenous lidocaine infusion for postoperative pain control: A meta-analysis of randomized controlled trials. Can. J. Anaesth. 2010, 58, 22–37. [Google Scholar] [CrossRef]
- Sun, Y.; Li, T.; Wang, N.; Yun, Y.; Gan, T.J. Perioperative systemic lidocaine for postoperative analgesia and recovery after abdominal surgery: A meta-analysis of randomized controlled trials. Dis. Colon. Rectum. 2012, 55, 1183–1194. [Google Scholar] [CrossRef]
- Kranke, P.; Jokinen, J.; Pace, N.L.; Schnabel, A.; Hollmann, M.W.; Hahnenkamp, K.; Eberhart, L.H.; Poepping, D.M.; Weibel, S. Continuous intravenous perioperative lidocaine infusion for postoperative pain and recovery. Cochrane Database Syst. Rev. 2015, CD009642. [Google Scholar] [CrossRef] [PubMed]
- Beaussier, M.; Delbos, A.; Maurice-Szamburski, A.; Ecoffey, C.; Mercadal, L. Perioperative Use of Intravenous Lidocaine. Drugs 2018, 78, 1229–1246. [Google Scholar] [CrossRef] [PubMed]
- Hermanns, H.; Hollmann, M.W.; Stevens, M.F.; Lirk, P.; Brandenburger, T.; Piegeler, T.; Werdehausen, R. Molecular mechanisms of action of systemic lidocaine in acute and chronic pain: A narrative review. Br. J. Anaesth. 2019, 123, 335–349. [Google Scholar] [CrossRef]
- Johns, R.A.; DiFazio, C.A.; Longnecker, D.E. Lidocaine Constricts or Dilates Rat Arterioles in a Dose-dependent Manner. Anesthesiology 1985, 62, 141–144. [Google Scholar] [CrossRef]
- Gottlieb, K.; Sherman, S. ERCP and biliary endoscopic sphincterotomy-induced pancreatitis. Gastrointest. Endosc. Clin. N. Am. 1998, 8, 87–114. [Google Scholar]
- Karlström, L.; Cassuto, J.; Jodal, M.; Lundgren, O. The Importance of the Enteric Nervous System for the Bile-Salt-Induced Secretion in the Small Intestine of the Rat. Scand. J. Gastroenterol. 1983, 18, 117–123. [Google Scholar] [CrossRef]
- Schwartz, J.J.; Lew, R.J.; Ahmad, N.A.; Shah, J.N.; Ginsberg, G.G.; Kochman, M.L.; Brensinger, C.M.; Long, W.B. The effect of lidocaine sprayed on the major duodenal papilla on the frequency of post-ERCP pancreatitis. Gastrointest. Endosc. 2004, 59, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Antkowiak, R.; Antkowiak, Ł.; Grzegorczyn, S.; Nalik-Iwaniak, K.; Kabała, N.; Arent, Z.; Warmusz-Reichman, E.; Stęplewska, K.; Domosławski, P. Efficacy of intra-arterial lidocaine infusion in the treatment of cerulein-induced acute pancreatitis. Adv. Clin. Exp. Med. 2020, 29, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, C.M.; Christophi, C. Disturbances of the microcirculation in acute pancreatitis. Br. J. Surg. 2006, 93, 518–530. [Google Scholar] [CrossRef] [PubMed]
- Jakobs, R.; Adamek, M.U.; Von Bubnoff, A.C.; Riemann, J.F. Buprenorphine or procaine for pain relief in acute pancreatitis. A prospective randomized study. Scand. J. Gastroenterol. 2000, 35, 1319–1323. [Google Scholar]
- Kahl, S.; Zimmermann, S.; Pross, M.; Schulz, H.-U.; Schmidt, U.; Malfertheiner, P. Procaine Hydrochloride Fails to Relieve Pain in Patients with Acute Pancreatitis. Digestion 2004, 69, 5–9. [Google Scholar] [CrossRef]
- Layer, P.; Bronisch, H.-J.; Henniges, U.M.; Koop, I.; Kahl, M.; Dignass, A.; Ell, C.; Freitag, M.; Keller, J. Effects of systemic administration of a local anesthetic on pain in acute pancreatitis: A randomized clinical trial. Pancreas 2011, 40, 673–679. [Google Scholar] [CrossRef]
- Bachmann, K.A.; Trepte, C.J.; Tomkötter, L.; Hinsch, A.; Stork, J.; Bergmann, W.; Heidelmann, L.; Strate, T.; Goetz, A.E.; Reuter, D.A.; et al. Effects of thoracic epidural anesthesia on survival and microcirculation in severe acute pancreatitis: A randomized experimental trial. Crit. Care 2013, 17, R281. [Google Scholar] [CrossRef] [Green Version]
- Landy, C.; Plancade, D.; Millot, I.; Gagnon, N.; Nadaud, J.; Favier, J.-C. Another use of the ultrasound-guided transversus abdominis plane block in the ED. Am. J. Emerg. Med. 2012, 30, 626–627. [Google Scholar] [CrossRef]
- Smith, D.I.; Hawson, A.; Correll, L. Transversus Abdominis Plane Block and Treatment of Viscerosomatic Abdominal Pain. Reg. Anesthesia Pain Med. 2015, 40, 731–732. [Google Scholar] [CrossRef] [PubMed]
- Ma, N.; Duncan, J.K.; Scarfe, A.J.; Schuhmann, S.; Cameron, A.L. Clinical safety and effectiveness of transversus abdominis plane (TAP) block in post-operative analgesia: A systematic review and meta-analysis. J. Anesthesia 2017, 31, 432–452. [Google Scholar] [CrossRef]
- Elkoundi, A.; Eloukkal, Z.; Bensghir, M.; Belyamani, L.; Lalaoui, S.J. Erector Spinae Plane Block for Hyperalgesic Acute Pancreatitis. Pain Med. 2018, 20, 1055–1056. [Google Scholar] [CrossRef]
- Chin, K.J.; El-Boghdadly, K. Mechanisms of action of the erector spinae plane (ESP) block: A narrative review. Can. J. Anaesth. 2021, 68, 387–408. [Google Scholar] [CrossRef] [PubMed]
- Swaroop, V.S.; Chari, S.T.; Clain, J.E. Severe acute pancreatitis. JAMA 2004, 291, 2865–2868. [Google Scholar] [CrossRef]
- Stein, C. Opioid Receptors. Annu. Rev. Med. 2016, 67, 433–451. [Google Scholar] [CrossRef]
- Zollner, C.; Stein, C. Opioids. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2007; Volume 177, pp. 31–63. [Google Scholar]
- Quigley, C. Hydromorphone for acute and chronic pain. Cochrane Database Syst. Rev. 2013, CD003447. [Google Scholar] [CrossRef]
- Chen, Z.R.; Irvine, R.J.; Somogyi, A.A.; Bochner, F. Mu receptor binding of some commonly used opioids and their metabolites. Life Sci. 1991, 48, 2165–2171. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, G.D. Buprenorphine in the Treatment of Chronic Pain. Phys. Med. Rehabilitation Clin. North Am. 2020, 31, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Gulen, B.; Dur, A.; Serinken, M.; Karcioglu, O.; Sonmez, E. Pain treatment in patients with acute pancreatitis: A randomized controlled trial. Turk. J. Gastroenterol. 2016, 27, 192–196. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.S.; Muktesh, G.; Samra, T.; Sharma, P.; Samanta, J.; Sinha, S.K.; Dhaka, N.; Yadav, T.D.; Gupta, V.; Kochhar, R. Comparison of efficacy of diclofenac and tramadol in relieving pain in patients of acute pancreatitis: A randomized parallel group double blind active controlled pilot study. Eur. J. Pain 2019, 24, 639–648. [Google Scholar] [CrossRef]
- Blamey, S.L.; Finlay, I.G.; Carter, D.C.; Imrie, C.W. Analgesia in acute pancreatitis: Comparison of buprenorphine and pethidine. Br. Med. J. (Clin. Res. Ed.) 1984, 288, 1494–1495. [Google Scholar] [CrossRef] [Green Version]
- Trujillo, K.A.; Akil, H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science 1991, 251, 85–87. [Google Scholar] [CrossRef] [PubMed]
- Juel, J.; Olesen, S.S.; Olesen, A.E.; Poulsen, J.L.; Dahan, A.; Wilder-Smith, O.; Madzak, A.; Frokjaer, J.B.; Drewes, A. Study protocol for a randomised, double-blinded, placebo-controlled, clinical trial of S-ketamine for pain treatment in patients with chronic pancreatitis (RESET trial). BMJ Open 2015, 5, e007087. [Google Scholar] [CrossRef] [Green Version]
- Rosenbaum, S.B.; Gupta, V.; Palacios, J.L. Ketamine; StatPearls: Treasure Island, FL, USA, 2022. [Google Scholar]
- Noppers, I.; Niesters, M.; Aarts, L.; Smith, T.; Sarton, E.; Dahan, A. Ketamine for the treatment of chronic non-cancer pain. Expert Opin. Pharmacother. 2010, 11, 2417–2429. [Google Scholar] [CrossRef]
- Davis, S.; Tomato, M.; Crino, L.; Colozza, M.A.; Lubansky, K.; Grignani, F. Cisplatin, etoposide, and mitomycin in the treatment of non-small cell carcinoma of the lung. A pilot study. Cancer 1986, 58, 1018–1019. [Google Scholar] [CrossRef] [PubMed]
- Malchow, R.J.; Black, I.H. The evolution of pain management in the critically ill trauma patient: Emerging concepts from the global war on terrorism. Crit. Care Med. 2008, 36, S346–S357. [Google Scholar] [CrossRef] [Green Version]
- Bell, R.F.; Kalso, E.A. Ketamine for pain management. PAIN Rep. 2018, 3, e674. [Google Scholar] [CrossRef]
- Sheehy, K.A.; Lippold, C.; Rice, A.L.; Nobrega, R.; Finkel, J.C.; Quezado, Z.M. Subanesthetic ketamine for pain management in hospitalized children, adolescents, and young adults: A single-center cohort study. J. Pain Res. 2017, 10, 787–795. [Google Scholar] [CrossRef] [Green Version]
- Schwenk, E.S.; Viscusi, E.R.; Buvanendran, A.; Hurley, R.W.; Wasan, A.D.; Narouze, S.; Bhatia, A.; Davis, F.N.; Hooten, W.M.; Cohen, S.P. Consensus Guidelines on the Use of Intravenous Ketamine Infusions for Acute Pain Management From the American Society of Regional Anesthesia and Pain Medicine, the American Academy of Pain Medicine, and the American Society of Anesthesiologists. Reg. Anesth. Pain Med. 2018, 43, 456–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bouwense, S.A.; Buscher, H.C.; Van Goor, H.; Wilder-Smith, O.H. S-Ketamine Modulates Hyperalgesia in Patients With Chronic Pancreatitis Pain. Reg. Anesthesia Pain Med. 2011, 36, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Mannion, S.; O’Brien, T. Ketamine in the management of chronic pancreatic pain. J. Pain Symptom Manag. 2003, 26, 1071–1072. [Google Scholar] [CrossRef]
- Mulder, D.; Sherlock, M.E.; Lysecki, D.L. NMDA-receptor Antagonism in Pediatric Pancreatitis: Use of Ketamine and Methadone in a Teenager With Refractory Pain. J. Craniofacial Surg. 2018, 66, e134–e136. [Google Scholar] [CrossRef] [PubMed]
- Agerwala, S.M.; Sundarapandiyan, D.; Weber, G. Ketamine Use for Successful Resolution of Post-ERCP Acute Pancreatitis Abdominal Pain. Case Rep. Anesthesiol. 2017, 2017, 7845358. [Google Scholar] [CrossRef] [Green Version]
- Cohen, S.P.; Bhatia, A.; Buvanendran, A.; Schwenk, E.S.; Wasan, A.D.; Hurley, R.W.; Viscusi, E.R.; Narouze, S.; Davis, F.N.; Ritchie, E.C.; et al. Consensus Guidelines on the Use of Intravenous Ketamine Infusions for Chronic Pain From the American Society of Regional Anesthesia and Pain Medicine, the American Academy of Pain Medicine, and the American Society of Anesthesiologists. Reg. Anesth. Pain Med. 2018, 43, 521–546. [Google Scholar] [CrossRef]
- Laulin, J.-P.; Maurette, P.; Corcuff, J.-B.; Rivat, C.G.; Chauvin, M.; Simonnet, G. The Role of Ketamine in Preventing Fentanyl-Induced Hyperalgesia and Subsequent Acute Morphine Tolerance. Obstet. Anesthesia Dig. 2002, 94, 1263–1269. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.F.; Angst, M.S.; Clark, D. Opioid-induced hyperalgesia in humans: Molecular mechanisms and clinical considerations. Clin. J. Pain 2008, 24, 479–496. [Google Scholar] [CrossRef]
- Domino, E.F.; Warner, D.S. Taming the Ketamine Tiger. Anesthesiology 2010, 113, 678–684. [Google Scholar] [CrossRef] [Green Version]
- White, M.C.; Karsli, C. Long-term use of an intravenous ketamine infusion in a child with significant burns. Pediatr. Anesthesia 2007, 17, 1102–1104. [Google Scholar] [CrossRef]
- Niesters, M.; Martini, C.; Dahan, A. Ketamine for chronic pain: Risks and benefits. Br. J. Clin. Pharmacol. 2014, 77, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, F.; Mohammed, A. Magnesium: The Forgotten Electrolyte—A Review on Hypomagnesemia. Med. Sci. 2019, 7, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elin, R.J. Magnesium metabolism in health and disease. Disease-A-Month 1988, 34, 166–218. [Google Scholar] [CrossRef] [PubMed]
- Krzewicki, J. Clinical study on magnesium and calcium level in the blood during the acute pancreatitis. Magnes. Res. 1998, 11, 19–23. [Google Scholar]
- Ryzen, E.; Rude, R.K. Low intracellular magnesium in patients with acute pancreatitis and hypocalcemia. West. J. Med. 1990, 152, 145–148. [Google Scholar]
- Mooren, F.C.; Turi, S.; Günzel, D.; Schlue, W.; Domschke, W.; Singh, J.; Lerch, M.M. Calcium-magnesium interactions in pancreatic acinar cells. FASEB J. 2001, 15, 659–672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schick, V.; Scheiber, J.A.; Mooren, F.C.; Turi, S.; Ceyhan, G.O.; Schnekenburger, J.; Sendler, M.; Schwaiger, T.; Omercevic, A.; Brandt, C.V.D.; et al. Effect of magnesium supplementation and depletion on the onset and course of acute experimental pancreatitis. Gut 2013, 63, 1469–1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fluhr, G.; Mayerle, J.; Weber, E.; Aghdassi, A.; Simon, P.; Gress, T.; Seufferlein, T.; Mössner, J.; Stallmach, A.; Rösch, T.; et al. Pre-Study protocol MagPEP: A multicentre randomized controlled trial of magnesium sulphate in the prevention of post-ERCP pancreatitis. BMC Gastroenterol. 2013, 13, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, E.T. Foreword: Opioid Use, Misuse and Abuse: The Rise and Fall of a National Opioid Epidemic. Clin. Obstet. Gynecol. 2019, 62, 1–2. [Google Scholar] [CrossRef]
- Delcher, C.; Pauly, N.; Moyo, P. Advances in prescription drug monitoring program research: A literature synthesis (June 2018 to December 2019). Curr. Opin. Psychiatry 2020, 33, 326–333. [Google Scholar] [CrossRef]
- Barlass, U.; Deshmukh, A.; Beck, T.; Bishehsari, F. Opioid use as a potential risk factor for pancreatic cancer in the United States: An analysis of state and national level databases. PLoS ONE 2021, 16, e0244285. [Google Scholar] [CrossRef] [PubMed]
- Dowell, D.; Haegerich, T.M.; Chou, R. CDC Guideline for Prescribing Opioids for Chronic Pain—United States, 2016. JAMA 2016, 315, 1624–1645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, B.U.; Butler, R.K.; Chen, W. Factors Associated With Opioid Use in Patients Hospitalized for Acute Pancreatitis. JAMA Netw. Open 2019, 2, e191827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salsitz, E.A. Chronic Pain, Chronic Opioid Addiction: A Complex Nexus. J. Med. Toxicol. 2015, 12, 54–57. [Google Scholar] [CrossRef] [Green Version]
- Volkow, N.D.; McLellan, A.T. Opioid Abuse in Chronic Pain—Misconceptions and Mitigation Strategies. N. Engl. J. Med. 2016, 374, 1253–1263. [Google Scholar] [CrossRef]
- Mercadante, S.; Arcuri, E.; Santoni, A. Opioid-Induced Tolerance and Hyperalgesia. CNS Drugs 2019, 33, 943–955. [Google Scholar] [CrossRef]
- Lee, M.; Silverman, S.M.; Hansen, H.; Patel, V.B.; Manchikanti, L. A comprehensive review of opioid-induced hyperalgesia. Pain Physician 2011, 14, 145–161. [Google Scholar] [CrossRef]
- Kasmin, F.E.; Siegel, J.H. Endoscopic Therapy for Chronic Pancreatitis. Surg. Clin. N. Am. 2001, 81, 421–430. [Google Scholar] [CrossRef]
- Delhaye, M.; Arvanitakis, M.; Verset, G.; Cremer, M.; Devière, J. Long-term clinical outcome after endoscopic pancreatic ductal drainage for patients with painful chronic pancreatitis. Clin. Gastroenterol. Hepatol. 2004, 2, 1096–1106. [Google Scholar] [CrossRef]
- Rösch, T.; Daniel, S.; Scholz, M.; Huibregtse, K.; Smits, M.; Schneider, T.; Ell, C.; Haber, G.; Riemann, J.-F.; Jakobs, R.; et al. Endoscopic Treatment of Chronic Pancreatitis: A Multicenter Study of 1000 Patients with Long-Term Follow-Up. Endoscopy 2002, 34, 765–771. [Google Scholar] [CrossRef]
- Dumonceau, J.-M.; Devière, J.; Le Moine, O.; Delhaye, M.; Vandermeeren, A.; Baize, M.; Van Gansbeke, D.; Cremer, M. Endoscopic pancreatic drainage in chronic pancreatitis associated with ductal stones: Long-term results. Gastrointest. Endosc. 1996, 43, 547–555. [Google Scholar] [CrossRef]
- Will, U.; Reichel, A.; Fueldner, F.; Meyer, F. Endoscopic ultrasonography-guided drainage for patients with symptomatic obstruction and enlargement of the pancreatic duct. World J. Gastroenterol. 2015, 21, 13140–13151. [Google Scholar] [CrossRef]
- Cahen, D.L.; Gouma, D.J.; Nio, Y.; Rauws, E.A.J.; Boermeester, M.A.; Busch, O.R.; Stoker, J.; Laméris, J.S.; Dijkgraaf, M.G.W.; Huibregtse, K.; et al. Endoscopic versus surgical drainage of the pancreatic duct in chronic pancreatitis. N. Engl. J. Med. 2007, 356, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Díte, P.; Ružicka, M.; Zboril, V.; Novotný, I. A Prospective, Randomized Trial Comparing Endoscopic and Surgical Therapy for Chronic Pancreatitis. Endoscopy 2003, 35, 553–558. [Google Scholar]
- Drewes, A.M.; Bouwense, S.A.W.; Campbell, C.M.; Ceyhan, G.O.; Delhaye, M.; Demir, I.E.; Garg, P.K.; Van Goor, H.; Halloran, C.; Isaji, S.; et al. Guidelines for the understanding and management of pain in chronic pancreatitis. Pancreatology 2017, 17, 720–731. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; de la Iglesia-García, D.; Baston-Rey, I.; Calviño-Suarez, C.; Lariño-Noia, J.; Iglesias-Garcia, J.; Shi, N.; Zhang, X.; Cai, W.; Deng, L.; et al. Exocrine Pancreatic Insufficiency Following Acute Pancreatitis: Systematic Review and Meta-Analysis. Dig. Dis. Sci. 2019, 64, 1985–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, J.; Layer, P. Human pancreatic exocrine response to nutrients in health and disease. Gut 2005, 54 (Suppl. 6), 1–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]

| Pancreatitis Flair Pathophysiology | |
|---|---|
| Trypsin | Rapidly increasing trypsin, a product of pancreatic acinar cells typically secreted into the pancreatic duct, induces early pancreatic damage [18] |
| Calcium Signaling | Endoplasmic reticulum surface ryanodine receptors (RyR) and membrane-bound Store-operated channels (SOC) increase acinar cytoplasmic calcium [22,25,26] |
| Calcineurin | Calcineurin plays a role downstream of calcium influx and upstream of trypsin activation in acute pancreatitis [27,28,29] |
| Colocalazation and Autophagy | Vacuole fusion with lysosomes activates trypsinogen—protease cathepsin B has been shown to play a role in premature trypsinogen activation [35,36] |
| Inflammatory Pathway | NF-κB cascade is initiated by calcium signaling and leads to increasing oxidative stress and apoptosis [37,38,39,40] |
| Neuropathic Pain | Dilation of intraparenchymal nerves with background fibrosis suggests that increased axonal diameter is likely responsible for initiating neuropathy [49,50,51,52,53,54] |
| Inflaammatory Pain | Stimuli destroy acinar cells and digestive enzymes and inflammatory mediators are released in the vicinity including K+, H+, ATP, histamine, substance P (SP), bradykinin, prostaglandins and norepinephrine [60,61,63] |
| Visceral Pain | Increased activity of dorsal root ganglia and relatively depolarized resting state and increased expression of TRPV1 [64,71,72] |
| Gut Flora Dysbiosis | Patients with AP are more likely to have gut dysbiosis, with higher populations of Enterobacteriaceae and Enterococcus and lower Bifidobacteria [95] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beiriger, J.; Khan, A.; Yan, B.; Ross, H.; Wang, M.; Carducci, M.; Parra, N.S.; Chowdhury, S.; Erwin, R.; Forrest, P.; et al. Comprehensive Review of Acute Pancreatitis Pain Syndrome. Gastrointest. Disord. 2023, 5, 144-166. https://doi.org/10.3390/gidisord5020014
Beiriger J, Khan A, Yan B, Ross H, Wang M, Carducci M, Parra NS, Chowdhury S, Erwin R, Forrest P, et al. Comprehensive Review of Acute Pancreatitis Pain Syndrome. Gastrointestinal Disorders. 2023; 5(2):144-166. https://doi.org/10.3390/gidisord5020014
Chicago/Turabian StyleBeiriger, Jacob, Adnan Khan, Brian Yan, Heather Ross, Makala Wang, Michael Carducci, Natalia Salinas Parra, Salil Chowdhury, Ryan Erwin, Paul Forrest, and et al. 2023. "Comprehensive Review of Acute Pancreatitis Pain Syndrome" Gastrointestinal Disorders 5, no. 2: 144-166. https://doi.org/10.3390/gidisord5020014